

Abstract
The nucleocapsid protein (NC) of HIV type 1 is a nucleic acid chaperone that facilitates the rearrangement of nucleic acids into conformations containing the maximum number of complementary base pairs. We use an optical tweezers instrument to stretch single DNA molecules from the helix to coil state at room temperature in the presence of NC and a mutant form (SSHS NC) that lacks the two zinc finger structures present in NC. Although both NC and SSHS NC facilitate annealing of complementary strands through electrostatic attraction, only NC destabilizes the helical form of DNA and reduces the cooperativity of the helix-coil transition. In particular, we find that the helix-coil transition free energy at room temperature is significantly reduced in the presence of NC. Thus, upon NC binding, it is likely that thermodynamic fluctuations cause continuous melting and reannealing of base pairs so that DNA strands are able to rapidly sample configurations to find the lowest energy state. The reduced cooperativity allows these fluctuations to occur in the middle of complex double-stranded structures. The reduced stability and cooperativity, coupled with the electrostatic attraction generated by the high charge density of NC, is responsible for the nucleic acid chaperone activity of this protein.
The nucleocapsid protein (NC) of HIV type 1 (HIV-1) is a small, highly basic nucleic acid binding protein that contains only 55 amino acids and two zinc finger motifs. NC possesses nucleic acid chaperone activity, by which it facilitates the rearrangement of nucleic acid molecules into conformations that contain the maximum number of complementary base pairs. To achieve such rearrangements, the base pairs of nucleic acid structures that are normally very stable must be broken, while other complementary structures must be formed (1, 2). Although it seems likely that NC acts by destabilizing the base pairs of nucleic acid structures, this destabilization does not explain the enhanced annealing of complementary structures observed in the presence of NC. Thus, although the chaperone activity of NC is of crucial importance in the life cycle of the retrovirus, the mechanism by which it achieves this unusual activity is not well understood. Here we directly measure the destabilization of nucleic acid base pairing by NC and explain the origin of the enhanced annealing of complementary structures in the presence of this small protein.
Wild-type NC (hereafter referred to as NC) contains two zinc finger motifs of the form CCHC. This highly conserved sequence is found either once or twice in all retroviral NCs (except those of the spumavirus class) (3, 4) and strongly affects the nucleic acid binding specificity of NC (5). In addition, two high-resolution NMR structures of NC bound to viral RNA stem-loop sequences indicate that the zinc fingers interact specifically with several purine bases in the loop (6, 7). Although the zinc fingers are known to be required for viral replication (8–13), their specific role in nucleic acid chaperone activity is less clear. In a recent study, Levin and coworkers (14) found that a mutant form of NC, in which both CCHC motifs had been changed to SSHS (SSHS NC), was significantly less active in facilitating minus strand transfer and in blocking nonspecific self-priming reactions than wild-type NC (14).
To investigate the capability of NC to alter the base pairing of nucleic acid structures, we used an optical tweezers instrument to measure the effect of NC on the force-induced helix-coil transition of single λ-DNA molecules. We find that the helix-coil transition free energy is significantly reduced in the presence of NC, while the transition free energy is increased in the presence of SSHS NC. We also show that only wild-type NC alters the cooperativity of the helix-coil transition, a property that is consistent with effective rearrangement of large nucleic acid structures.
Materials and Methods
The dual-beam optical tweezers instrument used in this study consists of two counter propagating 150-mW, 850-nm diode lasers (SDL, San Jose, CA) focused to a small spot inside a liquid flow cell with 1.0 NA Nikon water-immersion microscope objectives. One 4.4-μm diameter streptavidin-coated polystyrene bead (Bangs Laboratories, Fisher, IN) was held in the optical trap formed by the laser beams, as shown in Fig. 1. Another streptavidin-coated bead was held on the end of a glass micropipette. To obtain force-extension measurements, a single DNA molecule that had been labeled on each end of the same strand with biotin was captured between the two beads (15). The DNA molecule then was stretched by moving the pipette and measuring the resulting force on the bead in the trap, as described (15, 16). The tethering buffer used in this study was 10 mM Hepes with 245 mM NaCl and 5 mM NaOH, pH 7.5. For experiments in lower ionic strength, the amount of added NaCl was reduced.
Figure 1.

