

Fluorescence-based intravital imaging approaches have been essential for studying the function and distribution of biologically active small molecules and their molecular targets in vivo.[1] While optical imaging enables visualization over a broad scope of spatial resolution ranging from subcellular to whole body anatomical imaging, its application is constrained by the optical transparency of tissues. Optical imaging modalities are thus better suited for small animal imaging rather than for translational applications.[2] In contrast, Positron Emission Tomography (PET) is used routinely for clinical applications but its use is limited primarily to macroscopic spatial and temporal imaging. A strategy that would enable seamless switching between fluorescence and PET imaging would thus be highly desirable for several reasons. First, the ability to visualize PET probes at the microscopic level would allow more rapid validation of the former in vivo and consequently expedite the development of novel PET probes. Second, for macroscopic hybrid imaging[3], it would be possible for optical and PET signals to be superimposed onto structural information derived from magnetic resonance imaging (MRI) or computed tomography (CT). Likewise, the ability to image in more than one or two channels would be useful for visualizing underlying physiological events. Third, hits from microscopic screens of small molecule fluorochrome-adducts could be more rapidly translated into clinically relevant applications.
To date, the development of hybrid PET/optical probes has been hindered by various methodological limitations. As a result, a synthetic strategy that enables general access to such compounds has yet to be established. For any approach to be successful, however, two critical factors must be considered: 1) the short half-lives of clinically relevant PET radioisotopes, namely 18F (110 minutes) and 11C (20 minutes), both of which (ideally) require the introduction of the nuclide during the final synthetic steps; and 2) the pharmacodynamic changes that result from addition of the imaging tag(s). Because of these limiting factors, previous strategies have often relied on the use of distinct labels for each imaging modality; for example, PET ligands and fluorochromes have been used simultaneously on nanoparticles.[3] However, these methods result in chemically distinct compounds with non-identical pharmacological properties.
Here, we introduce a generic and widely applicable approach that enables boron-dipyrromethene (BODIPY)-based hybrid PET/fluorescence molecular tomography (FMT)-imaging agents to be accessed via the 19F/18F exchange of one of the fluorides within the canonical BF2-dipyrromethene core 1, a motif that is shared by most BODIPY dyes. This efficient method for incorporating a PET tracer thus evades the above-mentioned bottleneck in drug testing. We believe that this novel approach, which uses hybrid fluorescence/18F tags, will accelerate not only the development of imaging agents, but perhaps also the screening of therapeutic molecules.
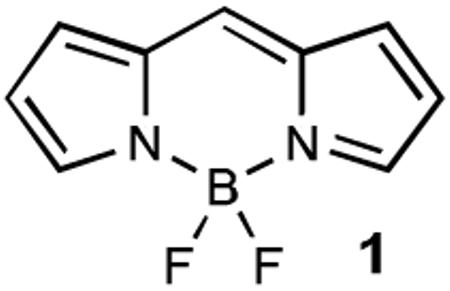
BODIPY dyes represent a unique class of fluorescent small molecules, which have recently received recognition for their extraordinary versatility as fluorescent tags in biological imaging applications.[4] The most widely appreciated characteristics of BODIPY dyes include their good photostability, high brightness, compatibility with biological media and their broad tunable color range that covers the green to near-infrared spectrum.[5] One particular advantage of BODIPY dyes over the majority of other commonly used fluorescent dyes is the neutral nature of their scaffold. This feature is critical for the efficient penetration of cell membranes, and as such BODIPY dyes remain one the few fluorophore classes suited to intracellular imaging within living cells.
These attractive features of the BODIPY dyes, combined with the presence of the canonical BF2-group within their central core, highlighted this fluorochrome class as a promising candidate for the development of hybrid 18F PET/FMT imaging agents via a 19F/18F exchange. This strategy would not only allow the wide variety of analogs already commercially available to be exploited, but also allows the desirable characteristics of the BODIPY dyes to be retained. Furthermore, this exchange system could potentially even allow the seamless conversion of BODIPY-tagged small molecule probe compounds, already validated for fluorescent imaging, into PET imaging probes.
