

Abstract
Retinogeniculate connections undergo postnatal refinement in the developing visual system. Here we report that non-ion channel epilepsy gene LGI1 (leucine-rich glioma-inactivated), mutated in human autosomal dominant lateral temporal lobe epilepsy (ADLTE), regulates postnatal pruning of retinal axons in visual relay thalamus. By introducing an ADLTE-associated truncated mutant LGI1 (836delC) or excess full-length LGI1 into transgenic mice, we found that mutant LGI1 blocks, whereas excess LGI1 accelerates, retinogeniculate axon pruning. The normal postnatal single fiber strengthening was arrested by mutant LGI1 and, contrastingly, was enhanced by excess wild-type LGI1. The maximum response of the retinogeniculate synapses, conversely, remained the same in mature LGI1 transgenic mice, indicating that mutant LGI1 blocks, whereas excess wild-type LGI1 promotes, weak axon fiber elimination. Heterozygous deletion of the LGI1 gene, as found in ADLTE patients, inhibited postnatal retinogeniculate synapse elimination, an effect similar to the ADLTE truncated mutant LGI1. The results identify sensory axon remodeling defects in a sensory aura-associated human epilepsy disorder.
Introduction
Brain circuit formation is a coordinated multistage process. Whereas the early stages of generating spines (spinogenesis) and synapses (synaptogenesis) are essential to the assembly of neural circuits, the later stages of activity-dependent elimination of early-formed redundant and inefficient synapses is central to creating the mature brain circuitry that can respond appropriately to sensory inputs (Lichtman and Colman, 2000; Luo and O'Leary, 2005; Kano and Hashimoto, 2009).
Synapse elimination has been widely studied using long-term time-lapse imaging methods in the developing neuromuscular junction and autonomic ganglia because of their large size (Gan et al., 2003; Walsh and Lichtman, 2003). In the CNS, however, elimination of initially exuberant synapses has only been investigated in a subset of central synapses that undergo drastic postnatal synapse refinement using mostly electrophysiological methods (Kano and Hashimoto, 2009), although morphological means have been used as well (Cheng et al., 2010; Dhande et al., 2011). In the cerebellum, Purkinje cells are initially innervated by multiple weak climbing fibers that originate from the inferior olive. This multiple axon innervation undergoes postnatal pruning so that the majority of mature Purkinje cells become innervated by only a single strengthened climbing fiber (Sugihara, 2006). The synapses between retinal ganglion cells (RGCs) and thalamic relay neurons of the dorsal lateral geniculate nucleus (dLGN) also undergo postnatal refinement (Guido, 2008; Bickford et al., 2010; Hong and Chen, 2011). Before eye opening, typical dLGN relay neurons receive approximately one dozen retinogeniculate inputs (Chen and Regehr, 2000; Jaubert-Miazza et al., 2005). With maturation, most dLGN relay neurons are innervated by only one to three RGC axonal inputs (Chen and Regehr, 2000). A similar postnatal refinement from many to only a few axonal inputs has also been observed recently in the somatosensory ventral basal nucleus of the thalamus (Arsenault and Zhang, 2006). Axon input refinement in the thalamic auditory relay nuclei has not yet been reported.
Although it is well established that central synapse elimination is activity dependent, detailed mechanisms underlying synapse elimination are still essentially unknown (Flavell and Greenberg, 2008). A recently discovered human epilepsy gene, LGI1 [leucine-rich glioma-inactivated; mutated to cause human autosomal dominant lateral temporal lobe epilepsy (ADLTE)] encodes a secreted protein that localizes to glutamatergic synapses (Kalachikov et al., 2002; Senechal et al., 2005; Fukata et al., 2006; Schulte et al., 2006; Nobile et al., 2009; Anderson, 2010). ADLTE is characterized by partial seizures and sensory auras that are predominantly auditory, although visual, vestibular, epigastric, and even psychic hallucinations are also reported (Nobile et al., 2009). We recently showed that LGI1 protein coordinates the functional maturation and dendritic pruning of glutamatergic synapses in the hippocampus during postnatal brain development (Zhou et al., 2009). Importantly, this same study also showed that LGI1 expression greatly increases postnatally, at the time period when visual thalamus retinogeniculate axons are remodeled (Hong and Chen, 2011). Thus, we asked whether LGI1 mediates this major axonal elimination event in the visual thalamus. Using two transgenic mouse lines [one carrying an ADLTE-associated truncated mutant form of LGI1 (mLGI1), 835delC; and the other overexpressing (OE) wild-type (WT) LGI1 protein] and one heterozygous LGI1 knock-out mouse, we found that LGI1 plays an important role in promoting the postnatal pruning of retinogeniculate fibers in the dLGN. LGI1 protein overexpression accelerated retinogeniculate axon pruning, resulting in fewer retinogeniculate fibers innervating each mature dLGN neuron. ADLTE truncated mLGI1 and heterozygous LGI1 knock-out, in contrast, inhibited axon elimination in the dLGN.
Materials and Methods
Animals
Mice were studied at ages postnatal day 8 (P8) to P46. mLGI1 (836delC) and WT LGI1 transgenic mice and their littermate controls were created as described previously (Zhou et al., 2009). These transgenic mice were generated from an FVB genetic background and were subsequently bred to a C57BL/6 for six or more generations. Heterozygous LGI1+/− mice were derived from a 75% C57BL/6 and 25% 129S2Sv/pas hybrid background (Chabrol et al., 2010). All protocols were approved by the Beth Israel Deaconess Medical Center Institutional Animal Care and Use Committee.
dLGN slice preparation
Angled parasagittal brain slices containing the dLGN were obtained from mice of various ages using a protocol adapted from Chen and Regehr (2000). Briefly, the brain was removed rapidly and immersed in 0–4°C, oxygenated cutting solution containing the following (in mm): 231 sucrose, 5 KCl, 26 NaHCO3, 1.25 NaH2PO4, 25 glucose, 1 CaCl2, and 5 MgSO4. Two sections were performed at ∼10° to the sagittal plane either side of the cerebral longitudinal fissure (Fig. 1A, left) and angled outward by 15–25° in the mediolateral plane (Fig. 1A, middle). The medial aspect of the brain was then glued onto the cutting stage of a vibratome (Vibratome 3000), and 250–300 μm sections were obtained. To prevent excitatory disynaptic connections (recurrent excitation between geniculate and corticothalamic neurons), the cortex was separated from the thalamus with a razor incision through the white matter tracts (Chen and Regehr, 2000). Slices were allowed to recover ∼30 min in oxygenated recording saline (in mm: 125 NaCl, 3 KCl, 26 NaHCO3, 1.25 NaH2PO4, 25 glucose, 2 CaCl2, and 1 MgCl2) at 35°C and subsequently for ≥1 h at room temperature before electrophysiological recordings.