Schematic drawing (not to scale) of an optical tweezers experiment in which a single DNA molecule is stretched between two 4.4-μm diameter polystyrene beads. One bead is held on the end of a glass micropipette by suction, while another bead is held in an optical trap.
The absolute extension of the molecule was estimated by measuring the distance between the centers of the two beads by using an image captured with a charge-coupled device camera (Edmund Industrial Optics, Barrington, NJ). The change in position of the pipette was measured by using a feedback-compensated piezoelectric translation stage that is accurate to 5 nm (Melles Griot, Irvine, CA). The position measurement was converted to a measurement of the molecular extension by correcting for the trap stiffness, which was 62 ± 3 pN/μm. For the measurements reported here, the pipette was moved in 100-nm steps, and after each step the force was measured 100 times and averaged, thus averaging out contributions of thermal motion to the force measurement. Each step took ≈0.5 s. At forces below 70 pN, the force-extension curves did not change significantly when the pulling rate was varied by changing the step size from 10 nm to 500 nm.
HIV-1 NC used in these experiments was prepared as described (17). All preparations were reconstituted with two molar equivalents of zinc. The preparation of SSHS mutant NC also has been described (14). After capturing a single DNA molecule in the tethering buffer, the molecule was stretched to verify that the usual force-extension curve was obtained. To measure the effect of the protein on this transition, a buffer solution with a reduced NaCl concentration containing a fixed amount of NC was added to the experimental cell until the buffer surrounding the captured DNA molecule was completely exchanged.
Results
The results of a typical experiment, in which λ-DNA is stretched in 100 mM ionic strength buffer at pH 7.5, are shown in Fig. 2. The force (F) vs. extension per base pair (b) curve begins to rise as the DNA helix is extended to near its normal B-form contour length of 0.34 nm/bp. At about 65 pN, a sharp overstretching transition occurs, where very little additional force is required to stretch the DNA molecule to 1.7 times its contour length. The DNA then is relaxed back to its initial structure (Fig. 2, open symbols). The relaxation curve resembles the stretch curve under these conditions, except for the region between b = 0.34 nm/bp and b = 0.42 nm/bp, where the data show some hysteresis (i.e., the stretch and relax curves to do not match exactly in this region).
Figure 2.

Typical room temperature force (F) as a function of extension per base pair (b) curve for a single dsDNA molecule in 10 mM Hepes, pH 7.5, 100 mM [Na+] (ionic strength). The data obtained while stretching (▴) and relaxing the λ-DNA (□) are very similar except for the region between b = 0.34 nm/bp and b = 0.42 nm/bp, which shows some hysteresis. The solid line on the left is the theoretical curve for an extensible wormlike chain (43) (dsDNA) with a persistence length of 50 nm, a contour length of 0.34 nm/bp, and an elastic stretch modulus of 1,000 pN. The solid line on the right is the curve for an extensible freely jointed chain (36) (ssDNA) with a persistence length of 0.75 nm, a contour length of 0.56 nm/bp, and an elastic stretch modulus of 800 pN (19). The thick solid line connecting the stretching data is the result of a fit to the Bragg-Zimm melting model by using the curves for dsDNA and ssDNA with σ = 10−3.
It has been suggested that the overstretching transition represents a structural change to a new form of double-stranded DNA, referred to as S-DNA (18, 19). However, we recently have shown that the overstretching force and thermal melting point of DNA exhibit similar trends as a function of pH, and that a model of the overstretching transition as force-induced melting (20, 21) accurately describes the dependence of the overstretching force on pH (16). In addition, our measurements of the temperature dependence of the overstretching transition force are well described by the force-induced melting model (15). Based on this work we were able to derive values for the entropy of DNA melting as well as the change in heat capacity of DNA upon melting that were in very good agreement with calorimetric measurements. Thus, this technique allows us to study the DNA helix-coil transition at very high resolution and at room temperature. The latter capability is particularly useful for studying protein-nucleic acid interactions, because temperature effects on protein structure are minimal at room temperature and do not complicate the analysis. We also are able to isolate the effects of protein-DNA interactions from interactions between DNA molecules.
