

Abstract
Several human genetic cataracts have been linked recently to point mutations in the γD crystallin gene. Here we provide a molecular basis for lens opacity in two genetic cataracts and suggest that the opacity occurs because of the spontaneous crystallization of the mutant proteins. Such crystallization of endogenous proteins leading to pathology is an unusual event. Measurements of the solubility curves of crystals of the Arg-58 to His and Arg-36 to Ser mutants of γD crystallin show that the mutations dramatically lower the solubility of the protein. Furthermore, the crystal nucleation rate of the mutants is enhanced considerably relative to that of the wild-type protein. It should be noted that, although there is a marked difference in phase behavior, there is no significant difference in protein conformation among the three proteins.
Human γD crystallin is a member of a highly homologous family of mammalian lens proteins called the γ crystallins (1). Together with the α and β crystallins, these proteins are essential for maintaining lens transparency. However, the γ crystallins differ from the α and β crystallins in one important respect: the interactions between the γ crystallins are attractive (2). This feature reduces the osmotic pressure in the lens, but it also makes the γ crystallins more susceptible to aggregation and phase separation, phenomena that diminish the homogeneity of the lens and lead to cataract (3). Yet, despite these attractive interactions, the γ crystallins remain soluble for many years at high concentrations and with little turnover, maintaining the proper refractive index gradient of the lens (4).
It is well known that random mutations in proteins dramatically affect their solubility (5). In this article, we show that the Arg-58 to His (R58H) mutant [linked to the aculeiform cataract (Fig. 1a; ref. 6)] and the Arg-36 to Ser (R36S) mutant [linked to another form of genetic (congenital) cataract (Fig. 1b; ref. 7)] are much less soluble than the wild-type human γD crystallin protein (HGD). We also show that these mutants are more prone to crystallize than the wild-type. Indeed, Kmoch et al. (7) recently extracted crystals of the R36S mutant from the eye of a young patient. To determine the mechanism of cataract formation caused by these mutations, we compared the conformation, stability, and phase behavior of the recombinant HGD, R58H, and R36S proteins in solution.
Figure 1.

Cataract phenotypes. (a) Aculeiform cataract (R58H) present in a 15-year-old patient (6). (b) Other genetic (congenital) cataract (R36S) present in a 5-year-old patient (7) .
Materials and Methods
Cloning, Expression, and Isolation of Proteins.
Recombinant human γD crystallin was prepared by the amplification of the coding sequence from a human fetal lens cDNA library as detailed (8). Overexpression of γD crystallin, and isolation and purification of the protein, were all done according to procedures as described (8). Mutant proteins were prepared as follows.
(i) The R58H mutant.
To introduce a histidine mutation in place of Arg-58, the following oligonucleotide primers were made: 5′-CCAGTACTTCCTGCACCGCGGCGACTATGC-3′ as the forward primer and 5′-GCATAGTCGCCGCGGTGCAGGAAGTACTGG-3′ as the reverse primer.
(ii) The R36S mutant.
To introduce a serine mutation in place of Arg-36, the following oligonucleotide primers were made: 5′-GCAACTCGGCGAGCGTGGACAGCGGC-3′ as the forward primer and 5′-GCCGCTGTCCACGCTCGCCGAGTTGC-3′ as the reverse primer.
All primers were synthesized by Life Technologies (Grand Island, NY). Mutagenesis was performed with the QuikChange site-directed mutagenesis kit from Stratagene. The plasmid DNA obtained after mutagenesis was sequenced with the T7 promoter primer and was found to contain the desired mutation but no other sequence changes. The mutant proteins were expressed in Escherichia coli and isolated as described for HGD (8). In all cases, the crystallins folded efficiently and fractionated almost exclusively (>95%) into the soluble fraction. The final product was analyzed by using electrospray ionization mass spectrometry. The concentration of human γD was determined by using an extinction coefficient of 41.4 mM−1⋅cm−1 at 280 nm (9). This value also was used for both mutant proteins.