Figure 1.
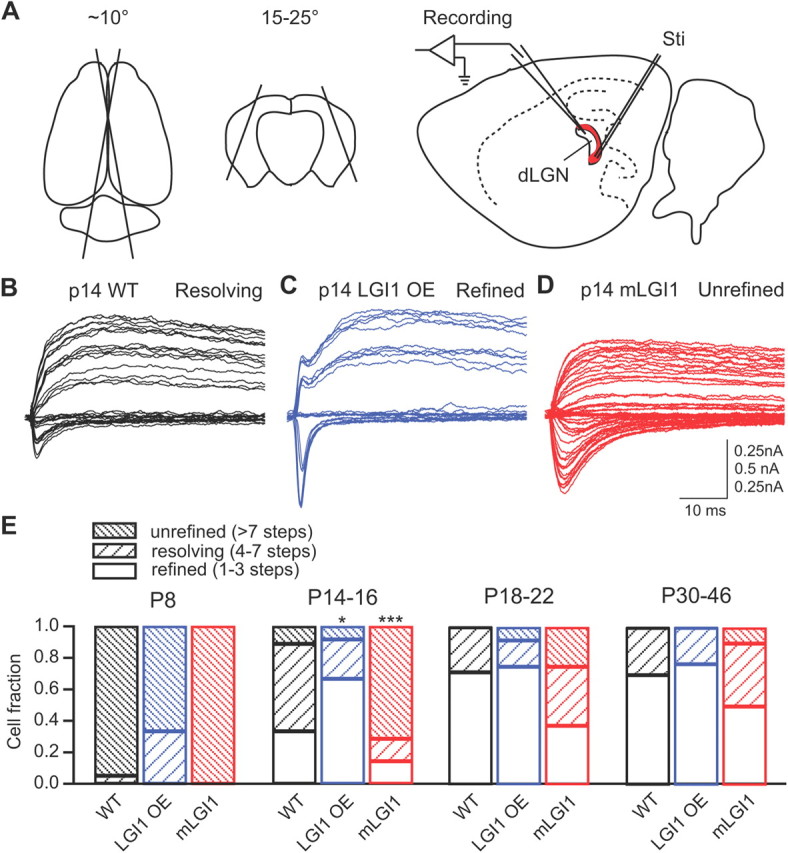
mLGI1 blocks, whereas excess WT promotes, retinogeniculate axon pruning during postnatal development. A, Schematics showing preparation of angled sagittal slices and recording of retinogeniculate synaptic transmission. Dorsal (left) and coronal (middle) views of the brain showing the position and angle of the two cuts made to produce brain blocks for subsequent slice cutting. We stimulated the optical tract (red) and recorded evoked responses in the dLGN (right). B, C, Representative traces showing retinogeniculate fiber refinement in P14 WT (B), LGI1 OE (C), and mLGI1 (D) mice. E, Stacked column histograms showing composition of unrefined (>7 steps), resolving (4–7 steps), and refined (1–3 steps) retinogeniculate inputs in developing WT, LGI1 OE, and mLGI1 mice (n = 7 to 20). *p < 0.05, ***p < 0.001.
Electrophysiology
Whole-cell recordings of dLGN relay neurons.
Slices were transferred to a recording chamber continuously perfused with a bubbled 95%O2/5% CO2 recording saline containing 20 μm bicuculline at 35°C. Dorsal lateral geniculate relay neurons were identified under infrared differential interference contrast optics based on their larger size (15–25 μm in diameter) and by the number of dendrites coming off of the cell body (three or more dendrites) (Chen and Regehr, 2000) and were patched with 3–5 MΩ glass pipettes filled with artificial intracellular fluid (in mm: 100 CsCH3SO3, 20 KCl, 10 HEPES, 4 Mg-ATP, 0.3 Tris-GTP, 7 Tris2-phosphocreatine, and 3 QX-314, pH 7.3). An extracellular glass electrode filled with recording saline was placed at the optic tract 200 μm away from the recording site. The optic tract was stimulated using current-pulse stimuli (duration, 180 μs; amplitude, 0.2–40 μA; frequency, 0.1 Hz for baseline condition) delivered via a constant-current stimulator. The membrane potential of the geniculate neuron was clamped at either −70 or +40 mV during stimulation of the optic tract.
Single-fiber recordings.
To establish a minimal stimulation, we first identified the highest stimulus that gave all failures. Then we slightly increased the stimulation intensity to lower the failure rate. In addition, we required that the EPSC latency remain stable throughout the experiments (<20% fluctuations).
dLGN microdissection and protein extraction
dLGN and brain tissue were isolated from P8, P17, P28, and adult CD1 mice. Four mice for each age group were used and treated separately. Mice were anesthetized with isoflurane. P28 and adult mice were transcardially perfused with cutting solution (see above, dLGN slice preparation). P8 and P17 mice were killed after anesthesia and were not perfused. Brains were removed rapidly and immersed in 0–4°C oxygenated cutting solution. Four to 6 coronal brain sections of 250–300 μm were made with a vibratome, corresponding to the location of dLGN (around bregma −1.60 to −2.90 mm in adult mice). The dLGNs were microdissected with ophthalmic scissors in cutting solution on ice. Remaining tissue from the slices after dLGN dissection was used as control samples (brain). The tissues were collected in 1.5 ml microfuge tubes and frozen on dry ice before additional processing.