We have used this technique to determine the effect of NC on the force versus extension curve of λ-DNA. When the DNA was stretched in the presence of NC, the cooperativity of the overstretching transition decreased significantly, as shown in Fig. 3. Cooperativity is a measure of the preference of the system for all helix or all coil states. In a highly cooperative system, as represented by the helix-coil transition in the absence of NC (Fig. 2), intermediate states between the helix and coil are difficult to achieve. Thus ≈65 pN of force are required to initiate the helix-coil transition, and the width of the transition is only ≈3 pN. At low ionic strength in the presence of NC, much lower force is required to initiate the transition and its width increases to a maximum value of ≈40 pN (Fig. 3). This increase in width is due to a decrease in cooperativity or an increase in the number of intermediate states that can be achieved by the system. As the experiments described below demonstrate, these changes are caused by NC binding rather than to low salt.
Figure 3.

DNA stretching curves in 10 mM Hepes, pH 7.5, containing 7 nM NC and 150 mM (▴), 75 mM (▵), 50 mM (■), and 25 mM [Na+] (□). b is the extension of the DNA molecule per base pair.
To interpret the changes observed in the stretching curves in the presence of NC, it is important to know the amount of NC bound per DNA nucleotide. Because there are no other DNA molecules to compete for binding to NC, the amount of NC bound to the λ-DNA molecule is determined by the ionic strength of the solution and the concentration of NC (22). The ratio of λ-DNA nucleotides to bound NC molecules (nt/NC ratio) in these experiments can be calculated based on the reported binding constant of NC to the SL3 DNA hairpin as a function of NaCl concentration (5). The NaCl dependence of the binding constant is given by
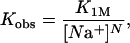 |
1 |
where K1M and N are the binding constant at 1 M NaCl and the effective charge of NC, respectively. The values have been fit to the binding constant measurements. The fraction θ of NC bound to the DNA is given by (22)
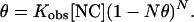 |
2 |
The nt/NC ratio is 1/θ. Here we solve this equation as a function of ionic strength by using K1M = 0.7 × 104 M−1 and n = 3.27, as determined for the binding of NC to the SL3 DNA hairpin (5) and an NC concentration of 7 nM used in our study. The values obtained are only approximate because the effective charge of NC is not well defined experimentally, and data for the binding of NC to polymeric DNA such as the λ-DNA used in these studies are not available. The parameters from binding measurements of NC to homopolymeric fluorescent RNA, poly(ɛA) (23), yield similar results for the calculation of θ by using Eq. 2.
With an NC concentration of 7 nM at 150 mM ionic strength, the nt/NC ratio is ≈50. Thus, under these conditions there was very little binding to the single DNA molecule (Fig. 3, filled triangles), and, as expected, the force-extension curve resembled that shown in Fig. 2. At 75 mM ionic strength, we observed a change in the cooperativity of the transition, indicated by an increase in the slope of the overstretching transition (Fig. 3, open triangles). At this ionic strength the nt/NC ratio is estimated to be ≈13. In 50 mM ionic strength, the slope of the overstretching transition was further increased (Fig. 3, filled squares), and the calculated nt/NC ratio is ≈8, which is similar to the ratio required for optimal nucleic acid chaperone activity (2). Finally, at 25 mM ionic strength very little additional change in slope is observed (Fig. 3, open squares), suggesting that the effect of NC on the overstretching transition saturates at this ionic strength. The effects we observe on the cooperativity of the helix-coil transition upon NC binding may, in part, be due to changes in helix geometry or local base stacking interactions. However, the fact that NC induces strand separation at much lower forces indicates that the protein facilitates destabilization of double-stranded DNA (dsDNA) rather than just changing the helix geometry.
Although the effects we observe on the cooperativity of the helix-coil transition in the presence of NC are amplified at low ionic strength, we can achieve the same effects shown here in physiological salt by increasing the concentration of NC. For example, when the NC concentration is increased to 15 nM, we observe the same change in the slope of the overstretching transition at 100 mM ionic strength as we observed with a 7 nM concentration of NC in 75 mM ionic strength solution (data not shown). This finding is consistent with the binding calculations above. However, changing the salt concentration allows us to explore a much wider range of binding.