Electrospray Ionization Mass Spectrometry.
Mass spectrometry was performed at the Biopolymers Laboratory at the Center for Cancer Research at the Massachusetts Institute of Technology. Six separate preparations of HGD, R58H, and R36S gave an average mass of 20,610 ± 2, 20,588 ± 2, and 20,537 ± 2, respectively. These results are consistent with previously published work for HGD to within 3 mass units (10), and with the R58H and R36S replacements in HGD.
Circular Dichroism (CD) Spectra.
CD spectra were obtained with an Aviv Associates (Lakewood, NJ) model 202 spectrometer. Protein concentrations of 0.5 mg/ml and 0.1 mg/ml in 5 mM phosphate buffer (pH 7) were used for near-UV CD spectra, and were used in 100 mM phosphate buffer for far-UV CD spectra. Far-UV spectra were normalized with respect to the concentration of peptide bonds, whereas near-UV spectra (not shown) were normalized with respect to protein concentration. Because R36S shows an atypical far-UV CD spectrum (Fig. 2a), the concentration dependence of the CD spectrum of this mutant was examined in the 0.07–1.0 mg/ml range. The spectrum was found to be independent of protein concentration.
Figure 2.

Conformation and stability studies of HGD, R58H, and R36S proteins. (a) Far-UV CD spectra. The spectra of HGD (blue) and R58H (red) are almost identical, whereas the spectrum of R36S (green) has an additional contribution from a band of negative ellipticity centered at 200 nm. This band is more readily seen in the difference spectrum R36S minus wild type (dashed black line). (b) Fluorescence emission spectra. The spectrum of R58H (red) protein is almost identical to that of HGD (blue), whereas that of R36S (green) shows a more open structure (17), possibly caused by the exposure of Trp-42, which is close to Ser-36 (7). The spectra are normalized with respect to protein concentration. (c) DSC. The thermal transitions shown are only partially reversible and hence preclude an equilibrium thermodynamic analysis. However, based on the apparent melting temperature, the stabilities of the HGD (blue), R58H (red), and R36S (green) proteins are very similar.
Fluorescence Spectra.
Fluorescence spectra were measured in a Hitachi (Tokyo) F-4500 spectrometer by using an excitation wavelength of 295 nm. The excitation and emission slits were set to 5 nm. Spectra were measured by under the same conditions and identical protein concentrations (0.1 mg/ml in 0.1 M phosphate buffer, pH 7) for HGD, R58H, and R36S.
Differential Scanning Calorimetry (DSC).
DSC measurements were made at Microcal (Amherst, MA) with a VP-DSC instrument. Protein concentrations were 0.1 mg/ml in 0.1 M NaCl/HCl buffer, pH 2. The scan rate was 1°C/min. These conditions were used for ease of comparison with previous work, and to minimize protein aggregation caused by disulfide cross-linking (11). The low pH does not adversely affect the proteins, because the γ crystallins are known to maintain their native conformation from pH 1 to 9 (11–13). Data were normalized with respect to protein concentration. DSC data were not used for an equilibrium thermodynamic analysis, because the thermal transition was not completely reversible. The endothermic transition observed in the DSC measurement paralleled the protein-unfolding transition observed in the near-UV CD spectra.
Solubility Measurements (Liquidus Lines).
Protein crystals were grown at room temperature from solutions of pure protein in 0.1 M phosphate buffer (pH 7) containing 20 mM DTT (to prevent thiol-mediated aggregation). Then these crystals were placed at a fixed temperature in fresh solutions of the same buffer. As the crystals melted, the concentration of protein in solution was monitored by removing aliquots of the supernatant and measuring the absorbance at 280 nm. The system was deemed to be in equilibrium when the concentration of the solution reached a constant value. The solution–crystal system was stirred continuously to ensure thorough mixing of the components. The DSC data assured us that, in the range of temperatures in which the solubility measurements were performed, the proteins were in the fully folded state.