Protein extracts from dLGN and brain tissues were prepared in RIPA buffer (50 mm Tris pH7.4, 150 mm NaCl, 1% Triton X-100, 0.1% SDS, 0.5% Na-deoxycholate, 1 mm EDTA, 1 mm PMSF, and protease inhibitors). Thirty to 40 μl of buffer for dLGN and 300–600 μl of buffer for brain slices were added, and the tissue was homogenized with a plastic pestle on ice. The homogenate was centrifuged at 13,200 rpm for 25 min at 4°C, and the supernatant was collected. Protein amounts were determined with BCA Protein Assay Kit and Nanodrop 1000 Spectrophotometer (both from Thermo Fisher Scientific).
Western blot
Mouse brain samples were obtained and homogenized using a chilled Tenbroeck tissue homogenizer containing 2 ml of chilled STE buffer (in mm: 320 sucrose, 20 Tris, 2 EDTA, and 200 μg/ml PMSF, pH 8.0) with protease and phosphatase inhibitors (1 μg/ml leupeptin, 1 mg/ml pepstatin, 1 μg/ml aprotinin, 1 mm NaF, and 1 mm Na3VO4). The homogenate was then centrifuged at 14,000 × g for 1 h at 4°C, and the supernatant was removed. The pellet was resuspended in chilled TET buffer (in mm: 20 Tris, 1 EDTA, and 1.3% Triton X-100, pH 8.0) with protease and phosphatase inhibitors and centrifuged at 100,000 × g for 1 h. Ten percent SDS-PAGE was used to separate protein samples. After electrophoresis, the gels were transferred to nitrocellulose membranes using a constant voltage of 70–80 V for 90 min. The membranes were then blocked in 5% milk in TTBS (50 mm Tris-HCl, pH 8, 150 mm NaCl, and 0.05% Tween 20) for 0.5 h at room temperature on a rocker and incubated in the primary antibody overnight on a rocker at 4°C. Primary antibodies were made with 5% milk in TTBS solutions [LGI1, 1:250, Santa Cruz Biotechnology (sc-28238) directed against the N-terminal amino acids 35–90, used in Fig. 3A; LGI1, 1:1000, Abcam (ab30868), rabbit polyclonal, used in Figure 3C; β-actin, 1:1000, Santa Cruz Biotechnology (sc-47778), mouse monoclonal]. Our previous studies confirmed specificity for these LGI1 antibodies with a complete loss of the single 64 kDa band in LGI1−/− mice (Chabrol et al., 2010). After primary antibody incubation, the membranes were rinsed three times in TTBS before being incubated with horseradish peroxidase-conjugated secondary antibody (1:10,000 in 5% milk in TTBS) for 1 h on a rocker at room temperature. After being washed in TTBS four times on a rocker at room temperature, the membranes were then developed using either ECL or ECL-Plus Western blotting detection reagents.
Figure 3.

Postnatal developmental increases of LGI1 protein are accelerated and magnified in LGI1 OE mice. A, Representative Western blots of full-length LGI1 protein in P8–P35 mice. The 64 kDa LGI1 protein band is indicated with an arrow in the P8 Western blots (A, C). Protein levels were adjusted to their corresponding β-actin levels and normalized to a positive control (+). B, Quantification of LGI1 protein density (normalized to the positive control, “+”) in WT, mLGI1, and LGI1 OE transgenic mice showing significant increases of LGI1 in LGI1 OE mice at P14 and P35 (n = 5 to 8). The top shows LGI1 Western blots from six LGI1 OE mice and four WT mice at P14. C, Western blots for LGI1 protein from P8, P17, P28, and adult (older than P50) WT mice. Lanes D1–D4 represent extracts obtained from microdissected dLGN; lanes B1–B4 represent extracts from the remaining brain slice tissue after dLGN microdissection. D, Quantification of the Western blots for LGI1 protein amount relative to β-actin (n = 4). LGI1 expression increases with age in both dLGN (p < 0.01) and surrounding brain regions (p < 0.001). *p < 0.05.
Statistics
Data are reported as means ± SEM. Two-tailed unpaired Student's t test was used to compare means of two groups. One-way ANOVA with Dunnett's post hoc test was used to compare multiple group means. Two-way ANOVA with Bonferroni's correction was used to compare group variance. χ2 test was used to compare the composition of different types of retinogeniculate synapses in WT and LGI1 transgenic and heterozygous mice. Single-fiber amplitude distributions were compared using Kolmogorov–Smirnov test.
Results
Bidirectional regulation of retinogeniculate pruning in LGI1 transgenic mice
To test the role of LGI1 in axon pruning, we stimulated the optical tract in angled sagittal brain slices while recording from relay neurons in the dLGN (Fig. 1A). We confirmed that there is a postnatal age-dependent decrease of axons innervating dLGN relay neurons in WT mice (Chen and Regehr, 2000). Stimulating the optical tract of P8 WT mice with increasing stimulation intensities resulted in graded responses in >90% of relay neurons, indicating that the majority of dLGN relay neurons are innervated by multiple retinogeniculate axons (unrefined; step number, more than seven). During the second and the third postnatal weeks, the number of unrefined relay neurons exhibiting graded responses decreased and the number showing distinct step responses (refined; step number, one to three) or mixed responses (resolving; step number, four to seven) was increased. By the end of the third postnatal week (P18–P22), ∼70% of the relay neurons were innervated by one to three axons. This developmental refinement of the axonal connections between the optical tract and dLGN relay neurons was accelerated in mice overexpressing WT LGI1 and was delayed in mice expressing the dominant-negative mutant form of LGI1 (835delC). Approximately 60% of dLGN relay neurons showed fully refined, one to three step responses to optical tract stimuli by P14–P16 in LGI1 OE mice (Fig. 1C,E). In comparison, only ∼40% of WT and 15% of mLGI1 dLGN relay neurons displayed fully refined, one to three step responses at P14–P16 (Fig. 1E). Instead, WT (Fig. 1B) and mLGI1 (Fig. 1D) relay neurons showed predominantly mixed and graded responses, respectively, at P14–P16. χ2 test revealed significant differences in the composition of cells received unrefined, resolving, and refined retinogeniculate inputs in both P14–P16 LGI1 OE (χ2 = 6.12, df = 2, p < 0.05) and mLGI1 (χ2 = 25.86, df = 2, p < 0.001) mice. The delayed elimination of dLGN axon inputs extended into young adulthood in mLGI1 mice (Fig. 1E). The results indicate developmental axon pruning in dLGN is accelerated in LGI1 OE mice and delayed in mLGI1 mice.