In the range of 50- to 150-mM ionic strength, all of the force-extension curves in the presence of NC matched at extensions less than the B-form contour length of dsDNA (0.34 nm/bp). There was no indication of NC or DNA aggregation under these conditions. However, as the ionic strength was lowered to 25 mM, a small force was required to stretch dsDNA even at extensions well below the contour length of the molecule (Fig. 3). This finding suggests that additional binding of positively charged NC at low ionic strength induces DNA aggregation, consistent with previous studies showing that high concentrations of NC cause RNA to aggregate (24, 25).
In the absence of a DNA binding protein, forces greater than 140 pN are required to completely separate two DNA strands by stretching (26). At ionic strength ≥50 mM, after a stretch/relax cycle where we stretched the DNA to 80 pN in the presence of NC, subsequent stretches always retraced the initial stretch curve. Although there was much greater hysteresis than without NC, the strands always reannealed when relaxed (data not shown). Thus, complete separation of the two DNA strands to obtain single-stranded DNA (ssDNA) was never achieved at ionic strength ≥50 mM. However, when we stretched dsDNA to only 80 pN in the presence of NC at 25 mM ionic strength, the relaxation curve resembled that of ssDNA (Fig. 4). When the DNA strand was stretched again in the presence of NC at 25 mM ionic strength, we obtained the same curve as the relaxation curve (Fig. 4 and data not shown). Taken together, these curves appear to represent the stretching and relaxation of ssDNA with NC bound and suggest that under low salt conditions in the presence of NC complete separation of the two DNA strands is achieved. This observation is in contrast to the stretching and relaxation curves obtained under the same conditions in the absence of NC, which resemble each other with a small amount of hysteresis due to nonuniform reannealing (Fig. 4 Inset). These data strongly support the notion that NC binding destabilizes dsDNA rather than just inducing a change in the helix geometry or base-stacking interactions. We also note that a higher force is necessary to extend the NC-bound ssDNA compared with normal ssDNA (Fig. 4). Therefore, NC binding reduces the ssDNA contour length and significantly alters its stretching behavior.
Figure 4.

NC binding decreases the effective length of ssDNA. The data shown (■) are for λ-DNA relaxed in 10 mM Hepes, pH 7.5, 25 mM [Na+], following a stretch of dsDNA to 80 pN in the presence of 7 nM NC. The solid line is a fit to the data of Smith et al. (19) in which ssDNA was stretched in 150 mM NaCl. b is the extension of the DNA molecule per base pair. (Inset) The stretch (□) and relaxation (▴) curve for dsDNA in 25 mM ionic strength without NC.
To investigate the role of the zinc fingers in NC's nucleic acid chaperone activity, we also carried out DNA stretching measurements by using SSHS NC. Representative force-extension curves for DNA in the presence of this mutant form of NC are shown in Fig. 5. Even at the lowest ionic strength used here (25 mM), SSHS NC did not affect the cooperativity of the overstretching transition. In addition, SSHS NC increased the overstretching force in low ionic strength. In 50 mM ionic strength, the overstretching force without NC is 60 pN. When SSHS NC is added to the solution, the transition force increases to 68 pN, while its cooperativity is unchanged (Fig. 5, filled triangles). As the ionic strength is lowered to 25 mM, a small force is required to stretch the DNA to its B-form contour length in the presence of SSHS NC (Fig. 5, open squares), indicating that enough protein has bound to dsDNA to cause aggregation. In this case, the force required to separate aggregated sections of DNA is constant at about 2–3 pN. This force is consistent with that required to stretch single DNA molecules that have been condensed with multivalent cations such as cobalt hexamine (CoHex3+) and spermidine (Spd3+) (27–29). It also has been shown that a mutant of NC without zinc fingers was able to induce dense compact aggregates of ssDNA (24). However, even when enough SSHS NC has bound to cause this condensation, the overstretching force is still 68 pN and the cooperativity remains constant. This finding indicates that SSHS NC binds preferentially to dsDNA, most likely due to the higher charge density of dsDNA compared with ssDNA. This conclusion is supported by the fact that very little hysteresis is observed in the presence of SSHS NC (data not shown), indicating that the double-stranded form of DNA is strongly favored in this case. In contrast, in the presence of NC there is significant hysteresis (Figs. 3 and 4), indicating stabilization of the single-stranded form of DNA by NC.
Figure 5.