Results and Discussion
An examination of the far-UV CD spectra (Fig. 2a) clearly shows that both mutants are very similar in secondary structure to HGD. The three proteins show a negative ellipticity at 218 nm, as do all of the γ crystallins (14). In the case of R36S, there is additional broadening and blueshift below 220 nm. A difference spectrum (HGD − R36S) shows a negative CD band centered around 200 nm (Fig. 2a). One possible explanation for this band is that the serine residue disrupts the normal β-sheet structure of the protein, leading to the formation of a “β -bulge” (15, 16). Should such a bulge form, the closest tryptophan (Trp-42) would become more solvent-exposed, resulting in a redshift of the emission maximum for the tryptophan fluorescence (14, 17). This redshift is indeed observed in the fluorescence spectrum of R36S (Fig. 2b). Even though our spectroscopic data reveal that R36S exhibits small, local conformational changes relative to HGD and R58H, the x-ray structure of Kmoch et al. (7) reveals no changes in the native γ-crystallin fold (1). An exact interpretation of our results on the secondary structure of R36S must await a higher resolution x-ray structure or a more detailed spectroscopic study.
We also compared the thermal stabilities of the three proteins (Fig. 2c), as measured by DSC. The apparent midpoint temperature of unfolding of the two mutants and the wild-type protein in the DSC data are within a 3°C range. This variation in unfolding temperature is typical of that found within the family of γ crystallin proteins (11). Thus, the three proteins are not only thermally stable, but also very similar in their thermal unfolding profiles.
Although these proteins are similar in structure, their phase behavior is quite different. We have shown that HGD exhibits the characteristic γ crystallin-phase behavior (8)—it crystallizes slowly enough so that metastable liquid–liquid phase separation occurs. However, this is not the case for the R58H and R36H mutants. Both crystallize rapidly when brought to the high-concentration low-temperature conditions under which liquid–liquid phase coexistence is expected (18, 19). Fig. 3 shows the crystals grown in pure solutions of these proteins. Crystals also formed easily in mixtures containing α and β crystallin and either R58H or R36S at compositions comparable to those found in the lens. No crystals were formed in a mixture containing HGD instead of the mutant protein.
Figure 3.

Crystals of the pure proteins produced in vitro. (a) HGD, (b) R58H, and (c) R36S. The crystals were grown in 0.1 M phosphate buffer (pH 7) containing 20 mM DTT.
The solubilities (the liquidus lines) of the three proteins are shown as a function of temperature in Fig. 4a. The solubility is defined as the concentration (at a given temperature) at which the protein solution is in equilibrium with the crystal phase. At body temperature, the mutants are an order of magnitude less soluble than the wild-type protein. In the range of concentrations studied, the protein solutions can be considered to be dilute (20). Therefore, the solubility curves could be adequately fit with the van't Hoff equation (21),
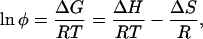 |
1 |
to determine the enthalpy (ΔH) and entropy (ΔS) of formation of the crystal phase for each protein (Fig. 4b and Table 1). In Eq. 1, φ is the volume fraction of protein in solution. [It is related to the concentration C (in mg/ml) by the expression φ = v̄C, where v̄ is the specific volume, taken to be 7.1 × 10−4 ml/mg (18).] ΔG is the Gibbs free energy of formation of the crystal, R is the universal gas constant, and T is the temperature.
Figure 4.

Protein solubilities (liquidus lines). (a) The solubility lines of HGD (open diamonds), R58H (circles), and R36S (squares) are shown together with the liquid–liquid coexistence curve of HGD (crossed diamonds). (b) Logarithmic-linear plot of the data in a was used to extract the enthalpies and entropies of the protein crystals. The lines are fits to Eq. 1. T0 is a reference temperature that was taken to be 298 K.
