

Abstract
The von Hippel–Lindau tumor suppressor gene (VHL) has attracted intensive interest not only because its mutations predispose carriers to devastating tumors, but also because it is involved in oxygen sensing under physiological conditions. VHL loss-of-function mutations result in organ-specific tumors, such as hemangioblastoma of the central nervous system and renal cell carcinoma, both untreatable with conventional chemotherapies. The VHL protein is best known as an E3 ubiquitin ligase that targets hypoxia-inducible factor-α (HIF-α), but many diverse, non-canonical cellular functions have also been assigned to VHL, mainly based on studies in cell culture systems. As such, although the HIF-dependent role of VHL is critical, the full spectrum of pathophysiological functions of VHL is still unresolved. Such understanding requires careful cross-referencing with physiologically relevant experimental models. Studies in model systems, such as Caenorhabditis elegans, Drosophila, zebrafish and mouse have provided critical in vivo confirmation of the VHL–HIF pathway, and verification of potentially important cellular functions including microtubule stabilization and epithelial morphogenesis. More recently, animal models have also suggested systemic roles of VHL in hematopoiesis, metabolic homeostasis and inflammation. In this review, the studies performed in model organisms will be summarized and placed in context with existing clinical and in vitro data.
Keywords: VHL, HIF, model organisms
Introduction
The von Hippel–Lindau tumor suppressor gene (VHL) mutations were the genetic defects in the familial VHL disease (Latif et al., 1993), which manifests in a limited number of organ-specific tumors (Maher et al., 2011), predominantly in the kidney (clear-cell renal cell carcinoma, ccRCC) and central nervous system (hemangioblastoma). Less frequent VHL tumors include those in pancreas (pancreatic cysts, serous cystadenoma and pancreatic neuroendocrine tumors), adrenal gland (pheochromocytoma) and testes (epididymal cystadenomas). The presence of these diverse but specific VHL tumor types of widely different tissue origins is just one of the intriguing facts about the VHL gene. Among these ‘VHL tumors’, ccRCC is the main cause of disease-related death. Up to 70% of the carriers of germ-line VHL mutations eventually develop ccRCC (Lonser et al., 2003). In addition, loss of VHL function, including somatic mutations and epigenetic defects, is found in 70–90% of the sporadic ccRCC (Herman et al., 1994; Kim and Kaelin, 2004; Banks et al., 2006). The pathophysiological mechanism for such strong association is currently unknown.
VHL has proved a unique and enigmatic tumor suppressor gene. Most of the tumor suppressor genes associated with familial cancer syndromes can be assigned specific cellular functions; for example, Rb is a cell cycle inhibitor, WT1 and TP53 are transcription factors, adenomatous polyposis coli is a Wnt signaling suppressor, and NF1 and NF2 are GTPase-activating protein regulator and cytoskeletal protein merlin, respectively. The role of VHL in tumorigenesis is more complex. The protein encoded by the VHL gene (pVHL) is best known as the substrate-binding subunit of a SCF (Skp1-Cdc53/Cul-1-F-box protein) type E3 ubiquitin ligase containing, besides pVHL, Cullin-2, elongin B and C and Rbx-1 (Figure 1; Pause et al., 1997; Lonergan et al., 1998; Kamura et al., 1999; Lisztwan et al., 1999; Stebbins et al., 1999). The best-known degradation target of VHL-containing E3 ligase is the α-subunit of hypoxia-inducible factor (HIF-α) in normal physiological conditions (Kaelin, 2005). At normal oxygen level, HIF-α is hydroxylated at the proline residues within an oxygen-dependent degradation domain. The prolyl-hydroxylated HIF-α is recognized by pVHL, leading to poly-ubiquitination and degradation. The hydroxylation reaction is mediated by the prolyl-hydroxylase domain proteins (PHDs; Berra et al., 2003). In hypoxic conditions, the prolyl hydroxylases are inactive and HIF-α-subunit is stabilized. HIF-α then dimerizes with the β-subunit (HIF-β), also termed aryl hydrocarbon receptor nuclear translocator (ARNT), which is constitutively expressed. The HIF heterodimer translocates to the nucleus where it functions as a transcription factor. Its best-known target genes encode proteins involved in glycolysis (phosphoglycerate kinase), glucose transport (Glut-1), angiogenesis (vascular endothelial growth factor (VEGF)) and erythropoiesis (erythropoietin); that is, proteins that mediate the cellular response and adaptation to hypoxic conditions. In addition, chemokine receptor CXCR4 was also identified as a HIF target (Staller et al., 2003), which indicates that HIF activation may contribute to the metastatic potential of cancer cells. These functions support a critical role of pVHL in regulating tumor progression, especially in hypervascularized tumors such as ccRCC. However, accumulated evidences have indicated that many HIF-independent activities of pVHL also exist (Figure 1), including regulation of extracellular matrix (Ohh et al., 1998; Tang et al., 2006; Feijoo-Cuaresma et al., 2008; Kurban et al., 2008), senescence (Young et al., 2008), apoptosis (Guo et al., 2009), phosphorylation enhancer (Yang et al., 2007), microtubule stability and cilia formation (Hergovich et al., 2003; Esteban et al., 2006; Schermer et al., 2006; Thoma et al., 2007, 2009), RNA stability (Datta et al., 2005; Danilin et al., 2009), endocytosis (Hsu et al., 2006), gene transcription (Mikhaylova et al., 2008) and many others (Frew and Krek, 2007, 2008). Interestingly, many of these HIFindependent functions of pVHL are mediated through stabilization of its binding proteins, contrary to its known E3 ligase activity. It therefore appears that pVHL is an adaptor protein that, depending on the interacting partners, can promote protein degradation or serve as a chaperon. Some of these diverse activities likely also contribute significantly to the tumor suppressor and other physiological functions.
Figure 1.
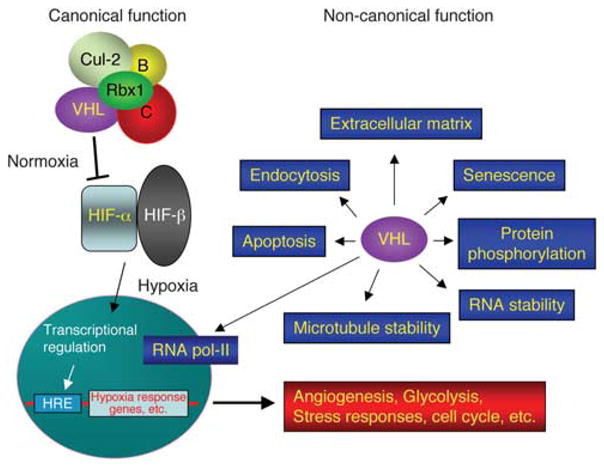
Pleiotropic VHL functions. See text for detail. Canonical VHL functions denote the degradation of HIF-α in normoxia through the E3 ubiquitin ligase activity. The E3 ligase complex contains VHL, Rbx1, cullin-2 (Cul-2) and elongin B and C (B, C). In hypoxia, HIF-α is stabilized, form active transcription factor complex with HIF-β and regulates gene expression via the hypoxia-responsive element (HRE). Non-canonical VHL functions denote those independent of HIF activity. Some, but not all, of these non-canonical functions are listed (in blue boxes).
Clinical observations support this view. Human mutations of PHD2, the major prolyl hydroxylase responsible for HIF-α degradation in normoxia, resulted in elevated expression of HIF-α in multiple tissues and a rare form of familial polycythemia, likely due to overexpression of erythropoietin, but without increased incidence of tumors (Berra et al., 2003). Also, the human Chuvash disease is caused by a specific homozygous VHL mutation (R200W) and elevated HIF activity (Gordeuk et al., 2004). The disease exhibits polycythemia without increased tumor incidence. These observations suggest that HIF activity is necessary but not sufficient for tumor formation. However, it is at times difficult to identify the most physiologically relevant functions of pVHL, particularly when the myriad of VHL functions is likely tissue and context dependent. To this end, the studies using model organisms have provided valuable insights. This review attempts to summarize these findings and place them in context with relevant data obtained from clinical and in vitro studies.
Caenorhabditis elegans
The first report of C. elegans VHL focused on evolutionary sequence conservation (Woodward et al., 2000). Because C. elegans VHL was the first non-mammalian VHL cloned, it provided valuable insights into the critical protein domains and amino-acid residues. For example, some of the disease-related mutation hot spots were suspected of being due to ‘founder effect’ within the patient population, not necessarily indicating a proportionally high degree of functional importance. The founder effect is the result of a large number of descendents from an ancestral carrier of a specific mutation. In a relatively rare genetic disease, a disproportionately large number of descendants from a single mutational event can bias the perception of functional importance (Zlotogora, 1994). Some of these hot spots, however, may indeed be critical for VHL function because they are conserved in C. elegans, including the ‘Black Forest mutation’ at Y98, which has been suspected of being a founder-effect outlier because almost all of the affected families can be traced to the Black Forest region of Germany (Brauch et al., 1995). This mutation was later shown to affect a critical amino-acid residue that confers a non-canonical function of VHL in promoting microtubule stability (Hergovich et al., 2003; more in the Drosophila section below). In addition to the specific hot spots, 60% of the human pVHL amino-acid residues that were predicted to be on the binding surface (Stebbins et al., 1999), and by extension important for protein–protein interaction, are conserved in C. elegans, thus supporting the molecular model.