Inspired by the recent report by Hudnall and Gabbaï, which described the development of a BODIPY-based fluorogenic sensor for fluoride ions, we chose to explore the use of a 4-dimethylaminopyridine (DMAP)-BODIPY intermediate as an activated reagent for the efficient incorporation of 18F.[6]
While the reported compound 3 was readily accessible following the published procedure, efficient incorporation of 18F to yield 4-[18F]-1,3,5,7,8-pentamethyl BODIPY [18F]2 could only be accomplished following microwave heating of the reaction mixture. Optimization of the reaction conditions subsequently allowed us to access the desired 18F-labeled [18F]2 in 67.6 ± 22.9 % radiochemical yield (Figure 1a). We next analyzed the stability of [18F]2 under physiological conditions to determine its applicability for pilot in vivo studies. As shown in Figure 1b, no radioactivity was released, even after 2 hours at 37°C in PBS buffer, validating the excellent hydrolytic stability of the BODIPY core under physiological conditions. We verified these findings by assessing the in vivo stability of [18F]2. The PET/CT scan (Figure 1c) demonstrates that [18F]2, as expected for a non-targeted small molecule, does not exhibit tissue specific labeling and is eliminated within 1 hour. BODIPY dyes have been used previously for in vivo imaging.[7] While some degree of metabolic modification is expected for virtually all small molecules including BODIPY labeled compounds, it is important to note that the BF2-core appears to be metabolically stable, as no measurable uptake of radioactivity in the skeleton was detected, which would sensitively indicate the release of free 18F−.[8]
Figure 1.
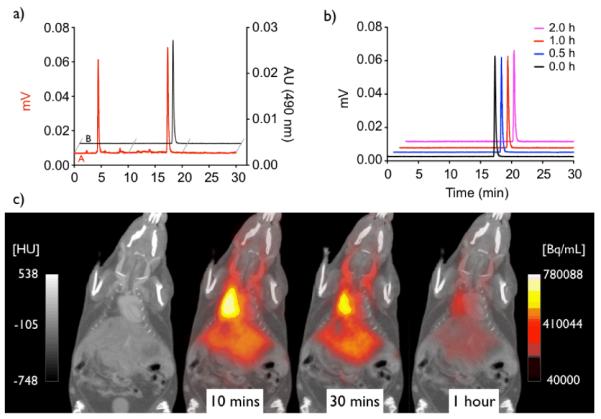
a) HPLC analyses of the labeling reaction mixture (red) and HPLC-purified 18F-BODIPY (black). b) Stability analyses of HPLC-purified [18F]2 in PBS buffer (pH = 7.4) at 37°C (individual traces are decay corrected). c) In vivo images of [18F]2 showing its systemic distribution, and rapid excretion within an hour, with no measurable release of free 18F . (left scale Hounsfield units [HU], right scale Becquerel/mL [Bq/mL])
While it was possible for us to synthesize [18F]2 successfully, we found that the specific reaction conditions necessary to do so had several shortcomings, which consequently limited the applicability of this approach. In particular, prolonged heating in the presence of the reactive reagents (to enable access to the DMAP-BODIPY intermediate), and the conditions required for efficient DMAP-18F exchange, are most likely to be incompatible with many common functional groups. This is especially true of more sensitive moieties, such as those typically present in complex biologically active small molecules or in activated BODIPY dyes, such as NHS-ester-bearing analogs. Reaction conditions that are compatible with the latter, however, are highly desirable since it would allow direct conversion of commercially available activated BODIPY esters into PET/FMT agents and consequently their immediate use for tagging amine-bearing small molecules or proteins. Moreover, upon heating, we observed that BODIPY dyes undergo a metathesis reaction in the presence of acid, where 19F is liberated from decomposed starting material and subsequently replaces 18F in [18F]2 (see Scheme 2). Following prolonged heating, this undesired side reaction results in low specific activity of the target compound.
Scheme 2.
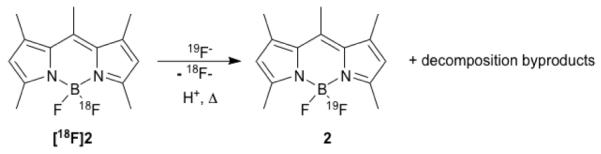
Acid and heat catalyzed 18F/19F metathesis.
During the course of our synthetic efforts, using 2 as a model substrate, we found that TMS-OTf efficiently abstracted one fluoride from the BODIPY core at 0°C within less than 5 minutes. This yielded the activated BODIPY-triflate 4 in near quantitative yields (Scheme 3).[9] The subsequent addition of free fluoride to 4 instantly and cleanly reconverted 4 back into its starting material 2, thus demonstrating the easily reversible, rapid and efficient incorporation of fluoride. Importantly, we discovered that the addition of a mild, non-nucluophilic, organic base such as diisopropylethylamine (DIPEA) or 2,6-lutidine 4 remained stable in solution at room temperature for prolonged time. In contrast, under unbuffered acidic conditions a metathesis reaction was seen to occur over time, causing the formation of 2 and decomposition byproducts. Thus, the efficient conversion of 4 into 2, under unbuffered conditions, consequently required immediate reaction with free fluoride following activation. While acidification of base-buffered 4 was required for the fluorination to proceed efficiently. Later reaction conditions therefore provide an additional level of reaction control. Furthermore, we found that addition of tert-butanol following TMS-OTf activation efficiently quenched excess TMS-OTf and reduced subsequent generation of TMS-18F to a minimum.