Interestingly, the proportions of cells receiving fully refined, one to three step inputs are similar in both P30–P46 LGI1 OE and WT mice (Fig. 1E). However, among the subset of neurons with fully refined, one to three step inputs, LGI1 OE mice display reduced numbers of axon inputs at P30–P46 (Fig. 2). This result indicates that excessive LGI1 protein causes overpruning of optical tract axon inputs to dLGN. In P30–P46 mLGI1 mice, a significant portion (∼50%) of dLGN relay neurons still received more than three optical inputs (Fig. 1E), although the average number of axons innervating the subset of neurons with fully refined, one to three step inputs was similar to WT mice (Fig. 2). Together, the results indicate LGI1 promotes, whereas mLGI1 inhibits, axon elimination in dLGN.
Figure 2.
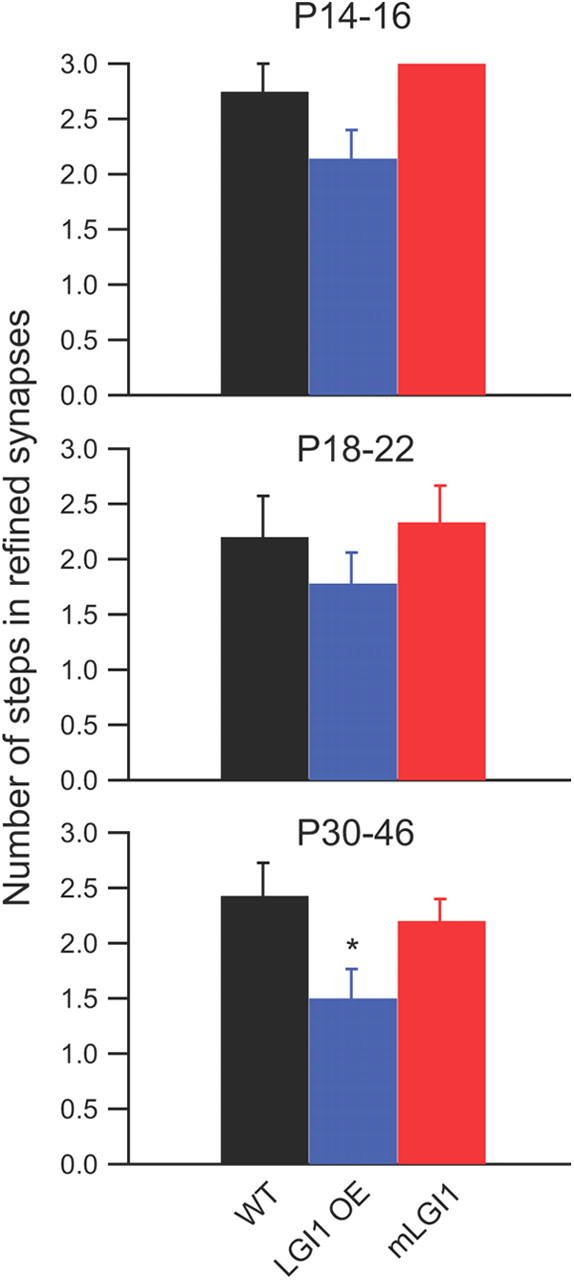
Excess WT LGI1 significantly decreased step number in refined synapses. Refined dLGN retinogeniculate synapses are defined as receiving one to three inputs. Quantification of the number of step responses in these refined synapses shows a significant decrease in LGI1 OE mice (n = 7 to 19). *p < 0.05.
Previous studies established LGI1 expression increases during postnatal development in the rodent brain (Zhou et al., 2009; Chabrol et al., 2010) (Fig. 3A). To test whether the accelerated axonal pruning in the dLGN of LGI1 OE mice correlates to an accelerated increase of LGI1 levels during early postnatal development, we examined LGI1 protein levels. To compare LGI1 levels across different blots, a common positive control was included in all immunoblots and used to normalize the data (Fig. 3A,B, +). LGI1 was significantly increased at P14 in LGI1 OE transgenic compared with WT mice (Fig. 3B). In contrast, both P14 WT and mLGI1 mice showed low LGI1 expression that was comparable with P8 animals. This magnified increase of LGI1 protein in young LGI1 OE mice persisted in the adulthood with an ∼50% increase of LGI1 protein in adult LGI1 OE mice (Fig. 3B). The timing of LGI1 overexpression correlates well with timing of accelerated axon elimination and the persistent reduction of total axon inputs to refined dLGN neurons of LGI1 OE mice. To further correlate LGI1 expression to postnatal retinogeniculate synapse pruning, we measured LGI1 levels specifically in dLGN (Fig. 3C). Although LGI1 levels were a little lower in the dLGN compared with the rest of the brain, the same age-dependent increase of LGI1 protein was observed (Fig. 3D). Together, the results indicate that LGI1 is expressed in the dLGN and is developmentally upregulated during retinogeniculate synapse elimination.
LGI1 promotes weak retinogeniculate fiber elimination
Axon elimination in the developing dLGN is associated with a loss of weak axonal inputs. We examined whether the bidirectional regulation of axon elimination by LGI1 transgenes also manifests as a preservation or excess elimination of weak optical tract axon inputs in dLGN. By measuring the minimum fiber stimulated AMPA receptor-mediated EPSC in fully refined (one to three steps) dLGN neurons, we observed that the minimum AMPA receptor-mediated currents are greatly increased in LGI1 OE mice and significantly decreased in mLGI1 mice (Fig. 4A). In contrast, maximum AMPA receptor-mediated currents were similar across the genotypes (Fig. 4B). This finding that mLGI1 causes dLGN relay neurons to retain weak retinal axon inputs suggests that weak axon pruning is blocked in mLGI1 transgenic mice. In contrast, excess LGI1 protein increased the minimum EPSC, suggesting that it promotes pruning of weak axon inputs.