The most significant early finding on pVHL function is the demonstration of its role in targeting prolyl-hydroxylated HIF-α (Ivan et al., 2001; Jaakkola et al., 2001). The VHL–HIF-prolyl hydroxylase triad was soon validated in C. elegans (Epstein et al., 2001). This study provided genetic and biochemical proof that prolyl-hydroxylated HIF-α is regulated by pVHL. More importantly, the study identified the nematode prolyl hydroxylase (encoded by the 2-oxoglutarate-dependent oxygenase gene egl-9) as the mediator of HIF-α degradation, as well as the mammalian ortholog EGLN/PHD (egl-9 homolog/prolyl hydroxylase domain). This cross-species validation was critical for the rapid acceptance of the model. Subsequent C. elegans studies of note came from whole-organism analysis of gene expression profiling. The C. elegans genome encodes a single VHL (vhl-1) and a single HIF-α (hif-1) gene. Interestingly, both vhl-1 and hif-1 loss-of-function mutants as well as double mutants are viable. The viability of these mutants made it possible to compare altered gene expression patterns in vhl-1-defective animals in the otherwise wild-type or hif-1 mutant backgrounds (Bishop et al., 2004). Thus, hif-dependent and independent vhl-responsive genes could be identified. Most of the hif-dependent genes upregulated in vhl-1 mutant are also upregulated by hypoxia. These include egl-9 that targets HIF-α during normoxia, indicating a possible feedback control. This mechanism is evolutionarily conserved because the mammalian ortholog of egl-9, the EGLN/PHD family of genes, was also upregulated in VHL mutant ccRCC cells (Harten et al., 2009). Another prolyl hydroxylase (encoded by phy-2) that modifies collagen is upregulated in both vhl-1 mutant and hypoxia. Curiously, no other familiar mammalian VHL–HIF target genes were revealed, such as homologs of VEGF, IGFBP3, Glut-1 and CXCR4, suggesting a different requirement for coping with hypoxia in nematodes as compared with mammals. Interestingly, among the vhl-dependent but hif-independent targets, one encodes an aldose 1-epimerase that is involved in gluconeogenesis, and three encode the solute carrier (slc) family of proteins that may be involved in sugar and lactate transport. These Vhl-dependent genes imply that the ancestral Vhl function may be involved in modulation of metabolic homeostasis.
A second whole-genome analysis focused on the hypoxia response (Shen et al., 2005). The analysis was designed to identify early response genes in acute hypoxia (4 h at 21°C in 0.1% oxygen). The analysis also compared three genotypes, wild type, hif-1 mutant and vhl-1 mutant, to identify hif-dependent and independent hypoxia-responsive genes. Among the hypoxia-inducible genes, only slightly more than half (63 out of 110) are hif-dependent. Most of the hif-dependent hypoxia-inducible genes are also vhl regulated, that is, they are upregulated in vhl-1 mutant and cannot be further induced by hypoxia. This indicates that in C. elegans, vhl is the major regulator of hif in normoxic cells and that hypoxia is the main activator of hif function. Prominent among the hypoxia-induced genes encode components of energy metabolism. For example, pyruvate carboxylase (pyc-1) is induced in a hif-1-independent (and vhl-dependent) manner. The enzyme provides oxaloacetate precursor for gluconeogenesis. gei-7 is induced in a hif-1- and vhl-dependent manner. It encodes isocitrate lyase that coverts isocitrate to succinate and glyoxylate in the glyoxylate cycle, a carbon-conserving pathway alternative to the carbon-consuming tricarboxylic acid cycle that converts isocitrate to succinyl-CoA. It is interesting to note that these gene products either promote energy store (glucose production) or conserve carbon sources, instead of the glycolysis switch observed in mammalian Vhl mutant (or hypoxic) cells. This may reflect the fact that there is no thermal homeostasis in nematodes, therefore, emergency energy production is not a priority. As in vhl-1 mutant gene profiling (Bishop et al., 2004), both egl-9 and phy-2 were upregulated in hypoxia in a hif-1- and vhl-1-dependent manner. Interestingly, phy-2 mutation shows <10% survival (to adults) in 0.5% oxygen. Thus, modification of extracellular matrix may be important for maintaining cellular function during hypoxic stress. Taken together, these studies demonstrated that the VHL–HIF axis is evolutionarily conserved to serve as a hypoxia responder, although the exact hypoxic response at the molecular level is specific to individual species.
Perhaps the most intriguing aspect of the C. elegans array analyses of VHL-responsive genes is what they did not show. Most of the VHL target genes in C. elegans are not prominent VHL targets found in the human ccRCC cell system. No growth factors/cytokines are identified and no obvious cell-cycle regulators are revealed. In fact, it appears that the VHL pathway in C. elegans regulates specific cell-autonomous functions that deal with environmental and metabolic stresses. These may be more closely aligned with the ancestral or the normal physiological functions of VHL. Interestingly, vhl-1 loss-of-function has recently been linked to hif-independent increased life span in C. elegans (Muller et al., 2009). It is intriguing to consider that stress responses may function at the expense of underlying health of the organism or vitality of individual cells. It is also instructive to consider that an ancestral gene that modulates stress responses can function as a tumor suppressor in complex organisms such as human. Whether the tumor suppressor function of VHL in human is acquired during evolution or is the manifestation of its ancestral function deserves careful investigation. Such understanding may influence our thinking of treating and preventing VHL tumors.
Drosophila
The Drosophila VHL (dVHL) was identified by amino-acid sequence homology query against the newly deposited fly genomic sequence in the late 1990s (Adryan et al., 2000). The dVHL protein was shown to interact with Drosophila elongin C in vitro. A similar strategy was used and the same dVHL sequence was identified independently at about the same time (Aso et al., 2000). The latter study showed that dVHL protein could form complex with the human Cul-2, human elongin B and C and mouse Rbx-1, but not with the human elongin A. Thus, the ubiquitin ligase function of pVHL is conserved in insects. Later, Arquier et al. (2006) showed that dVHL is involved in degradation of hydroxylated oxygen-dependent degradation domain of either the human HIF-1α or the Drosophila homolog Sima in normoxia, and that the oxygen-dependent degradation–GFP fusion protein is stabilized in hypoxia in vivo.
The first phenotypic analyses (Adryan et al., 2000) utilized the then new but promising RNA interference technology. dVHL was in fact one of the first Drosophila genes studied using siRNA-mediated knockdown. Drosophila contains a vascular system, the trachea, for transporting oxygen, which is capable of oxygen sensing (Jarecki et al., 1999; Wingrove and O’Farrell, 1999). On the other hand, the Malpighian tubes perform excretory and osmoregulatory functions similar to those of the vertebrate kidneys. There was therefore much speculation as to which of these systems will be affected in dVHL mutant. As it turned out, injection of dVHL-specific siRNA into pre-blastoderm embryos resulted in ectopic looping and branching in the trachea, but not in the Malpighian tubes (Adryan et al., 2000). Importantly, injection of either dVHL or human VHL sense RNA into wild-type embryos blocked tracheal branch migration. These reciprocal experiments demonstrated that VHL could negatively regulate vascular cell motility.
The function of VHL was further explored in the Drosophila system by analyzing the genomic knockout of the endogenous dVHL gene (Hsouna et al., 2010). Homozygous dVHL mutant is lethal at the early larval stage and exhibits profound tracheal phenotypes, consistent with the earlier RNA interference study. Surprisingly, the lethality could be rescued by re-expressing dVHL specifically in the trachea, indicating that modulation of vascular development is the main developmental function of dVHL. This notion is consistent with the earlier mouse knockout phenotype (Gnarra et al., 1997) and was later confirmed in zebrafish (van Rooijen et al., 2010). In both flies and vertebrates, the key defining process in vasculogenesis is the outgrowth of branches, regulated by growth factor signaling pathways. In Drosophila embryo, the fibroblast growth factor (FGF) signaling pathway is utilized (Ghabrial et al., 2003; Uv et al., 2003; Akis and Madaio, 2004) to pattern the trachea in a stereotyped fashion. The tracheal phenotype in dVHL mutant resembled that resulting from over-active FGF receptor (FGFR) signaling (Dammai et al., 2003). Indeed, FGFR-ERK-Ets1 signaling pathway is over-activated in dVHL mutant tracheal cells, which is the result of accumulated FGFR on the mutant tracheal cell surface (Hsouna et al., 2010). Genetic epistasis analysis indicates that the surface accumulation of FGFR is the result of defective endocytic pathway and that dVHL functionally interacts with and stabilizes Abnormal Wing Discs (AWD), the homolog of human anti-metastasis factor Nm23, which has been shown to regulate endocytosis (Dammai et al., 2003; Hsu et al., 2006; Nallamothu et al., 2008, 2009; Woolworth et al., 2009) in part by stabilizing Rab5 (Woolworth et al., 2009). The detailed phenotypic analysis also revealed that the endocytic function of dVHL not only regulates cell motility but also controls the size and length of the tubule lumen (Hsouna et al., 2010). Formation of the tracheal lumen, and likely of other tubule systems in mammals, is controlled by secretion (exocytosis) of matrix material and, at the maturation phase, reabsorption through endocytosis of these structural materials (Behr et al., 2007; Tsarouhas et al., 2007). Mutations in dVHL result in defective endocytosis of the lumenal chitin and consequently, enlarged and tortuous lumen of the trachea. This phenotype can be reduced or exacerbated by expression of constitutively active Rab5 or dominant-negative Rab5, respectively. Interestingly, ectopic branching and dilated tubule phenotypes were also observed in the mouse Vhl knockout kidney tubules (Hsouna et al., 2010).