Binding of SSHS NC to DNA increases the overstretching force and does not affect the cooperativity of the overstretching transition. The stretching curves for λ-DNA in 10 mM Hepes, pH 7.5, in the presence of 7 nM SSHS NC are shown for 50 mM (▴) and 25 mM (□) [Na+]. For comparison, we also show the data for stretching DNA in 50 mM ionic strength without NC (▵). b is the extension of the DNA molecule per base pair.
Discussion
In this study, we use an optical tweezers instrument to measure the effect of NC on the helix-coil transition of single DNA molecules at room temperature. We show that NC binding alters the cooperativity of the transition, whereas the SSHS mutant form of NC, which lacks the zinc fingers structures, does not have this effect. Furthermore, whereas NC destabilizes the double-stranded form of DNA, the SSHS mutant stabilizes the double helix.
These results can be quantified by fitting the force-extension behavior to the standard Bragg–Zimm model for a helix-coil transition (Table 1) (20, 30). Theoretical force-extension curves for dsDNA (left solid line) and ssDNA (right solid line) are shown in Fig. 2. We use these curves to predict the shape of the overstretching transition. In this model, the fraction of base pairs in the helical state is given by
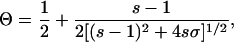 |
3 |
where s is the equilibrium constant for conversion of a base pair from single-stranded to double-stranded form, given by
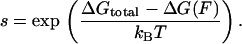 |
4 |
The free energy of the helix-coil transition as a function of force ΔG(F) is obtained from the theoretical force-extension curves (20), where
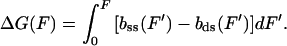 |
5 |
bss(F) and bds(F) describe the theoretical force-extension curves of ssDNA and dsDNA, respectively. ΔGtotal is obtained by directly calculating the area between the experimental stretching curves for dsDNA and ssDNA. The DNA extension as a function of force then is given by
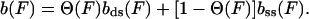 |
6 |
By fitting these relations to our experimental force-extension curves, we obtain both s and an estimate of the cooperativity parameter σ. The value of this parameter increases as the transition cooperativity decreases. The average number of bases that must be simultaneously melted to nucleate a melted domain within a double-stranded structure is approximately σ−1/2 (31).
Table 1.
Free energy (ΔGtotal) and cooperativity (σ) of the helix-coil transition of a single λ-DNA molecule as a function of ionic strength in the absence of protein and in the presence of 7 nM SSHS NC or wild-type (wt) NC
| Ionic strength, mM | No NC
|
SSHS
NC
|
wt NC
|
|
|---|---|---|---|---|
| ΔGtotal, kcal/mol⋅bp | ΔGtotal, kcal/mol⋅bp | ΔGtotal, kcal/mol⋅bp | σ | |
| 150 | 1.5 ± 0.1 | 1.5 ± 0.1 | 1.5 ± 0.1 | 0.001 |
| 100 | 1.5 ± 0.1 | 1.4 ± 0.1 | 1.4 ± 0.1 | 0.001 |
| 75 | 1.4 ± 0.1 | 1.4 ± 0.1 | 1.2 ± 0.3 | 0.05 |
| 50 | 1.3 ± 0.1 | 1.6 ± 0.1 | 0.6 ± 0.2 | 0.13 |
| 25 | 1.2 ± 0.1 | 1.6 ± 0.2 | 0.6 ± 0.1 | 0.25 |
Cooperativity results are shown only for wt NC, because this value remains essentially constant at 0.001 in the absence of NC or with SSHS NC. Reported error is calculated as the root mean square of the error due to reproducibility of the force measurement for at least three stretches and estimated error due to interpolation of ssDNA curves.
Fig. 6 shows representative fits to the model at 50 and 150 mM ionic strength in the presence of NC (filled triangles and filled squares, respectively). The values of σ obtained from our fits, as well as the helix-coil transition free energies ΔGtotal in the absence of NC and in the presence of wild-type or SSHS NC are given in Table 1. The model gives a very good fit to the data. The transition free energy at ionic strength ≤50 mM with wild-type NC is half the free energy of the transition without NC. These data are consistent with the preferential binding of NC to ssDNA (32). At room temperature, the helix-coil transition free energy in 50 mM ionic strength is reduced from 1.3 kcal/mol bp, or ≈2 kBT, to 0.6 kcal/mol bp, or ≈1 kBT. Under these conditions, we estimated that the bound nt/NC ratio is ≈8. Thus, at this critical binding density, thermal fluctuations are sufficient to melt single base pairs, allowing a DNA molecule to rapidly sample various base-paired configurations at room temperature.