Figure 4.

LGI1 regulates minimum AMPA receptor-mediated currents at refined retinogeniculate synapses. A, B, Histograms showing minimum (A) and maximum (B) AMPA receptor-mediated currents in mature retinogeniculate synapses of WT, mLGI1, and LGI1 OE mice (n = 7 to 19). Recordings from refined dLGN neurons (1–3 steps). *p < 0.05.
To further test whether weak optical tract axon inputs are preserved in mLGI1and overpruned in LGI1 OE mice relative to WTs, we examined the amplitude distribution of minimum EPSCs recorded from dLGN relay neurons in various age groups of WT and LGI1 transgenic mice. All three types of animals showed a progressive increase of the fraction of dLGN relay neuron fibers with a large EPSC to minimal optical tract stimulation during postnatal development (Fig. 5A–C). However, when compared with WT (Fig. 5A), LGI1 overexpression increased the number of large EPSC responses to minimum optical tract stimulation (Fig. 5B). In contrast, the majority of dLGN neurons in mLGI1 mice responded to minimum optical tract stimulation with predominantly small EPSCs (Fig. 5C), indicating a preservation of weak single fibers. Cumulative amplitude histograms clearly reveal the bidirectional effect of the LGI1 transgenes on single-fiber strength during postnatal development (Fig. 5D). These results indicate that single-axon strength is increased by excess LGI1 and decreased by dominant-negative LGI1.
Figure 5.

LGI1 regulates single-fiber strengthening during postnatal development. A–C, Histograms of single-fiber AMPA receptor-mediated currents in 100 pA bins for WT (A), LGI1 OE (B), and mLGI1 (C) mice in three age groups (P14–16, P18–22, P30–46). D, Cumulative frequency (CF) plots of single-fiber AMPA receptor-mediated currents for WT (black), LGI1 OE (blue), and mLGI1 (red) mice in three age groups (n = 7 to 13).
Halving LGI1 gene copy number inhibits postnatal retinogeniculate fiber elimination
We recently created a heterozygous knock-out mouse model of ADLTE that displays an increased susceptibility to auditory-stimulus kindled audiogenic seizures analogous to the susceptibility to auditory stimulus (e.g., telephone ring) induced seizures in ADLTE humans (Chabrol et al., 2010). The bidirectional regulation of axonal pruning in dLGN by LGI1 transgenes had suggested a dominant-negative effect of mLGI1 (Zhou et al., 2009). To further establish that LGI1 is involved in retinogeniculate axonal pruning, we investigated whether heterozygous LGI1 gene deletion causes abnormal pruning defects similar to mLGI1. More than 50% of dLGN relay neurons received at least four retinogeniculate inputs in adult LGI1+/− mice, whereas <30% of dLGN cells showed similar multiple axon innervation in adult WT littermates (Fig. 6A,B). The percentages of cells that received at least four retinogeniculate inputs were indeed very similar in both adult LGI1+/− and mLGI1 mice (LGI1+/−, 54.5%; mLGI1, 50%). In addition, cumulative single-fiber amplitude distributions revealed an increase of weak retinogeniculate axon inputs in LGI1+/− mice (Fig. 6C), as observed in mLGI1 mice. In contrast, AMPA/NMDA receptor current ratio was unaffected (Fig. 6D). Although homozygous LGI1 deletion is likely to be more severe, these mice die prematurely beginning at 10 d of age, precluding studies of this postnatal developmental process. These results indicate that postnatal axon pruning is defective in LGI1+/− mice, further confirming the role of LGI1 in axonal elimination.
Figure 6.
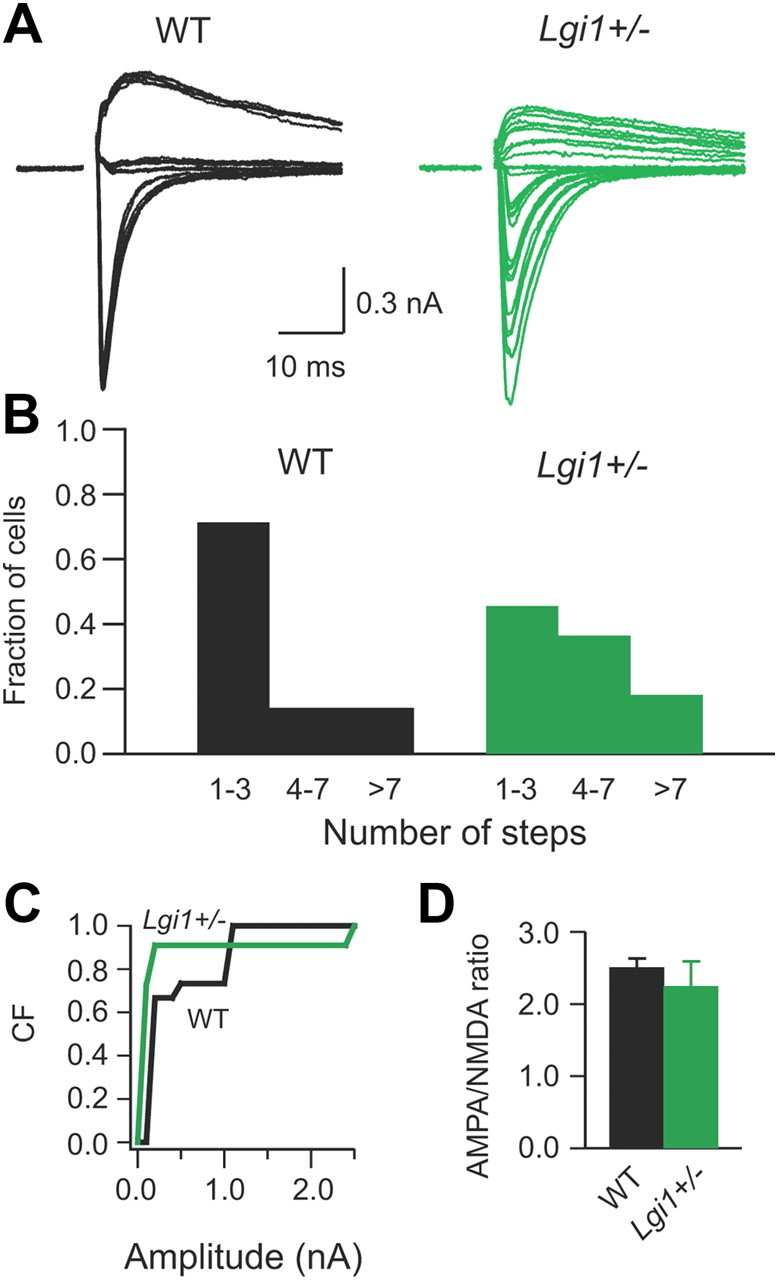
Heterozygous deletion of LGI1 blocks complete retinogeniculate synapse pruning. A, Representative traces showing evoked retinogeniculate synaptic currents in response to optical tract stimulation with increasing stimulation intensities in WT and LGI1 heterozygous mice. B, Histograms of relative fraction of unrefined (>7 step), resolving (4–7 steps), and refined (1–3 steps) retinogeniculate synapses for P30–P46 WT and LGI1 heterozygous mice (n = 7 to 11). C, Cumulative frequency (CF) plots of single-fiber AMPA receptor-mediated currents for WT (black) and LGI1 heterozygous (green) mice (n = 7 to 11). D, Plot of AMPA receptor/NMDA receptor current ratio for P30–P46 WT and LGI1 heterozygous mice (n = 7 to 11).