We also showed that specific regulation of FGFR internalization is evolutionarily conserved. In VHL mutant human ccRCC cells and primary human endothelial cells knocked down with VHL-specific RNA duplex, FGFR over-accumulates on the cell surface, leading to increased FGFR, ERK and Ets1 transcription factor activation, elevated cell motility and increased angiogenic potential in vitro (Hsu et al., 2006; Champion et al., 2008). In addition, heterozygous Vhl knockout mouse shows increased angiogenic response toward bFGF in vivo (Champion et al., 2008). Importantly, this endocytic function is at least partly independent of HIF function, as Hif-α knockdown could not rescue the cell motility phenotypes in multiple cell systems. However, pVHL has recently been shown to regulate endocytosis of EGF receptor via HIF-dependent suppression of a Rab5 effector, rabaptin-5, in ccRCC cell lines (Wang et al., 2009). It is not known whether these different mechanisms are specific for different surface receptors.
In a separate study (Mortimer and Moberg, 2009), the tracheal branching phenotype in dVHL knockdown flies (mediated by trachea-specific expression of dVHL shRNA) was shown to be partly the result of over-expression of sima, the Drosophila homolog of HIF-1α. sima in turn stimulates the transcription of btl (FGFR), leading to ectopic branching. sima-dependent btl transcription has also been demonstrated in terminal branching in the larval trachea, which is induced by localized hypoxia (Centanin et al., 2008). Thus, both canonical and non-canonical functions of dVHL are involved in the tubule formation in Drosophila.
The role of dVHL in regulating motile epithelial cells was further studied in Drosophila border cells (Doronkin et al., 2010), which have been considered an in vivo model for epithelial cell invasion and epithelial–to–mesenchymal transition (Duchek et al., 2001; McDonald et al., 2003). During oogenesis, a specialized group of follicular epithelial cells (border cells) delaminates from the epithelium and invades through the germ cell complex until they reach the anterior end of the oocyte (Rorth, 2002; Montell, 2003). The precise movement of the border cells is guided by the Drosophila platelet-derived growth factor/VEGF signaling pathway. Interestingly, Doronkin et al. (2010) showed that the dVHL-sima system is involved in border cell migration. However, the exact correlation between border cell migration and dVHL function appears to be complicated, because the extents of hypoxia and the expression levels of sima or dVHL could cause either slowed or accelerated migration. Nonetheless, this study presented the first in vivo demonstration of the role of pVHL in regulating epithelial cell invasion.
Because VHL mutant ccRCC involves pathological transformation of epithelial tissues, whether and how VHL regulates epithelial morphogenesis has been of great interest. One useful epithelial model is again the Drosophila follicular epithelium in the egg chamber (Duchi et al., 2010). Epithelial cells are characterized by asymmetrical specification of membrane domains. One crucial step in establishing epithelial polarity is the specification of the apical domain, which is defined by the localized accumulation of a complex containing atypical PKC (aPKC), Bazooka (Baz; mammalian and worm PAR-3) and PAR-6 (Suzuki and Ohno, 2006). The PAR complex is required for subsequent localization of the basolateral complex that consists of known tumor suppressors Discs Large, Lethal Giant Larvae and Scribble (Bilder et al., 2003; Betschinger et al., 2005), and the adherens junction (Yamanaka et al., 2003). In mutant dVHL clones of follicle cells, microtubule bundles are disrupted and aPKC is mislocalized, leading to adenoma-like piling up of the epithelium (Duchi et al., 2010). Consistent with the known human VHL activity (Hergovich et al., 2003), wild-type dVHL can bind to microtubules. The direct influence of microtubule stability by dVHL leading to aPKC localization is supported by the ex vivo culture of dVHL mutant egg chambers, in which the aPKC mislocalization and epithelial phenotypes could be rescued by treatment with microtubule stabilizing agent paclitaxel, whereas treatment of wild-type egg chambers with microtubule destabilizing agent nocodazole could recapitulate the dVHL mutant phenotypes (Duchi et al., 2010). Furthermore, although wild-type dVHL could rescue the phenotypes, the type 2A mutant Y98H (Y51H in Drosophila), which has been shown to lose the microtubule-stabilizing function but retain partly the HIF-degradation function (Hergovich et al., 2003), could not. Therefore, the initial apical localization of aPKC requires intact microtubule bundles, which is maintained by dVHL protein (Figure 2). The Drosophila study is the first demonstration of physiological and developmental significance of the microtubule-stabilizing function of VHL in vivo.
Figure 2.

Microtubule-stabilizing function of dVHL contributes to epithelial morphogenesis. See Duchi et al. (2010) for detailed experimental evidence. Schematics showing polarized follicle cells. In many organ-associated epithelial cells such as the follicle cells, microtubules are not centrosome anchored, but are instead organized in cortical bundles parallel to the apicobasal axis, with the plus-end localized basally (Bartolini and Gundersen, 2006). VHL interacts with microtubule and aPKC. Microtubule bundles are stabilized by VHL and are important for proper localization of the aPKC–Par6–Baz complex. aPKC–Par6–Baz complex prevents apical spreading of the Dlg–Lgl–Scrib complex, which in turn restricts aPKC–Par6–Baz complex to the apical region. The opposing actions of aPKC–Par6–Baz complex and Dlg–Lgl–Scrib complex help define the location of adherens junction (AJ). AJ, adherens junction; Arm, Armadillo (Drosophila homolog of β-catenin); α-Cat, α-catenin; Baz, Bazooka; Dlg, discs large; Lgl, lethal giant larvae; Scrib, scribble. Septate junction is an adhesion complex similar in function to the mammalian tight junction.
Taken together, the Drosophila system has confirmed the HIF regulatory pathway, but unbiased genetic studies also revealed novel and important HIF-independent functions in endocytosis and microtubule-stabilizing functions in morphogenesis of both epithelial tubule system and polarized epithelial tissue. Also important, the Drosophila system demonstrated clearly tissue-specific functions of VHL; that is, the endocytic function of dVHL is required in tubule epithelial cells whereas the microtubule-stabilizing activity is functional in organcovering epithelium.
Zebrafish
Zebrafish genome encodes a vhl ortholog and a vhl-like gene; the latter is fish-specific (van Rooijen et al., 2009). Two lines of zvhl nonsense mutants were isolated using the Targeting Induced Local Lesions in Genomes method (van Rooijen et al., 2009). The mutant embryos develop to term and survive up to a week at the larval stage. These mutants exhibit behavioral and physiological hypoxic response in non-hypoxic conditions, including hyperventilation and increased cardiophysiological responses such as elevated heart rate (50% higher) and cardiac output (15-fold). As a result, the mutant larvae display cardiomegaly and stretched cardiomyocytes. They eventually develop edema and die. These symptoms are similar to but more severe than those in the familial Chuvash polycythemia patients, who carry a homozygous point mutation in the VHL gene (R200W; Gordeuk et al., 2004). Molecularly, whole-embryo gene expression profiling using 7-d.p.f. (days post fertilization) animals showed increased expression of genes involved in anaerobic metabolism, oxygen sensing and angiogenesis, including glut1, PHD3, Epo, Epo receptor, transferrin and vegf-a. In zvhl mutant most of these genes are hypoxia-inducible and are HIF targets. Therefore, the main physiological defects in zvhl mutant are mediated by the canonical VHL–HIF axis. Significantly, as in nematodes and mammals, homologs of egl-9 and phy-2 are both prominently overexpressed in zvhl mutant. In zvhl mutant fish as in Chuvash disease patients, the polycythemic phenotype is mainly attributable to the increased HIF-α activity. The pseudo-hypoxic response in zvhl mutant is later confirmed, because the phenotypes of cardiovascular flexibility and oxygen consumption in zvhl mutant in normoxia could be reproduced by placing wild-type animal in extreme hypoxic conditions (Yaqoob and Schwerte, 2010).
More detailed phenotypic analyses revealed that the homozygous mutants display increased blood vessel formation (van Rooijen et al., 2010). Significant increase in neovascularization was observed in the brain, eye and trunk. These abnormal vessels are leaky and can lead to macular edema and retinal detachment. Increased neovascularization is at least in part mediated by increased VEGF signaling, because treatment with VEGFR tyrosine kinase inhibitors could relieve the phenotypes. One important novel observation in the fish model is the increased activity of hematopoietic stem cells (HSCs), which not only increases the number of red blood cells, but also results in increased levels of endothelial progenitors in peripheral blood. This is highly interesting because in human hemangioblastoma, loss of heterozygosity of VHL occurs in stromal cells neighboring the proliferating blood vessels (Vortmeyer et al., 1997) and these VHL mutant stromal cells display multipotent cell characteristics (Park et al., 2007). The fish model may provide a clue to the origin of these mutant stromal cells.