Table 1.
Thermodynamic and kinetic parameters for the crystallization of the HGD, R58H, and R36S proteins
| Protein | Ceq(T0), mg/ml | ΔG(T0), kJ⋅mol−1 | Ci(T0), mg/ml | Ci/Ceq | ΔH, kJ⋅mol−1 |
|---|---|---|---|---|---|
| HGD | 12.6 | −11.7 | 60–300 | 4.8–24 | −61.5 |
| R58H | 0.78 | −18.6 | 25–95 | 32–122 | −76.9 |
| R36S | 0.72 | −18.8 | 0.8–0.9 | 1.1–1.25 | −40.4 |
Ceq(T0) is the solubility at T0 (a reference temperature that was taken to be 298 K). ΔG and ΔH are, respectively, the Gibbs free energy and enthalpy of formation of the crystal phase for each protein. Ci(T0) is the range of initial protein concentrations required to produce crystals at 298 K from pure solutions in approximately 24 h. Ci/Ceq is the corresponding supersaturation, which represents the extent to which the solubility limit must be exceeded for crystals to form. The large difference in ΔH between R58H and R36S reflects the different space groups of these two crystals (ref. 7; C. Slingsby, personal communication).
Table 1 shows that the free energy change on crystallization of the two mutants is much lower than that of the wild-type protein. A change in solubility is to be expected, because the mutations are believed to occur at crystal contacts (ref. 7; C. Slingsby, personal communication). Despite having similar solubilities, R36S and R58H have different kinetics of crystallization. We find that R36S crystallizes on reaching the solubility limit, whereas R58H can be solubilized up to more than 100 times its solubility limit (Table 1). Because we observed that the growth rates of the two crystals are comparable, the difference in the supersaturations is most likely caused by a difference in nucleation rates between the two mutants. This conclusion is consistent with the values of the enthalpies of formation (ΔH) shown in Table 1. Because both mutants have the same enthalpy in solution (the solutions are dilute), the difference between the enthalpies of formation of R58H and R36S is a measure of the difference in the enthalpies of the crystal phase. Thus, a protein molecule in a crystal of R36S is less strongly bound to its neighbors than one in R58H. Furthermore, because the free energies are the same for both crystals, a protein molecule in a crystal of R36S also has larger entropy than one in a crystal of R58H. The lower enthalpy and larger entropy should allow the R36S proteins to rearrange more easily and thus facilitate nucleation of these crystals.
Our findings show that although HGD can be solubilized to concentrations as high as 300 mg/ml, neither mutant protein can be solubilized to such a high concentration. The lowered solubility of the mutants is expected because arginine, a highly polar and charged residue, is replaced by less polar residues in the mutants, as pointed out by Kmoch et al. (7) for R36S. We also found that these mutations have an effect on the kinetics of nucleation. This finding is significant not only for protein condensation diseases such as cataract, but also for the efficient production of protein crystals for x-ray structure determination.
The work presented here also shows that protein destabilization or unfolding is not necessary for the formation of the genetic cataracts examined. We have shown (8) that this is the case for yet another genetic cataract, namely the juvenile hereditary cataract (22) caused by the Arg-14 to Cys (R14C) mutation in γD crystallin. It is important to determine whether other forms of cataract occur without significant destabilization or unfolding of the proteins involved.