Discussion
We have established that LGI1 is a key molecule in axonal elimination during postnatal development. mLGI1 and heterozygous LGI1 blocked axonal pruning in the dLGN, resulting in a larger fraction of unrefined and resolving retinogeniculate synapses in these mice. In contrast, excess full-length LGI1 accelerated synaptic pruning, which is correlated to an accelerated upregulation of LGI1 protein in LGI1 OE mice.
Three phases of retinogeniculate synapse refinement have been observed in mice (Hong and Chen, 2011). During phase I (P0–P8), the optical tract projects to the dLGN and there is eye-specific segregation of retinogeniculate inputs. Retinal waves are essential for this eye-specific segregation because animals lacking an early phase of retinal activity exhibit failed segregation (Torborg and Feller, 2005). A typical dLGN relay neuron receives ∼12 RGC inputs at the end of this phase. Phase II (P8–P16, at approximately eye opening) is the key refinement period during which more than half of the retinogeniculate synapses are eliminated and the remaining synapses are strengthened eightfold. Spontaneous retinal activity is critical in driving the drastic retinogeniculate pruning (Hooks and Chen, 2006) and in the maintenance of eye-specific segregation (Demas et al., 2006) during this phase. Phase III (P20–P34), starting ∼1 week after eye opening, is the maintenance period of refined retinogeniculate circuitry (Hooks and Chen, 2006, 2008). During this phase, vision-dependent stabilization of the refined circuitry leads to an additional elimination of one to three RGC inputs. Visual deprivation during this phase causes weakening of individual retinogeniculate axon inputs and reversion to an increased number of axonal inputs (Hooks and Chen, 2006, 2008). Our results suggest that LGI1 is a key molecule promoting synapse refinement in the second phase and possibly in maintaining this state during the third phase. The most significant differences in retinogeniculate synapse pruning were observed in the P14–P16 LGI1 transgenic mice compared with WT (Fig. 1). The role of LGI1 was diminished in the third-phase refinement period, but in refined dLGN neurons (one to three steps), excess LGI1 did significantly reduce the average step number (Fig. 2, P30–46) and the minimum AMPA current amplitude was significantly increased with excess LGI1 and reduced with mLGI1 (Fig. 4). Furthermore, the distribution of minimum fiber amplitudes was altered during the later ages (Fig. 5, P18–22 and P30–46). These observations suggest that LGI1 may continue to play a role in maintaining the refined state of the circuit. This is in contrast to visual experience-dependent insertion of GluR1 subunits (Kielland et al., 2009) and the transcription regulator MeCP2 (Noutel et al., 2011), both of which appear to preferentially affect the third phase of retinogeniculate circuitry refinement. Thus, different synapse refinement phases may be mediated by different set of molecules.
Our previous work indicated that LGI1 was involved in developmental downregulation of both presynaptic and postsynaptic functions (Zhou et al., 2009). At the cellular level, the downregulation of presynaptic release probability and postsynaptic NR2B subunit-containing NMDA receptors weakens synaptic strength and favors synaptic depression (Collingridge et al., 2010), which may contribute to the elimination of retinogeniculate inputs. In addition, the stabilization of synapses requires forming synapses onto stable dendrites (Li et al., 2011), suggesting a cooperative interaction in dendritic and axon maturation during postnatal development. Because we have now established that LGI1 plays a critical role in both dendritic and axonal pruning, we identify a novel molecular pathway in synapse and circuit remodeling.
Although the detailed molecular mechanisms whereby LGI1 promotes axon pruning still remain essentially unknown, many signaling molecules associated with LGI1 could potentially be involved. Gene expression profile analysis of culture cells overexpressing LGI1 revealed that LGI1 mainly regulates genes in canonical axon guidance pathways (Kunapuli et al., 2010). For instance, LGI1 expression downregulates ROBO1 (Kunapuli et al., 2010), the receptor for the SLIT protein that functions as a repulsive cue for neurite branching and outgrowth. LGI1 also reduces matrix metalloproteinases (Kunapuli et al., 2004), which have been implicated in axon outgrowth (Gonthier et al., 2007). In addition, LGI1 was shown recently to have a high binding affinity for a Nogo receptor (NogoR1) (Thomas et al., 2010). Nogo-66 receptor-mediated signaling has already been implicated in consolidating neural circuitry during postnatal development and in limiting axonal regeneration and functional recovery in pathological conditions (McGee et al., 2005). It will be interesting to study how LGI1 regulates axon guidance cues, extracellular matrix proteins, and cytoskeletal molecules in neurons to promote axonal elimination in the future.