Another recent study of zvhl provided some important insights into potential cancer therapeutics. Inhibitors of mammalian target of rapamycin (mTOR) signaling have been used in clinical trials for treating ccRCC. This is based on the observation that mTOR can promote HIF-α translation (Thomas et al., 2006; Wilhelm et al., 2006; Cho et al., 2007; Kaelin, 2009) and that mTOR signaling is upregulated in some cultured ccRCC cells because VHL mutant cells secret VEGF and platelet-derived growth factor-β, which activate PI3K-Akt-mTOR signaling (Wilhelm et al., 2006; Clark, 2009). Therefore, inhibiting mTOR signaling may potentially reduce the expression level of HIF-α in VHL mutant cells. However, the efficacy of mTOR inhibitors has not proven satisfactory. The zebrafish model may provide a plausible answer to this discrepancy. TOR signaling is the main regulator of protein synthesis and is the primary target for control during energy stress (Ma and Blenis, 2009). One of the major energy stress response pathway is mediated by the liver kinase β1 (LKβ1) and AMP-activated protein kinase cascade (Alexander and Walker, 2011). A primary inhibitory target of AMP-activated protein kinase is TOR. Thus, during energy starvation AMP-activated protein kinase is activated by LKβ1 and it reduces protein synthesis by inhibiting TOR activity. Interestingly zvhl mutation can suppress the hypermetabolism phenotype of lkβ mutant, due to reduced TOR signaling in zvhl mutant (van der Velden et al., 2011). This is presumably because persistent pseudo-hypoxic response in zvhl mutant cells leads to reduced TOR signaling via a yet unidentified alternative pathway. Therefore, if in physiological conditions, TOR signaling is already reduced in VHL mutant, additional TOR inhibitors is not likely to exert significant effects. This may explain the unsatisfactory efficacy of mTOR inhibitors in treating ccRCC.
Mouse
The mouse Vhl cDNA sequence was published in 1995 soon after the cloning of human VHL (Gao et al., 1995). In situ RNA analysis showed that Vhl is expressed ubiquitously in embryogenesis but with enrichment in the epithelial tissues of the lung, kidney and eye (Kessler et al., 1995). There are a number of mouse knockout models for Vhl, which have been described in an excellent review (Kapitsinou and Haase, 2008). Here I will highlight a few more recent attempts and discuss how they relate to the previous models. The first mouse knockout was generated as a constitutively null allele, which resulted in homozygous lethality at 10.5–12.5 days of gestation (Gnarra et al., 1997). The lethality was attributed to defects in vasculogenesis in the placenta. This is the first indication that VHL is indeed a factor in vasculoangiogenesis, as predicted. One seemingly counter-intuitive nature of this phenotype is that the outcome is a lack of vasculature, instead of the expected increased vasculogenesis (due to over-expressed VEGF). This is likely because the Vhl mutant vascular cells in the placenta, as in Drosophila trachea (Adryan et al., 2000), exhibit ectopic migratory activity, thus failing to interconnect and form vascular labyrinth.
The next knockout was a conditional allele (Haase et al., 2001). Surprisingly, both heterozygous null (one lox allele knockout in embryonic stem cells) and hepatocyte-specific knockout (using albumin promoter-directed Cre) resulted in cavernous hemangiomas of the liver, which can be correlated with increased level of HIF-2α and increased expression of VEGF. In addition, hepatocyte-specific knockout mutant mice developed polycythemia due to increased erythropoietin production in the liver. Although no hyperplasia was induced in these early Vhl knockout models, they did provide the in vivo confirmation of the VHL–HIF pathway. Subsequently, the first kidney proximal tubule conditional knockout was generated using the phosphoenolpyruvate carboxykinase (PEPCK) driver. Proximal tubule-specific knockout was attempted because proximal tubules have long been considered the origin of ccRCC. Somewhat surprisingly, this model generated only modest phenotype of renal microcysts in about 25% of >12-month old mice but no tumors. This was nonetheless significant because cysts are believed to be the precursor of ccRCC. It is formally possible that other genetic factors are needed for tumorigenesis in Vhl mutants. On the other hand, a recent seminal study suggests that proximal tubule cells may not be the origin of ccRCC, because in human VHL patient samples, VHL mutant proximal tubule cells do not show capacity to proliferate (Mandriota et al., 2002). More recently, Ksp1.3-Cre (expressed in distal tubules, ascending and descending loops of Henle and collecting ducts, but rarely in proximal tubules) was used to knockout Vhl outside of the proximal tubule domain (Frew et al., 2008b). This conditional knockout alone yielded hydronephrosis but no abnormalities in the tubule epithelium. However, when combined with another tumor suppressor gene knockout Pten, the kidney displayed, in addition to hydronephrosis, hyperproliferation of the urothelium and high penetrance of enlarged kidneys due to multiple epithelial tubule cysts in the cortex and medulla. This model strengthened the notion that loss of Vhl alone is insufficient for tumor initiation. Similarly, Vhl-Pten double conditional knockout in male genital track generated cystadenoma and squamous metaplasia (Frew et al., 2008a).
The vascular function of VHL was verified using endothelial cell-specific (Tie2-Cre-driven) knockout (Tang et al., 2006). The endothelial knockout also confirmed the defect in fibronectin deposition previously demonstrated in cell culture (Ohh et al., 1998; Feijoo-Cuaresma et al., 2008). Acute tamoxifen-induced mosaic inactivation of the Vhl gene at E10.5 resulted in embryonic lethality between E14.5 and E15.0 with extensive hemorrhage and necrosis (Hong et al., 2006). Liver damage and placental abnormalities were also observed, which confirmed the phenotypes of the previous Vhl knockouts. A number of subsequent conditional knockouts in other organs have shown defects in spermatogenesis, thymus cell survival and bone development due to reduced cell proliferation and/ or increased cell death (Ma et al., 2003; Biju et al., 2004; Pfander et al., 2004). More recently, Vhl has also been shown to influence the activity of HSCs (Takubo et al., 2010), which reside in hypoxic niches in the bone marrow and maintain a quiescent state. In Hif-1α-conditional allele (driven by interferon-inducible Mx1-Cre) mice, the HSCs lost cell cycle quiescence and their numbers decreased during various stress settings including bone marrow transplantation. Over-stabilization of HIF-1α in conditional biallelic loss of Vhl induced cell cycle quiescence in HSCs and their progenitors, as expected, but unexpectedly also resulted in impairment in bone marrow transplantation capacity, probably due to over-quiescence. In contrast, heterozygous loss of Vhl induced cell cycle quiescence in HSCs but improved (over-wild type) bone marrow engraftment after transplantation. It is possible that VHL modulates HSC activity by both HIF-dependent and independent mechanisms, and that the precise levels of its target proteins are critical. In this regard, it will be important to take into account the heterozygosity of VHL in the VHL disease patients, especially concerning the hematopoietic and vascular components of the disease manifestations.
The metabolic imbalance in VHL mutant cells due to HIF-mediated glycolic switch has attracted intensive interest, because this may present a new therapeutic approach by forcing oxidative metabolism on the mutant cells. A recent knockout model provided the physiological proof of glycolic switch in Vhl mutant cells (Zehetner et al., 2008). In this study, Vhl was conditionally knocked out in β cells of the pancreas. The mice developed impaired systemic glucose tolerance. This, intriguingly, can be attributed to the false sense of glucose shortage by the β cells because these Vhl mutant cells undergo glycolic switch and respond by increasing insulin secretion. In another study (Shen et al., 2009), intending to model the pancreatic cystadenoma of VHL disease, Vhl knockout in either α or β cells did not result in cysts formation. However, when Vhl was knocked out in the pancreatic progenitor cells using Pdx1-Cre, highly vascularized microcystic adenomas and hyperplastic islets did develop, thus representing the success modeling of one VHL disease-associated tumor phenotype.
One other intriguing disease model is the human polycythemic Chuvash disease. These patients inherit a homozygous VHL mutation that is incapable of down-regulate HIF-α, but they show no increased tumor incidences (Gordeuk et al., 2004). A mouse model for the Chuvash disease was generated by knocking in the R200W (R166W in mouse) mutant Vhl allele (Hickey et al., 2010). This mouse recapitulates the polycythemic phenotype of the human disease and exhibits pulmonary hypertension and lung fibrosis in a Hif-2α-dependent manner. The latter phenotypes also manifest in some Chuvash patients (Bushuev et al., 2006; Smith et al., 2006), which can be induced in hypoxic conditions (Stenmark et al., 2006). The fibrotic phenotype recalled an earlier knockout strain specific for the podocyte in kidney, which exhibited glomerulomegaly and occasional glomerulosclerosis (Brukamp et al., 2007). Interestingly, the earlier PEPCK-driven Vhl knockout could be induced to develop renal fibrosis after subtotal nephrectomy of one kidney and complete removal of the other (Kimura et al., 2008). In addition, hypoxia and increased Hif-1α activity have been linked to kidney fibrosis in mouse (Higgins et al., 2007). The connection between Vhl mutant cells and inflammation is worthy of more in-depth investigation, because prolonged inflammation can promote proliferation through the action of secreted cytokines, and importantly, can induce genetic changes in pre-cancerous cells by induction of reactive oxygen species or by oxidative inactivation of mismatch repair enzymes (Hussain et al., 2003; Colotta et al., 2009; Kraus and Arber, 2009; Grivennikov et al., 2010).
The information obtained from the above model organisms relevant to the epithelial and vascular phenotypes in VHL disease patients is summarized in Figure 3.
Figure 3.