Cataract is but one member of a group of protein condensation diseases in which the pathology is caused by a loss of solubility. The archetypal protein condensation disease is sickle-cell anemia (23). In this disease, the mutant protein hemoglobin S (also a product of a single point mutation) has a tertiary structure that is almost identical to that of wild-type hemoglobin A. Nevertheless, in the deoxygenated form, the solubility of hemoglobin S is less than that of hemoglobin A, leading to the formation of crystals or fibers (24). Although the formation of pure protein crystals within tissues is a rare event, it has been reported in eosinophil-rich diseases in the human lung (25). In the case of cataract, to our knowledge, the only unambiguous discovery of crystals in vivo has been that of Kmoch et al. (7) for the cataract associated with the R36S mutation. The parallel between our in vitro findings and the in vivo behavior of R36S leads us to predict that the aculeiform cataract caused by the R58H mutation also results from the crystallization of the mutant protein in the lens. There are reports of crystalline deposits in the lens in several other nongenetic forms of cataract, such as the “Christmas tree” cataract (26), the “uncombable hair” cataract (27), and the hyperferritinemia-related cataract (28). Given that crystallization is a possible mechanism for cataract formation, the aforementioned reports (26–28) should be reexamined to see whether crystallization is a more general phenomenon involved in nongenetic cataracts as well.
Acknowledgments
We thank Dr. Nicolette Lubsen (University of Nijmegen, Nijmegen, The Netherlands) for the gift of the human γD crystallin cDNA clone, Drs. Mohan Chellani and Lung-Nan Lin (Microcal Inc.) for the DSC data, and Dr. Yevgeniya Zastavker for assistance with Fig. 3. We are indebted to Drs. Elise Héon and Jirí Brynda for providing photographs for Fig. 1 a and b, respectively. This work was supported by National Institutes of Health Grants EY10535 (to J.P.), GM17980 (to J.K.), and EY05127 (to G.B.B.).
Abbreviations
- HGD
wild-type human γD crystallin
- R58H
Arg-58 to His mutant
- R36S
Arg-36 to Ser mutant
- DSC
differential scanning calorimetry
References
- 1.Lindley P F, Narebor M E, Summers L J, Wistow G J. In: The Ocular Lens: Structure, Function and Pathology. Maisel H, editor. New York: Dekker; 1985. pp. 123–167. [Google Scholar]
- 2.Tardieu A, Vérétout F, Krop B, Slingsby C. Eur Biophys J. 1992;21:1–21. doi: 10.1007/BF00195438. [DOI] [PubMed] [Google Scholar]
- 3.Clark J I. In: Principles and Practice of Ophthalmology. Alberts E M, Jacobiec F A, editors. Philadelphia: Saunders; 1994. pp. 114–123. [Google Scholar]
- 4.Zigler J S., Jr . In: Principles and Practice of Ophthalmology. Alberts E M, Jacobièc F A, editors. Philadelphia: Saunders; 1994. pp. 97–113. [Google Scholar]
- 5.Dale G E, Broger C, Langen H, D'Arcy A, Stüber D. Protein Eng. 1994;7:933–939. doi: 10.1093/protein/7.7.933. [DOI] [PubMed] [Google Scholar]
- 6.Héon E, Priston M, Schorderet D F, Billingsley G D, Girard P O, Lubsen N, Munier F L. Am J Hum Genet. 1999;65:1261–1267. doi: 10.1086/302619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kmoch S, Brynda J, Befekadu A, Bezouška K, Novák P, Rezácová P, Ondrová L, Filipec M, Sedlácek J, Elleder M. Hum Mol Genet. 2000;9:1779–1786. doi: 10.1093/hmg/9.12.1779. [DOI] [PubMed] [Google Scholar]