ADLTE patients develop frequent partial seizures accompanied by auditory and visual hallucinations (Nobile et al., 2009). Even tonic–clonic seizures can be triggered by sound and light in some ADLTE patients. Arrested synapse refinement in the dLGN of LGI1 mutant and heterozygous mice could provide one potential explanation for aberrant sensory processing in these epilepsy patients. Impaired axon elimination may induce imprecise wiring and unrefined receptive fields in the thalamus. Sensory signal distortion induced by imprecise sensory inputs (Grubb et al., 2003; Grubb and Thompson, 2004) in the thalamus and relayed to the cortex may contribute to sensory abnormalities found in ADLTE patients. Although bridging the defects in sensory input refinement to abnormal cortical sensory processing needs additional examination, blocking axon and synapse elimination appears sufficient to render the circuitry prone to epileptogenesis, as evidenced by our previous work and more recently by studies examining seizure susceptibility in mice deficient for the classic complement pathway component C1q (Stevens et al., 2007; Chu et al., 2010).
References
- Anderson MP. Arrested glutamatergic synapse development in human partial epilepsy. Epilepsy Curr. 2010;10:153–158. doi: 10.1111/j.1535-7511.2010.01386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arsenault D, Zhang ZW. Developmental remodelling of the lemniscal synapse in the ventral basal thalamus of the mouse. J Physiol. 2006;573:121–132. doi: 10.1113/jphysiol.2006.106542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bickford ME, Slusarczyk A, Dilger EK, Krahe TE, Kucuk C, Guido W. Synaptic development of the mouse dorsal lateral geniculate nucleus. J Comp Neurol. 2010;518:622–635. doi: 10.1002/cne.22223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabrol E, Navarro V, Provenzano G, Cohen I, Dinocourt C, Rivaud-Péchoux S, Fricker D, Baulac M, Miles R, Leguern E, Baulac S. Electroclinical characterization of epileptic seizures in leucine-rich, glioma-inactivated 1-deficient mice. Brain. 2010;133:2749–2762. doi: 10.1093/brain/awq171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Regehr WG. Developmental remodeling of the retinogeniculate synapse. Neuron. 2000;28:955–966. doi: 10.1016/s0896-6273(00)00166-5. [DOI] [PubMed] [Google Scholar]
- Cheng TW, Liu XB, Faulkner RL, Stephan AH, Barres BA, Huberman AD, Cheng HJ. Emergence of lamina-specific retinal ganglion cell connectivity by axon arbor retraction and synapse elimination. J Neurosci. 2010;30:16376–16382. doi: 10.1523/JNEUROSCI.3455-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu Y, Jin X, Parada I, Pesic A, Stevens B, Barres B, Prince DA. Enhanced synaptic connectivity and epilepsy in C1q knockout mice. Proc Natl Acad Sci USA. 2010;107:7975–7980. doi: 10.1073/pnas.0913449107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collingridge GL, Peineau S, Howland JG, Wang YT. Long-term depression in the CNS. Nat Rev Neurosci. 2010;11:459–473. doi: 10.1038/nrn2867. [DOI] [PubMed] [Google Scholar]
- Demas J, Sagdullaev BT, Green E, Jaubert-Miazza L, McCall MA, Gregg RG, Wong RO, Guido W. Failure to maintain eye-specific segregation in nob, a mutant with abnormally patterned retinal activity. Neuron. 2006;50:247–259. doi: 10.1016/j.neuron.2006.03.033. [DOI] [PubMed] [Google Scholar]
- Dhande OS, Hua EW, Guh E, Yeh J, Bhatt S, Zhang Y, Ruthazer ES, Feller MB, Crair MC. Development of single retinofugal axon arbors in normal and β2 knock-out mice. J Neurosci. 2011;31:3384–3399. doi: 10.1523/JNEUROSCI.4899-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavell SW, Greenberg ME. Signaling mechanisms linking neuronal activity to gene expression and plasticity of the nervous system. Annu Rev Neurosci. 2008;31:563–590. doi: 10.1146/annurev.neuro.31.060407.125631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukata Y, Adesnik H, Iwanaga T, Bredt DS, Nicoll RA, Fukata M. Epilepsy-related ligand/receptor complex LGI1 and ADAM22 regulate synaptic transmission. Science. 2006;313:1792–1795. doi: 10.1126/science.1129947. [DOI] [PubMed] [Google Scholar]
- Gan WB, Kwon E, Feng G, Sanes JR, Lichtman JW. Synaptic dynamism measured over minutes to months: age-dependent decline in an autonomic ganglion. Nat Neurosci. 2003;6:956–960. doi: 10.1038/nn1115. [DOI] [PubMed] [Google Scholar]
- Gonthier B, Nasarre C, Roth L, Perraut M, Thomasset N, Roussel G, Aunis D, Bagnard D. Functional interaction between matrix metalloproteinase-3 and semaphorin-3C during cortical axonal growth and guidance. Cereb Cortex. 2007;17:1712–1721. doi: 10.1093/cercor/bhl082. [DOI] [PubMed] [Google Scholar]
- Grubb MS, Thompson ID. Visual response properties in the dorsal lateral geniculate nucleus of mice lacking the β2 subunit of the nicotinic acetylcholine receptor. J Neurosci. 2004;24:8459–8469. doi: 10.1523/JNEUROSCI.1527-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubb MS, Rossi FM, Changeux JP, Thompson ID. Abnormal functional organization in the dorsal lateral geniculate nucleus of mice lacking the beta 2 subunit of the nicotinic acetylcholine receptor. Neuron. 2003;40:1161–1172. doi: 10.1016/s0896-6273(03)00789-x. [DOI] [PubMed] [Google Scholar]