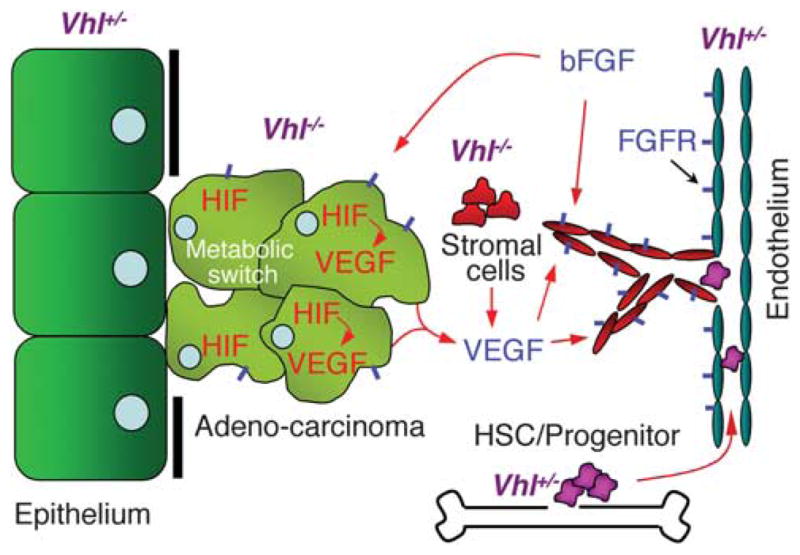
Epithelial–stromal interaction in VHL disease. The model is based on VHL functions verified in the model organisms and is focused on the epithelial and vascular phenotypes. Homozygous VHL mutant epithelial cells are shown losing epithelial characteristics (Duchi et al., 2010; Hsouna et al., 2010). These cells as well as heterozygous VHL mutant endothelial cells overexpress FGFR (short blue bar) on the surface, contributing to their increased response to the chemotactic bFGF ligand (Dammai et al., 2003; Champion et al., 2008; Hsouna et al., 2010). Mutant VHL cells overexpress HIF-α, leading to overproduction of VEGF, which in turn induces angiogenesis (van Rooijen et al., 2009, 2010) as well as metabolic switch that mimic hypoxic response (Bishop et al., 2004; Shen et al., 2005; Zehetner et al., 2008; van Rooijen et al., 2009). Homozygous VHL mutant ‘stromal cells’ neighboring the blood vessels, although not yet identified in model organisms, have been shown in human patients, presumably inducing angiogenesis via increased VEGF secretion (Vortmeyer et al., 1997; Park et al., 2007). Heterozygosity of VHL in the HSCs and resultant endothelial progenitor cells exhibit increased activity (Takubo et al., 2010; van Rooijen et al., 2010) and potentially can contribute to neovasculogenesis.
Conclusion and perspective
The studies in model organisms are summarized in Table 1. They have provided physiological proof of the VHL–HIF regulatory pathway. The ancestral function of C. elegans vhl suggests a role as a metabolic regulator in normal physiological condition and in hypoxia. Also importantly, potentially significant VHL functions in microtubule stabilization and endocytosis found physiological relevance in morphogenesis of organ-covering and tubular epithelia in the Drosophila system. New insights such as a potential role in hematopoietic maintenance and metabolic homeostasis were revealed in zebrafish and mouse systems. VHL has proved a unique and enigmatic tumor suppressor gene. Research in the past 20 years consisting of over 2000 publications has revealed that pVHL is a multi-functional protein. Accumulated evidence also suggests that, although the apparent pleiotropic functions of pVHL may have been the hindrance to a full understanding of its role in tumorigenesis, it is likely the combined loss of some of these functions that underlie the initiation of VHL tumors. To fully resolve the VHL disease mechanism, a systemic view of the disease process is essential to understand such complex problems as the tissue specificity of VHL tumors, metabolic abnormality, hematopoietic over-activation, and inflammation. Undoubtedly model systems will continue to have a critical role in elucidating the functions of this challenging but fascinating tumor suppressor gene.
Table 1.
Summary of VHL functions revealed in model organisms
| Species | Functions | References |
|---|---|---|
| Caenorhabditis elegans | Evolutionary conservation | (Woodward et al., 2000) |
| Identification of HIF prolyl hydroxylase | (Epstein et al., 2001) | |
| Regulation of hypoxic response | (Bishop et al., 2004; Shen et al., 2005) | |
| Regulation of energy-conserving metabolic pathways | (Bishop et al., 2004; Shen et al., 2005) | |
| Drosophila | VHL-elongin B/C complex formation | (Adryan et al., 2000; Aso et al., 2000) |
| Regulation of vasculogenesis | (Adryan et al., 2000; Centanin et al., 2008; Mortimer and Moberg, 2009; Hsouna et al., 2010) | |
| Regulation of hypoxic response | (Arquier et al., 2006; Centanin et al., 2008; Mortimer and Moberg, 2009) | |
| Regulation of endocytosis | (Hsouna et al., 2010) | |
| Regulation of epithelial cell motility | (Doronkin et al., 2010; Hsouna et al., 2010) | |
| Microtubule stabilization in epithelial morphogenesis | (Duchi et al., 2010) | |
| Zebrafish | Regulation of vasculo/angiogenesis | (van Rooijen et al., 2009, 2010) |
| Regulation of hypoxic response | (van Rooijen et al., 2009, 2010; Yaqoob and Schwerte, 2010) | |
| Polycythemic phenotype in mutant | (van Rooijen et al., 2009) | |
| Regulation of hematopoietic stem cell activity | (van Rooijen et al., 2009, 2010) | |
| Reduced TOR signaling in mutant | (van der Velden et al., 2011) | |
| Mouse | Regulation of vasculo/angiogenesis | (Gnarra et al., 1997; Hong et al., 2006; Tang et al., 2006) |
| Regulation of fibronectin deposition | (Tang et al., 2006) | |
| Cystic/adenoma growth in mutant | (Rankin et al., 2006; Frew et al., 2008a, b; Shen et al., 2009) | |
| Metabolic homeostasis | (Zehetner et al., 2008) | |
| Polycythemic phenotype | (Haase et al., 2001; Hickey et al., 2010) | |
| Regulation of hematopoietic stem cell activity | (Takubo et al., 2010) | |
| Inflammation/fibrosis phenotypes | (Brukamp et al., 2007; Kimura et al., 2008; Hickey et al., 2010) |
Abbreviations: HIF, hypoxia-inducible factor; TOR, target of rapamycin; VHL, von Hippel–Lindau tumor suppressor gene.
Acknowledgments
This work was funded by the National Cancer Institute (USA) grant (RO1CA109860).
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- Adryan B, Decker HJ, Papas TS, Hsu T. Tracheal development and the von Hippel-Lindau tumor suppressor homolog in Drosophila. Oncogene. 2000;19:2803–2811. doi: 10.1038/sj.onc.1203611. [DOI] [PubMed] [Google Scholar]
- Akis N, Madaio MP. Isolation, culture, and characterization of endothelial cells from mouse glomeruli. Kidney Int. 2004;65:2223–2227. doi: 10.1111/j.1523-1755.2004.00634.x. [DOI] [PubMed] [Google Scholar]
- Alexander A, Walker CL. The role of LKB1 and AMPK in cellular responses to stress and damage. FEBS Lett. 2011;585:952–957. doi: 10.1016/j.febslet.2011.03.010. [DOI] [PubMed] [Google Scholar]
- Arquier N, Vigne P, Duplan E, Hsu T, Therond PP, Frelin C, et al. Analysis of the hypoxia-sensing pathway in Drosophila melanogaster. Biochem J. 2006;393:471–480. doi: 10.1042/BJ20050675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aso T, Yamazaki K, Aigaki T, Kitajima S. Drosophila von Hippel-Lindau tumor suppressor complex possesses E3 ubiquitin ligase activity. Biochem Biophys Res Commun. 2000;276:355–361. doi: 10.1006/bbrc.2000.3451. [DOI] [PubMed] [Google Scholar]
- Banks RE, Tirukonda P, Taylor C, Hornigold N, Astuti D, Cohen D, et al. Genetic and epigenetic analysis of von Hippel-Lindau (VHL) gene alterations and relationship with clinical variables in sporadic renal cancer. Cancer Res. 2006;66:2000–2011. doi: 10.1158/0008-5472.CAN-05-3074. [DOI] [PubMed] [Google Scholar]
- Bartolini F, Gundersen GG. Generation of noncentrosomal microtubule arrays. J Cell Sci. 2006;119:4155–4163. doi: 10.1242/jcs.03227. [DOI] [PubMed] [Google Scholar]
- Behr M, Wingen C, Wolf C, Schuh R, Hoch M. Wurst is essential for airway clearance and respiratory-tube size control. Nat Cell Biol. 2007;9:847–853. doi: 10.1038/ncb1611. [DOI] [PubMed] [Google Scholar]
- Berra E, Benizri E, Ginouves A, Volmat V, Roux D, Pouyssegur J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1alpha in normoxia. EMBO J. 2003;22:4082–4090. doi: 10.1093/emboj/cdg392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betschinger J, Eisenhaber F, Knoblich JA. Phosphorylationinduced autoinhibition regulates the cytoskeletal protein Lethal (2) giant larvae. Curr Biol. 2005;15:276–282. doi: 10.1016/j.cub.2005.01.012. [DOI] [PubMed] [Google Scholar]
- Biju MP, Neumann AK, Bensinger SJ, Johnson RS, Turka LA, Haase VH. Vhlh gene deletion induces Hif-1-mediated cell death in thymocytes. Mol Cell Biol. 2004;24:9038–9047. doi: 10.1128/MCB.24.20.9038-9047.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilder D, Schober M, Perrimon N. Integrated activity of PDZ protein complexes regulates epithelial polarity. Nat Cell Biol. 2003;5:53–58. doi: 10.1038/ncb897. [DOI] [PubMed] [Google Scholar]