- 8.Pande A, Pande J, Asherie N, Lomakin A, Ogun O, King J A, Lubsen N H, Walton D, Benedek G B. Proc Natl Acad Sci USA. 2000;97:1993–1998. doi: 10.1073/pnas.040554397. . (First Published February 25, 2000; 10.1073/pnas.040554397) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Andley U P, Mathur S, Griest T A, Petrash J M. J Biol Chem. 1996;271:31973–31980. doi: 10.1074/jbc.271.50.31973. [DOI] [PubMed] [Google Scholar]
- 10.Hanson S R A, Smith D L, Smith J B. Exp Eye Res. 1998;67:301–312. doi: 10.1006/exer.1998.0530. [DOI] [PubMed] [Google Scholar]
- 11.Sen A C, Walsh M T, Chakrabarti B. J Biol Chem. 1992;267:11898–11907. [PubMed] [Google Scholar]
- 12.Rudolf R, Sienbendritt R, Nesslauer G, Sharma A, Jaenicke R. Proc Natl Acad Sci USA. 1990;87:4625–4629. doi: 10.1073/pnas.87.12.4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mayr E-V, Jaenicke R, Glockshuber R. J Mol Biol. 1997;269:260–269. doi: 10.1006/jmbi.1997.1033. [DOI] [PubMed] [Google Scholar]
- 14.Mandal K, Bose S K, Chakrabarti B, Siezen R J. Biochim Biophys Acta. 1985;832:156–164. doi: 10.1016/0167-4838(85)90327-9. [DOI] [PubMed] [Google Scholar]
- 15.Besley N A, Hirst J D. J Am Chem Soc. 1999;121:9636–9644. [Google Scholar]
- 16.Wan W-Y, Milner-White E J. J Mol Biol. 1999;286:1651–1662. doi: 10.1006/jmbi.1999.2551. [DOI] [PubMed] [Google Scholar]
- 17.Mandal K, Chakrabarti B, Thomson J, Siezen R J. J Biol Chem. 1987;262:8096–8102. [PubMed] [Google Scholar]
- 18.Broide M L, Berland C R, Pande J, Ogun O O, Benedek G B. Proc Natl Acad Sci USA. 1991;88:5660–5664. doi: 10.1073/pnas.88.13.5660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berland C R, Thurston G M, Kondo M, Broide M L, Pande J, Ogun O, Benedek G B. Proc Natl Acad Sci USA. 1992;89:1214–1218. doi: 10.1073/pnas.89.4.1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lomakin A, Asherie N, Benedek G B. J Chem Phys. 1996;104:1646–1656. [Google Scholar]
- 21.van Holde K E. Physical Biochemistry. 2nd. Ed. Englewood Cliffs, NJ: Prentice–Hall; 1985. [Google Scholar]
- 22.Stephan D A, Gillanders E, Vanderveen D, Freas-Lutz D, Wistow G, Baxevanis A D, Robbins C M, VanAuken A, Quesenberry M I, Bailey-Wilson J, et al. Proc Natl Acad Sci USA. 1999;96:1008–1012. doi: 10.1073/pnas.96.3.1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eaton W A, Hofrichter J. Adv Protein Chem. 1990;40:63–279. doi: 10.1016/s0065-3233(08)60287-9. [DOI] [PubMed] [Google Scholar]
- 24.Noguchi C T, Schechter A N. Annu Rev Biophys Biophys Chem. 1985;14:239–263. doi: 10.1146/annurev.bb.14.060185.001323. [DOI] [PubMed] [Google Scholar]
- 25.Guo L, Johnson R S, Schuh J C L. J Biol Chem. 2000;275:8032–8037. doi: 10.1074/jbc.275.11.8032. [DOI] [PubMed] [Google Scholar]
- 26.Shun-Shin G A, Vrensen G F, Brown N P, Willekens B, Smeets M H, Bron A J. Invest Ophthalmol Visual Sci. 1993;34:3489–3496. [PubMed] [Google Scholar]
- 27.de Jong P T, Bleeker-Wagemakers E M, Vrensen G F, Broekhuyse R M, Peereboom-Wynia J D, Delleman J W. Ophthalmology. 1990;97:1181–1187. doi: 10.1016/s0161-6420(90)32438-7. [DOI] [PubMed] [Google Scholar]
- 28.Mumford A D, Cree I A, Arnold J D, Hagan M C, Rixon K C, Harding J J. Br J Ophthalmol. 2000;84:697–700. doi: 10.1136/bjo.84.7.697. [DOI] [PMC free article] [PubMed] [Google Scholar]