Scheme 3.
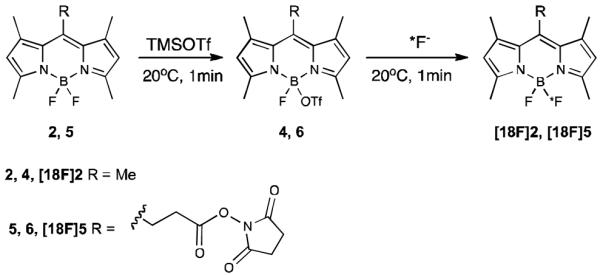
One-pot synthesis of 18F -labelled BODIPY dyes via activated BODIPY triflates.
We next explored the applicability of the above method to more sensitive molecules such as the BODIPY-NHS ester 5, which proved incompatible with the previously described DMAP-based approach. As expected, the NHS-ester was not only stable under the new reaction conditions but also allowed clean TMS-OTf activation and efficient re-fluorination following the simple one-pot strategy.
Following its validation under cold conditions, we next tested the applicability of the new strategy to the 18F-labeling of model substrate 2. We thus found that the methodology developed under non-radioactive conditions could readily be adapted to radiochemical synthesis. We observed, however, that a 4-5 fold molar excess of TMS-OTf was required for high radiochemical yields. We speculate that this is a consequence of the relatively small quantities of reagents needed for the radiosynthesis, which renders the reaction more sensitive to both residual water (present in the reaction vessel) and to the dried 18F starting material. Optimization of the reaction conditions allowed us to cleanly converted 2 to [18F]2 within 2 minutes in 67% radiochemical yield with a specific activity of 0.96Ci/umol (see Experimental Section and Supporting Information). Following the same strategy, [18F]5 was accessed via solid phase extraction in 73% radiochemical yield, with excellent purity (see Supporting Information). To subsequently demonstrate efficient biomolecule labeling, we selected Trastuzumab (Herceptin©, Genentech) as a clinically relevant model system.[10] Trastuzumab is a monoclonal antibody that binds to the HER2/neu receptor and is routinely used for the treatment of HER2-positive breast cancer.
Incubation of a PBS-solution of Trastuzumab with [18F]5 for 15 minutes at room temperature, followed by centrifugal ultrafiltration-dialysis, demonstrated that the monoclonal antibody could be efficiently labeled using our PET/FMT ligand (Figure 2).
Figure 2.
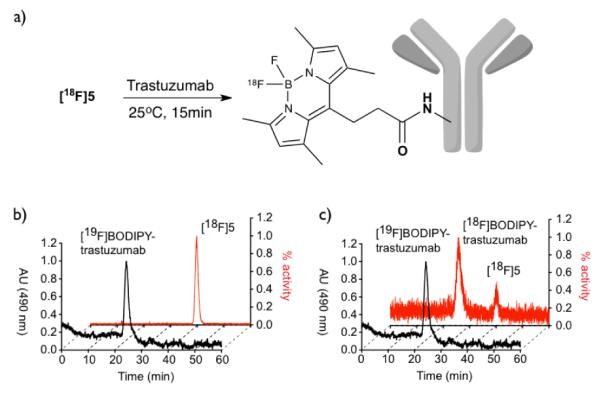
a) Labeling of Trastuzumab with [18F]5. b) Reference HPLC-traces of Trastuzumab labeled with cold 5 (front) and free [18F]5 (back). c) HPLC-traces of the reaction mixture.
In summary, we have developed an easily amenable and efficient approach for converting BODIPY dyes into hybrid PET/FMT imaging agents. We believe this approach is well suited for the labelling of small molecules as well as biological macromolecules. Given the stability of the BODIPY scaffold in vivo, small BODIPY-NHS esters or similar BODIPY dyes, functionalized with maleimides and click-tags, represent promising reagents that can now be readily converted into [18F]-PET probes and used to modify biologically relevant molecules for biomedical imaging.