- Bishop T, Lau KW, Epstein AC, Kim SK, Jiang M, O’Rourke D, et al. Genetic analysis of pathways regulated by the von Hippel-Lindau tumor suppressor in Caenorhabditis elegans. PLoS Biol. 2004;2:e289. doi: 10.1371/journal.pbio.0020289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brauch H, Kishida T, Glavac D, Chen F, Pausch F, Hofler H, et al. Von Hippel-Lindau (VHL) disease with pheochromocytoma in the Black Forest region of Germany: evidence for a founder effect. Hum Genet. 1995;95:551–556. doi: 10.1007/BF00223868. [DOI] [PubMed] [Google Scholar]
- Brukamp K, Jim B, Moeller MJ, Haase VH. Hypoxia and podocyte-specific Vhlh deletion confer risk of glomerular disease. Am J Physiol Renal Physiol. 2007;293:F1397–F1407. doi: 10.1152/ajprenal.00133.2007. [DOI] [PubMed] [Google Scholar]
- Bushuev VI, Miasnikova GY, Sergueeva AI, Polyakova LA, Okhotin D, Gaskin PR, et al. Endothelin-1, vascular endothelial growth factor and systolic pulmonary artery pressure in patients with Chuvash polycythemia. Haematologica. 2006;91:744–749. [PubMed] [Google Scholar]
- Centanin L, Dekanty A, Romero N, Irisarri M, Gorr TA, Wappner P. Cell autonomy of HIF effects in Drosophila: tracheal cells sense hypoxia and induce terminal branch sprouting. Dev Cell. 2008;14:547–558. doi: 10.1016/j.devcel.2008.01.020. [DOI] [PubMed] [Google Scholar]
- Champion KJ, Guinea M, Dammai V, Hsu T. Endothelial function of von Hippel-Lindau tumor suppressor gene: control of fibroblast growth factor receptor signaling. Cancer Res. 2008;68:4649–4657. doi: 10.1158/0008-5472.CAN-07-6003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho D, Signoretti S, Regan M, Mier JW, Atkins MB. The role of mammalian target of rapamycin inhibitors in the treatment of advanced renal cancer. Clin Cancer Res. 2007;13:758s–7763s. doi: 10.1158/1078-0432.CCR-06-1986. [DOI] [PubMed] [Google Scholar]
- Clark PE. The role of VHL in clear-cell renal cell carcinoma and its relation to targeted therapy. Kidney Int. 2009;76:939–945. doi: 10.1038/ki.2009.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis. 2009;30:1073–1081. doi: 10.1093/carcin/bgp127. [DOI] [PubMed] [Google Scholar]
- Dammai V, Adryan B, Lavenburg KR, Hsu T. Drosophila awd, the homolog of human nm23, regulates FGF receptor levels and functions synergistically with shi/dynamin during tracheal development. Genes Dev. 2003;17:2812–2824. doi: 10.1101/gad.1096903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danilin S, Sourbier C, Thomas L, Rothhut S, Lindner V, Helwig JJ, et al. von Hippel-Lindau tumor suppressor gene-dependent mRNA stabilization of the survival factor parathyroid hormonerelated protein in human renal cell carcinoma by the RNA-binding protein HuR. Carcinogenesis. 2009;30:387–396. doi: 10.1093/carcin/bgn275. [DOI] [PubMed] [Google Scholar]
- Datta K, Mondal S, Sinha S, Li J, Wang E, Knebelmann B, et al. Role of elongin-binding domain of von Hippel Lindau gene product on HuR-mediated VPF/VEGF mRNA stability in renal cell carcinoma. Oncogene. 2005;24:7850–7858. doi: 10.1038/sj.onc.1208912. [DOI] [PubMed] [Google Scholar]
- Doronkin S, Djagaeva I, Nagle ME, Reiter LT, Seagroves TN. Dose-dependent modulation of HIF-1alpha/sima controls the rate of cell migration and invasion in Drosophila ovary border cells. Oncogene. 2010;29:1123–1134. doi: 10.1038/onc.2009.407. [DOI] [PubMed] [Google Scholar]
- Duchek P, Somogyi K, Jekely G, Beccari S, Rorth P. Guidance of cell migration by the Drosophila PDGF/VEGF receptor. Cell. 2001;107:17–26. doi: 10.1016/s0092-8674(01)00502-5. [DOI] [PubMed] [Google Scholar]
- Duchi S, Fagnocchi L, Cavaliere V, Hsouna A, Gargiulo G, Hsu T. Drosophila VHL tumor-suppressor gene regulates epithelial morphogenesis by promoting microtubule and aPKC stability. Development. 2010;137:1493–1503. doi: 10.1242/dev.042804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O’Rourke J, Mole DR, et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107:43–54. doi: 10.1016/s0092-8674(01)00507-4. [DOI] [PubMed] [Google Scholar]
- Esteban MA, Harten SK, Tran MG, Maxwell PH. Formation of primary cilia in the renal epithelium is regulated by the von Hippel-Lindau tumor suppressor protein. J Am Soc Nephrol. 2006;17:1801–1806. doi: 10.1681/ASN.2006020181. [DOI] [PubMed] [Google Scholar]
- Feijoo-Cuaresma M, Mendez F, Maqueda A, Esteban MA, Naranjo-Suarez S, Castellanos MC, et al. Inadequate activation of the GTPase RhoA contributes to the lack of fibronectin matrix assembly in von Hippel-Lindau protein-defective renal cancer cells. J Biol Chem. 2008;283:24982–24990. doi: 10.1074/jbc.M709390200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frew IJ, Krek W. Multitasking by pVHL in tumour suppression. Curr Opin Cell Biol. 2007;19:685–690. doi: 10.1016/j.ceb.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Frew IJ, Krek W. pVHL: a multipurpose adaptor protein. Sci Signal. 2008;1:pe30. doi: 10.1126/scisignal.124pe30. [DOI] [PubMed] [Google Scholar]
- Frew IJ, Minola A, Georgiev S, Hitz M, Moch H, Richard S, et al. Combined VHLH and PTEN mutation causes genital tract cystadenoma and squamous metaplasia. Mol Cell Biol. 2008a;28:4536–4548. doi: 10.1128/MCB.02132-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frew IJ, Thoma CR, Georgiev S, Minola A, Hitz M, Montani M, et al. pVHL and PTEN tumour suppressor proteins cooperatively suppress kidney cyst formation. EMBO J. 2008b;27:1747–1757. doi: 10.1038/emboj.2008.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Naglich JG, Laidlaw J, Whaley JM, Seizinger BR, Kley N. Cloning and characterization of a mouse gene with homology to the human von Hippel-Lindau disease tumor suppressor gene: implications for the potential organization of the human von Hippel-Lindau disease gene. Cancer Res. 1995;55:743–747. [PubMed] [Google Scholar]
- Ghabrial A, Luschnig S, Metzstein MM, Krasnow MA. Branching morphogenesis of the Drosophila tracheal system. Annu Rev Cell Dev Biol. 2003;19:623–647. doi: 10.1146/annurev.cellbio.19.031403.160043. [DOI] [PubMed] [Google Scholar]
- Gnarra JR, Ward JM, Porter FD, Wagner JR, Devor DE, Grinberg A, et al. Defective placental vasculogenesis causes embryonic lethality in VHL-deficient mice. Proc Natl Acad Sci USA. 1997;94:9102–9107. doi: 10.1073/pnas.94.17.9102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordeuk VR, Sergueeva AI, Miasnikova GY, Okhotin D, Voloshin Y, Choyke PL, et al. Congenital disorder of oxygen sensing: association of the homozygous Chuvash polycythemia VHL mutation with thrombosis and vascular abnormalities but not tumors. Blood. 2004;103:3924–3932. doi: 10.1182/blood-2003-07-2535. [DOI] [PubMed] [Google Scholar]
- Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Schoell MC, Freeman RS. The von Hippel-Lindau protein sensitizes renal carcinoma cells to apoptotic stimuli through stabilization of BIM(EL) Oncogene. 2009;28:1864–1874. doi: 10.1038/onc.2009.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haase VH, Glickman JN, Socolovsky M, Jaenisch R. Vascular tumors in livers with targeted inactivation of the von Hippel-Lindau tumor suppressor. Proc Natl Acad Sci USA. 2001;98:1583–1588. doi: 10.1073/pnas.98.4.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harten SK, Shukla D, Barod R, Hergovich A, Balda MS, Matter K, et al. Regulation of renal epithelial tight junctions by the von Hippel-Lindau tumor suppressor gene involves occludin and claudin 1 and is independent of E-cadherin. Mol Biol Cell. 2009;20:1089–1101. doi: 10.1091/mbc.E08-06-0566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hergovich A, Lisztwan J, Barry R, Ballschmieter P, Krek W. Regulation of microtubule stability by the von Hippel-Lindau tumour suppressor protein pVHL. Nat Cell Biol. 2003;5:64–70. doi: 10.1038/ncb899. [DOI] [PubMed] [Google Scholar]
- Herman JG, Latif F, Weng Y, Lerman MI, Zbar B, Liu S, et al. Silencing of the VHL tumor-suppressor gene by DNA methylation in renal carcinoma. Proc Natl Acad Sci USA. 1994;91:9700–9704. doi: 10.1073/pnas.91.21.9700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickey MM, Richardson T, Wang T, Mosqueira M, Arguiri E, Yu H, et al. The von Hippel-Lindau Chuvash mutation promotes pulmonary hypertension and fibrosis in mice. J Clin Invest. 2010;120:827–839. doi: 10.1172/JCI36362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins DF, Kimura K, Bernhardt WM, Shrimanker N, Akai Y, Hohenstein B, et al. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J Clin Invest. 2007;117:3810–3820. doi: 10.1172/JCI30487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong SB, Furihata M, Baba M, Zbar B, Schmidt LS. Vascular defects and liver damage by the acute inactivation of the VHL gene during mouse embryogenesis. Lab Invest. 2006;86:664–675. doi: 10.1038/labinvest.3700431. [DOI] [PubMed] [Google Scholar]