- Guido W. Refinement of the retinogeniculate pathway. J Physiol. 2008;586:4357–4362. doi: 10.1113/jphysiol.2008.157115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong YK, Chen C. Wiring and rewiring of the retinogeniculate synapse. Curr Opin Neurobiol. 2011;21:228–237. doi: 10.1016/j.conb.2011.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooks BM, Chen C. Distinct roles for spontaneous and visual activity in remodeling of the retinogeniculate synapse. Neuron. 2006;52:281–291. doi: 10.1016/j.neuron.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Hooks BM, Chen C. Vision triggers an experience-dependent sensitive period at the retinogeniculate synapse. J Neurosci. 2008;28:4807–4817. doi: 10.1523/JNEUROSCI.4667-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaubert-Miazza L, Green E, Lo FS, Bui K, Mills J, Guido W. Structural and functional composition of the developing retinogeniculate pathway in the mouse. Vis Neurosci. 2005;22:661–676. doi: 10.1017/S0952523805225154. [DOI] [PubMed] [Google Scholar]
- Kalachikov S, Evgrafov O, Ross B, Winawer M, Barker-Cummings C, Martinelli Boneschi F, Choi C, Morozov P, Das K, Teplitskaya E, Yu A, Cayanis E, Penchaszadeh G, Kottmann AH, Pedley TA, Hauser WA, Ottman R, Gilliam TC. Mutations in LGI1 cause autosomal-dominant partial epilepsy with auditory features. Nat Genet. 2002;30:335–341. doi: 10.1038/ng832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano M, Hashimoto K. Synapse elimination in the central nervous system. Curr Opin Neurobiol. 2009;19:154–161. doi: 10.1016/j.conb.2009.05.002. [DOI] [PubMed] [Google Scholar]
- Kielland A, Bochorishvili G, Corson J, Zhang L, Rosin DL, Heggelund P, Zhu JJ. Activity patterns govern synapse-specific AMPA receptor trafficking between deliverable and synaptic pools. Neuron. 2009;62:84–101. doi: 10.1016/j.neuron.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunapuli P, Kasyapa CS, Hawthorn L, Cowell JK. LGI1, a putative tumor metastasis suppressor gene, controls in vitro invasiveness and expression of matrix metalloproteinases in glioma cells through the ERK1/2 pathway. J Biol Chem. 2004;279:23151–23157. doi: 10.1074/jbc.M314192200. [DOI] [PubMed] [Google Scholar]
- Kunapuli P, Lo K, Hawthorn L, Cowell JK. Reexpression of LGI1 in glioma cells results in dysregulation of genes implicated in the canonical axon guidance pathway. Genomics. 2010;95:93–100. doi: 10.1016/j.ygeno.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Erisir A, Cline H. In vivo time-lapse imaging and serial section electron microscopy reveal developmental synaptic rearrangements. Neuron. 2011;69:273–286. doi: 10.1016/j.neuron.2010.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtman JW, Colman H. Synapse elimination and indelible memory. Neuron. 2000;25:269–278. doi: 10.1016/s0896-6273(00)80893-4. [DOI] [PubMed] [Google Scholar]
- Luo L, O'Leary DD. Axon retraction and degeneration in development and disease. Annu Rev Neurosci. 2005;28:127–156. doi: 10.1146/annurev.neuro.28.061604.135632. [DOI] [PubMed] [Google Scholar]
- McGee AW, Yang Y, Fischer QS, Daw NW, Strittmatter SM. Experience-driven plasticity of visual cortex limited by myelin and Nogo receptor. Science. 2005;309:2222–2226. doi: 10.1126/science.1114362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobile C, Michelucci R, Andreazza S, Pasini E, Tosatto SC, Striano P. LGI1 mutations in autosomal dominant and sporadic lateral temporal epilepsy. Hum Mutat. 2009;30:530–536. doi: 10.1002/humu.20925. [DOI] [PubMed] [Google Scholar]
- Noutel J, Hong YK, Leu B, Kang E, Chen C. Experience-dependent retinogeniculate synapse remodeling is abnormal in MeCP2-deficient mice. Neuron. 2011;70:35–42. doi: 10.1016/j.neuron.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte U, Thumfart JO, Klöcker N, Sailer CA, Bildl W, Biniossek M, Dehn D, Deller T, Eble S, Abbass K, Wangler T, Knaus HG, Fakler B. The epilepsy-linked Lgi1 protein assembles into presynaptic Kv1 channels and inhibits inactivation by Kvbeta1. Neuron. 2006;49:697–706. doi: 10.1016/j.neuron.2006.01.033. [DOI] [PubMed] [Google Scholar]
- Senechal KR, Thaller C, Noebels JL. ADPEAF mutations reduce levels of secreted LGI1, a putative tumor suppressor protein linked to epilepsy. Hum Mol Genet. 2005;14:1613–1620. doi: 10.1093/hmg/ddi169. [DOI] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SW, Barres BA. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131:1164–1178. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- Sugihara I. Organization and remodeling of the olivocerebellar climbing fiber projection. Cerebellum. 2006;5:15–22. doi: 10.1080/14734220500527385. [DOI] [PubMed] [Google Scholar]
- Thomas R, Favell K, Morante-Redolat J, Pool M, Kent C, Wright M, Daignault K, Ferraro GB, Montcalm S, Durocher Y, Fournier A, Perez-Tur J, Barker PA. LGI1 is a Nogo receptor 1 ligand that antagonizes myelin-based growth inhibition. J Neurosci. 2010;30:6607–6612. doi: 10.1523/JNEUROSCI.5147-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torborg CL, Feller MB. Spontaneous patterned retinal activity and the refinement of retinal projections. Prog Neurobiol. 2005;76:213–235. doi: 10.1016/j.pneurobio.2005.09.002. [DOI] [PubMed] [Google Scholar]
- Walsh MK, Lichtman JW. In vivo time-lapse imaging of synaptic takeover associated with naturally occurring synapse elimination. Neuron. 2003;37:67–73. doi: 10.1016/s0896-6273(02)01142-x. [DOI] [PubMed] [Google Scholar]
- Zhou YD, Lee S, Jin Z, Wright M, Smith SE, Anderson MP. Arrested maturation of excitatory synapses in autosomal dominant lateral temporal lobe epilepsy. Nat Med. 2009;15:1208–1214. doi: 10.1038/nm.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]