Experimental Section
General procedure: At room temperature 5 μL of a 250 mM solution (1.25 μmol, 5 mol equivalents) trimethylsilyl triflate in acetonitrile were added to 0.25 μmol 4,4-Difluoro-1,3,5,7,8-pentamethyl-bodipy 2 in 25 μL acetonitrile/dichloromethane (2:1). After 1 min tert-butanol (5 μL, 250 mM in acetonitrile) was added to quench residual trimethylsilyl triflate, followed by 2,6-lutidine (5 μL, 250 mM in acetonitrile) to stabilize the BODIPY-triflate 4 intermediate. 1.6 μL (0.01 μmol) of this solution were combined with a solution of azeotropically dried 18F/TBAB (15 μL, 0.9 (33.3 MBq) ± 0.3 mCi) in acetontitrile, that had been pretreated with triflic anhydride (10 μL, 100 mM in acetonitrile) to remove residual water, followed by tert-butanol (5 μL, 250 mM in acetonitrile) to quench excess triflic anhydride and to acidify the reaction mixture (as result of the generated triflic acid). The reaction was complete after stirring for 1 min at room temperature. Radiochemical yield was determined by radio-HPLC and radio-TLC (10:1 acetonitrile:toluene, ITLC; Rf (18F− ) = 0.0; Rf(4-[18F]-1,3,5,7,8-pentamethyl bodipy [18F]2) = 0.91) dcRCY 66.7% (HPLC), 67.9% (TLC) with an specific activity of 0.96 Ci/μmol.
Supplementary Material
Scheme 1.
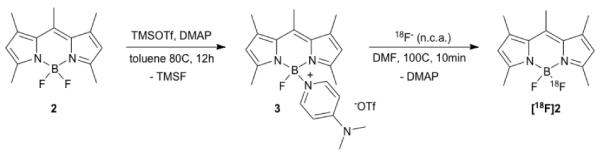
Synthesis of [18F]-BODIPY [18F]2 via DMAP activated BODIPY 3
Footnotes
Financial support from the NCI (P50CA086355, U24CA092782, T32-CA079443) is gratefully acknowledged
Supporting information for this article is available on the WWW Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.((Please delete if not appropriate))
References
- [1].Ntziachristos V, Ripoll J, Wang LV, Weissleder R. Nat. Biotechnol. 2005;23:313. doi: 10.1038/nbt1074. [DOI] [PubMed] [Google Scholar]; Pittet MJ, Weissleder R. Cell. 2011;147:983. doi: 10.1016/j.cell.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]; Weissleder R, Pittet MJ. Nature. 2008;452:580. doi: 10.1038/nature06917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Orth JD, Kohler RH, Foijer F, Sorger PK, Weissleder R, Mitchison TJ. Cancer Res. 2011;71:4608. doi: 10.1158/0008-5472.CAN-11-0412. [DOI] [PMC free article] [PubMed] [Google Scholar]; Reiner T, Thurber G, Gaglia J, Vinegoni C, Liew CW, Upadhyay R, Kohler RH, Li L, Kulkarni RN, Benoist C, Mathis D, Weissleder R. Proc. Natl. Acad. Sci. U S A. 2011;108:12815. doi: 10.1073/pnas.1109859108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Nahrendorf M, Keliher E, Marinelli B, Waterman P, Feruglio PF, Fexon L, Pivovarov M, Swirski FK, Pittet MJ, Vinegoni C, Weissleder R. Proc. Natl. Acad. Sci. U S A. 2010;107:7910. doi: 10.1073/pnas.0915163107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ulrich G, Ziessel R, Harriman A. Angew. Chem. Int. Ed. Engl. 2008;47:1184. doi: 10.1002/anie.200702070. [DOI] [PubMed] [Google Scholar]
- [5].Lavis LD, Raines RT. ACS Chem. Biol. 2008;3:142. doi: 10.1021/cb700248m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hudnall TW, Gabbaï FP. Chem. Commun. 2008:4596. doi: 10.1039/b808740g. [DOI] [PubMed] [Google Scholar]
- [7].Urano Y, Asanuma D, Hama Y, Koyama Y, Barrett T, Kamiya M, Nagano T, Watanabe T, Hasegawa A, Choyke PL, Kobayashi H. Nat. Med. 2009;15:104. doi: 10.1038/nm.1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Even-Sapir E, Mishani E, Flusser G, Metser U. Semin. Nucl. Med. 2007;37:462. doi: 10.1053/j.semnuclmed.2007.07.002. [DOI] [PubMed] [Google Scholar]; Hawkins RA, Choi Y, Huang SC, Hoh CK, Dahlbom M, Schiepers C, Satyamurthy N, Barrio JR, Phelps ME. J. Nucl. Med. 1992;33:633. [PubMed] [Google Scholar]
- [9].Activation was monitored by LC/MS. See supporting information
- [10].Lee-Hoeflich ST, Crocker L, Yao E, Pham T, Munroe X, Hoeflich KP, Sliwkowski MX, Stern HM. Cancer Res. 2008;68:5878. doi: 10.1158/0008-5472.CAN-08-0380. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.