- Hsouna A, Nallamothu G, Kose N, Guinea M, Dammai V, Hsu T. Drosophila von Hippel-Lindau tumor suppressor gene function in epithelial tubule morphogenesis. Mol Cell Biol. 2010;30:3779–3794. doi: 10.1128/MCB.01578-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu T, Adereth Y, Kose N, Dammai V. Endocytic function of von Hippel-Lindau tumor suppressor protein regulates surface localization of fibroblast growth factor receptor 1 and cell motility. J Biol Chem. 2006;281:12069–12080. doi: 10.1074/jbc.M511621200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain SP, Hofseth LJ, Harris CC. Radical causes of cancer. Nat Rev Cancer. 2003;3:276–285. doi: 10.1038/nrc1046. [DOI] [PubMed] [Google Scholar]
- Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- Jarecki J, Johnson E, Krasnow MA. Oxygen regulation of airway branching in Drosophila is mediated by branchless FGF. Cell. 1999;99:211–220. doi: 10.1016/s0092-8674(00)81652-9. [DOI] [PubMed] [Google Scholar]
- Kaelin WG. The von Hippel-Lindau tumor suppressor protein: roles in cancer and oxygen sensing. Cold Spring Harb Symp Quant Biol. 2005;70:159–166. doi: 10.1101/sqb.2005.70.001. [DOI] [PubMed] [Google Scholar]
- Kaelin WG., Jr Treatment of kidney cancer: insights provided by the VHL tumor-suppressor protein. Cancer. 2009;115:2262–2272. doi: 10.1002/cncr.24232. [DOI] [PubMed] [Google Scholar]
- Kamura T, Koepp DM, Conrad MN, Skowyra D, Moreland RJ, Iliopoulos O, et al. Rbx1, a component of the VHL tumor suppressor complex and SCF ubiquitin ligase. Science. 1999;284:657–661. doi: 10.1126/science.284.5414.657. [DOI] [PubMed] [Google Scholar]
- Kapitsinou PP, Haase VH. The VHL tumor suppressor and HIF: insights from genetic studies in mice. Cell Death Differ. 2008;15:650–659. doi: 10.1038/sj.cdd.4402313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler PM, Vasavada SP, Rackley RR, Stackhouse T, Duh FM, Latif F, et al. Expression of the Von Hippel-Lindau tumor suppressor gene, VHL, in human fetal kidney and during mouse embryogenesis. Mol Med. 1995;1:457–466. [PMC free article] [PubMed] [Google Scholar]
- Kim WY, Kaelin WG. Role of VHL gene mutation in human cancer. J Clin Oncol. 2004;22:4991–5004. doi: 10.1200/JCO.2004.05.061. [DOI] [PubMed] [Google Scholar]
- Kimura K, Iwano M, Higgins DF, Yamaguchi Y, Nakatani K, Harada K, et al. Stable expression of HIF-1alpha in tubular epithelial cells promotes interstitial fibrosis. Am J Physiol Renal Physiol. 2008;295:F1023–F1029. doi: 10.1152/ajprenal.90209.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus S, Arber N. Inflammation and colorectal cancer. Curr Opin Pharmacol. 2009;9:405–410. doi: 10.1016/j.coph.2009.06.006. [DOI] [PubMed] [Google Scholar]
- Kurban G, Duplan E, Ramlal N, Hudon V, Sado Y, Ninomiya Y, et al. Collagen matrix assembly is driven by the interaction of von Hippel-Lindau tumor suppressor protein with hydroxylated collagen IV alpha 2. Oncogene. 2008;27:1004–1012. doi: 10.1038/sj.onc.1210709. [DOI] [PubMed] [Google Scholar]
- Latif F, Tory K, Gnarra J, Yao M, Duh FM, Orcutt ML, et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science. 1993;260:1317–1320. doi: 10.1126/science.8493574. [DOI] [PubMed] [Google Scholar]
- Lisztwan J, Imbert G, Wirbelauer C, Gstaiger M, Krek W. The von Hippel-Lindau tumor suppressor protein is a component of an E3 ubiquitin-protein ligase activity. Genes Dev. 1999;13:1822–1833. doi: 10.1101/gad.13.14.1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonergan KM, Iliopoulos O, Ohh M, Kamura T, Conaway RC, Conaway JW, et al. Regulation of hypoxia-inducible mRNAs by the von Hippel-Lindau tumor suppressor protein requires binding to complexes containing elongins B/C and Cul2. Mol Cell Biol. 1998;18:732–741. doi: 10.1128/mcb.18.2.732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonser RR, Glenn GM, Walther M, Chew EY, Libutti SK, Linehan WM, et al. von Hippel-Lindau disease. Lancet. 2003;361:2059–2067. doi: 10.1016/S0140-6736(03)13643-4. [DOI] [PubMed] [Google Scholar]
- Ma W, Tessarollo L, Hong SB, Baba M, Southon E, Back TC, et al. Hepatic vascular tumors, angiectasis in multiple organs, and impaired spermatogenesis in mice with conditional inactivation of the VHL gene. Cancer Res. 2003;63:5320–5328. [PubMed] [Google Scholar]
- Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol. 2009;10:307–318. doi: 10.1038/nrm2672. [DOI] [PubMed] [Google Scholar]
- Maher ER, Neumann HP, Richard S. von Hippel-Lindau disease: a clinical and scientific review. Eur J Hum Genet. 2011;19:617–623. doi: 10.1038/ejhg.2010.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandriota SJ, Turner KJ, Davies DR, Murray PG, Morgan NV, Sowter HM, et al. HIF activation identifies early lesions in VHL kidneys: evidence for site-specific tumor suppressor function in the nephron. Cancer Cell. 2002;1:459–468. doi: 10.1016/s1535-6108(02)00071-5. [DOI] [PubMed] [Google Scholar]
- McDonald JA, Pinheiro EM, Montell DJ. PVF1, a PDGF/VEGF homolog, is sufficient to guide border cells and interacts genetically with Taiman. Development. 2003;130:3469–3478. doi: 10.1242/dev.00574. [DOI] [PubMed] [Google Scholar]
- Mikhaylova O, Ignacak ML, Barankiewicz TJ, Harbaugh SV, Yi Y, Maxwell PH, et al. The von Hippel-Lindau tumor suppressor protein and Egl-9-Type proline hydroxylases regulate the large subunit of RNA polymerase II in response to oxidative stress. Mol Cell Biol. 2008;28:2701–2717. doi: 10.1128/MCB.01231-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montell DJ. Border-cell migration: the race is on. Nat Rev Mol Cell Biol. 2003;4:13–24. doi: 10.1038/nrm1006. [DOI] [PubMed] [Google Scholar]
- Mortimer NT, Moberg KH. Regulation of Drosophila embryonic tracheogenesis by dVHL and hypoxia. Dev Biol. 2009;329:294–305. doi: 10.1016/j.ydbio.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller RU, Fabretti F, Zank S, Burst V, Benzing T, Schermer B. The von Hippel Lindau tumor suppressor limits longevity. J Am Soc Nephrol. 2009;20:2513–2517. doi: 10.1681/ASN.2009050497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nallamothu G, Dammai V, Hsu T. Developmental function of Nm23/awd: a mediator of endocytosis. Mol Cell Biochem. 2009;329:35–44. doi: 10.1007/s11010-009-0112-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nallamothu G, Woolworth JA, Dammai V, Hsu T. Awd, the homolog of metastasis suppressor gene Nm23, regulates Drosophila epithelial cell invasion. Mol Cell Biol. 2008;28:1964–1973. doi: 10.1128/MCB.01743-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohh M, Yauch RL, Lonergan KM, Whaley JM, Stemmer-Rachamimov AO, Louis DN, et al. The von Hippel-Lindau tumor suppressor protein is required for proper assembly of an extracellular fibronectin matrix. Mol Cell. 1998;1:959–968. doi: 10.1016/s1097-2765(00)80096-9. [DOI] [PubMed] [Google Scholar]
- Park DM, Zhuang Z, Chen L, Szerlip N, Maric I, Li J, et al. von Hippel-Lindau disease-associated hemangioblastomas are derived from embryologic multipotent cells. PLoS Med. 2007;4:e60. doi: 10.1371/journal.pmed.0040060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pause A, Lee S, Worrell RA, Chen DY, Burgess WH, Linehan WM, et al. The von Hippel-Lindau tumor-suppressor gene product forms a stable complex with human CUL-2, a member of the Cdc53 family of proteins. Proc Natl Acad Sci USA. 1997;94:2156–2161. doi: 10.1073/pnas.94.6.2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfander D, Kobayashi T, Knight MC, Zelzer E, Chan DA, Olsen BR, et al. Deletion of Vhlh in chondrocytes reduces cell proliferation and increases matrix deposition during growth plate development. Development. 2004;131:2497–2508. doi: 10.1242/dev.01138. [DOI] [PubMed] [Google Scholar]
- Rankin EB, Tomaszewski JE, Haase VH. Renal cyst development in mice with conditional inactivation of the von Hippel-Lindau tumor suppressor. Cancer Res. 2006;66:2576–2583. doi: 10.1158/0008-5472.CAN-05-3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rorth P. Initiating and guiding migration: lessons from border cells. Trends Cell Biol. 2002;12:325–331. doi: 10.1016/s0962-8924(02)02311-5. [DOI] [PubMed] [Google Scholar]
- Schermer B, Ghenoiu C, Bartram M, Muller RU, Kotsis F, Hohne M, et al. The von Hippel-Lindau tumor suppressor protein controls ciliogenesis by orienting microtubule growth. J Cell Biol. 2006;175:547–554. doi: 10.1083/jcb.200605092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen C, Nettleton D, Jiang M, Kim SK, Powell-Coffman JA. Roles of the HIF-1 hypoxia-inducible factor during hypoxia response in Caenorhabditis elegans. J Biol Chem. 2005;280:20580–20588. doi: 10.1074/jbc.M501894200. [DOI] [PubMed] [Google Scholar]
- Shen HC, Adem A, Ylaya K, Wilson A, He M, Lorang D, et al. Deciphering von Hippel-Lindau (VHL/Vhl)-associated pancreatic manifestations by inactivating Vhl in specific pancreatic cell populations. PLoS One. 2009;4:e4897. doi: 10.1371/journal.pone.0004897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TG, Brooks JT, Balanos GM, Lappin TR, Layton DM, Leedham DL, et al. Mutation of von Hippel-Lindau tumour suppressor and human cardiopulmonary physiology. PLoS Med. 2006;3:e290. doi: 10.1371/journal.pmed.0030290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staller P, Sulitkova J, Lisztwan J, Moch H, Oakeley EJ, Krek W. Chemokine receptor CXCR4 downregulated by von Hippel-Lindau tumour suppressor pVHL. Nature. 2003;425:307–311. doi: 10.1038/nature01874. [DOI] [PubMed] [Google Scholar]
- Stebbins CE, Kaelin WG, Jr, Pavletich NP. Structure of the VHL-ElonginC–ElonginB complex: implications for VHL tumor suppressor function. Science. 1999;284:455–461. doi: 10.1126/science.284.5413.455. [DOI] [PubMed] [Google Scholar]
- Stenmark KR, Fagan KA, Frid MG. Hypoxia-induced pulmonary vascular remodeling: cellular and molecular mechanisms. Circ Res. 2006;99:675–691. doi: 10.1161/01.RES.0000243584.45145.3f. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Ohno S. The PAR-aPKC system: lessons in polarity. J Cell Sci. 2006;119:979–987. doi: 10.1242/jcs.02898. [DOI] [PubMed] [Google Scholar]
- Takubo K, Goda N, Yamada W, Iriuchishima H, Ikeda E, Kubota Y, et al. Regulation of the HIF-1alpha level is essential for hematopoietic stem cells. Cell Stem Cell. 2010;7:391–402. doi: 10.1016/j.stem.2010.06.020. [DOI] [PubMed] [Google Scholar]
- Tang N, Mack F, Haase VH, Simon MC, Johnson RS. pVHL function is essential for endothelial extracellular matrix deposition. Mol Cell Biol. 2006;26:2519–2530. doi: 10.1128/MCB.26.7.2519-2530.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoma CR, Frew IJ, Hoerner CR, Montani M, Moch H, Krek W. pVHL and GSK3beta are components of a primary ciliummaintenance signalling network. Nat Cell Biol. 2007;9:588–595. doi: 10.1038/ncb1579. [DOI] [PubMed] [Google Scholar]
- Thoma CR, Toso A, Gutbrodt KL, Reggi SP, Frew IJ, Schraml P, et al. VHL loss causes spindle misorientation and chromosome instability. Nat Cell Biol. 2009;11:994–1001. doi: 10.1038/ncb1912. [DOI] [PubMed] [Google Scholar]
- Thomas GV, Tran C, Mellinghoff IK, Welsbie DS, Chan E, Fueger B, et al. Hypoxia-inducible factor determines sensitivity to inhibitors of mTOR in kidney cancer. Nat Med. 2006;12:122–127. doi: 10.1038/nm1337. [DOI] [PubMed] [Google Scholar]
- Tsarouhas V, Senti KA, Jayaram SA, Tiklova K, Hemphala J, Adler J, et al. Sequential pulses of apical epithelial secretion and endocytosis drive airway maturation in Drosophila. Dev Cell. 2007;13:214–225. doi: 10.1016/j.devcel.2007.06.008. [DOI] [PubMed] [Google Scholar]
- Uv A, Cantera R, Samakovlis C. Drosophila tracheal morphogenesis: intricate cellular solutions to basic plumbing problems. Trends Cell Biol. 2003;13:301–309. doi: 10.1016/s0962-8924(03)00083-7. [DOI] [PubMed] [Google Scholar]
- van der Velden YU, Wang L, Zevenhoven J, van Rooijen E, van Lohuizen M, Giles RH, et al. The serine-threonine kinase LKB1 is essential for survival under energetic stress in zebrafish. Proc Natl Acad Sci USA. 2011;108:4358–4363. doi: 10.1073/pnas.1010210108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rooijen E, Voest EE, Logister I, Bussmann J, Korving J, van Eeden FJ, et al. von Hippel-Lindau tumor suppressor mutants faithfully model pathological hypoxia-driven angiogenesis and vascular retinopathies in zebrafish. Dis Model Mech. 2010;3:343–353. doi: 10.1242/dmm.004036. [DOI] [PubMed] [Google Scholar]
- van Rooijen E, Voest EE, Logister I, Korving J, Schwerte T, Schulte-Merker S, et al. Zebrafish mutants in the von Hippel-Lindau tumor suppressor display a hypoxic response and recapitulate key aspects of Chuvash polycythemia. Blood. 2009;113:6449–6460. doi: 10.1182/blood-2008-07-167890. [DOI] [PubMed] [Google Scholar]
- Vortmeyer AO, Gnarra JR, Emmert-Buck MR, Katz D, Linehan WM, Oldfield EH, et al. von Hippel-Lindau gene deletion detected in the stromal cell component of a cerebellar hemangioblastoma associated with von Hippel-Lindau disease. Hum Pathol. 1997;28:540–543. doi: 10.1016/s0046-8177(97)90075-7. [DOI] [PubMed] [Google Scholar]
- Wang Y, Roche O, Yan MS, Finak G, Evans AJ, Metcalf JL, et al. Regulation of endocytosis via the oxygen-sensing pathway. Nat Med. 2009;15:319–324. doi: 10.1038/nm.1922. [DOI] [PubMed] [Google Scholar]
- Wilhelm S, Carter C, Lynch M, Lowinger T, Dumas J, Smith RA, et al. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat Rev Drug Discov. 2006;5:835–844. doi: 10.1038/nrd2130. [DOI] [PubMed] [Google Scholar]
- Wingrove JA, O’Farrell PH. Nitric oxide contributes to behavioral, cellular, and developmental responses to low oxygen in Drosophila. Cell. 1999;98:105–114. doi: 10.1016/S0092-8674(00)80610-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodward ER, Buchberger A, Clifford SC, Hurst LD, Affara NA, Maher ER. Comparative sequence analysis of the VHL tumor suppressor gene. Genomics. 2000;65:253–265. doi: 10.1006/geno.2000.6144. [DOI] [PubMed] [Google Scholar]
- Woolworth JA, Nallamothu G, Hsu T. The Drosophila metastasis suppressor gene Nm23 homolog, awd, regulates epithelial integrity during oogenesis. Mol Cell Biol. 2009;29:4679–4690. doi: 10.1128/MCB.00297-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka T, Horikoshi Y, Sugiyama Y, Ishiyama C, Suzuki A, Hirose T, et al. Mammalian Lgl forms a protein complex with PAR-6 and aPKC independently of PAR-3 to regulate epithelial cell polarity. Curr Biol. 2003;13:734–743. doi: 10.1016/s0960-9822(03)00244-6. [DOI] [PubMed] [Google Scholar]
- Yang H, Minamishima YA, Yan Q, Schlisio S, Ebert BL, Zhang X, et al. pVHL acts as an adaptor to promote the inhibitory phosphorylation of the NF-kappaB agonist Card9 by CK2. Mol Cell. 2007;28:15–27. doi: 10.1016/j.molcel.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaqoob N, Schwerte T. Cardiovascular and respiratory developmental plasticity under oxygen depleted environment and in genetically hypoxic zebrafish (Danio rerio) Comp Biochem Physiol A Mol Integr Physiol. 2010;156:475–484. doi: 10.1016/j.cbpa.2010.03.033. [DOI] [PubMed] [Google Scholar]
- Young AP, Schlisio S, Minamishima YA, Zhang Q, Li L, Grisanzio C, et al. VHL loss actuates a HIF-independent senescence programme mediated by Rb and p400. Nat Cell Biol. 2008;10:361–369. doi: 10.1038/ncb1699. [DOI] [PubMed] [Google Scholar]
- Zehetner J, Danzer C, Collins S, Eckhardt K, Gerber PA, Ballschmieter P, et al. PVHL is a regulator of glucose metabolism and insulin secretion in pancreatic beta cells. Genes Dev. 2008;22:3135–3146. doi: 10.1101/gad.496908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlotogora J. High frequencies of human genetic diseases: founder effect with genetic drift or selection? Am J Med Genet. 1994;49:10–13. doi: 10.1002/ajmg.1320490104. [DOI] [PubMed] [Google Scholar]