Figure 6.

Representative fits of the data to the Bragg-Zimm melting model. The solid line on the left represents the elasticity of dsDNA according to the wormlike chain model (43). The solid line on the right is stretching data for ssDNA in 150 mM ionic strength without NC. The data for stretching dsDNA in the presence of NC at 150 mM ionic strength is shown (■) along with the fit to the melting model indicated as a line between these data points. The thick solid line is our data from stretching ssDNA (generated after the initial stretching and relaxation of dsDNA with NC) at 25 mM ionic strength in the presence of NC. The data for stretching dsDNA in 50 mM ionic strength with NC is also shown (▴), as well as a fit to that data, indicated as a line between the data points. b is the extension of the DNA molecule per base pair.
For wild-type NC, the cooperativity parameter increases from σ = 0.001 to σ = 0.25 as ionic strength is lowered from 150 mM to 25 mM (Table 1). This difference represents an apparent change in the average number of base pairs required to initiate a melted domain within a double-stranded nucleic acid structure from 32 to 2, which most likely results from a decrease in the entropy of ssDNA due to the binding of NC. A decrease in the entropy of the helix-coil transition should increase the free energy of the transition, given by ΔG = ΔH − TΔS. However, because NC binds preferentially to ssDNA, ΔH also is expected to decrease. Therefore, the total transition free energy decreases in the presence of NC (Table 1). The change in cooperativity may allow complementary strands to sample states that differ by only a few base pairs. The cooperativity parameter did not change significantly in the presence of SSHS NC and remained similar to its value of σ = 0.001 in the absence of NC.
The striking difference between our results for NC and SSHS NC offers insights into the role of zinc fingers in the nucleic acid chaperone activity of NC, as well as revealing details about the mechanism by which NC interacts with nucleic acids. It has been shown that NC facilitates annealing of tRNALys,3 to the primer binding site (PBS) of the HIV-1 genomic RNA (33–35). Similarly, SSHS NC also facilitates tRNA annealing (M. R. S. Hargittai, A. Mangla, R.G., and K.M.-F., unpublished work). In addition, previous studies with NC zinc finger mutants have concluded that the zinc fingers are relatively unimportant for genomic placement of tRNALys,3 in vivo (37) and for tRNA primer annealing in vitro (33, 38).
In vitro experiments measuring the effect of SSHS NC on annealing in plus- and minus-strand transfer also have been reported (14). Elimination of the zinc finger structure by the SSHS mutation showed no effect on the rates of annealing of the complementary 18-nt PBS sequences in plus-strand transfer. In contrast, annealing of the complementary repeat (R) region in minus-strand transfer is much less effective with SSHS NC than with wild-type NC (14). In this reaction, two strands that are both predicted to contain at least 22 intramolecular base pairs within the highly structured complementary TAR stem loops present in the R regions are annealed, resulting in a thermodynamically more stable RNA-DNA hybrid structure (1). Thus, the zinc finger structures apparently are needed to unfold highly structured RNA and DNA intermediates formed before minus-strand transfer, but are not absolutely required for some of the chaperone activities of NC, such as annealing of the tRNA primer and annealing in plus-strand transfer.
NC promotes efficient minus-strand transfer, in part, due to its ability to inhibit a self-priming reaction, which occurs due to intramolecular TAR-dependent secondary structure formation (39). This chaperone function of wild-type NC is in accordance with its preference for binding ssDNA (Fig. 4). In contrast, SSHS NC is unable to effectively block the TAR-induced self-priming reaction (14), an observation that is consistent with our data showing that this mutant form of NC stabilizes dsDNA (Fig. 5).
The data presented here help to explain the apparent paradox surrounding the role of the zinc fingers in various chaperone functions of NC. Our results support the conclusion that the zinc fingers are indeed important for nucleic acid chaperone function involving complex nucleic acid rearrangements. We have shown that both NC and SSHS NC act as multivalent cations and cause DNA aggregation or condensation. It is known that multivalent cations at concentrations that are sufficient to cause DNA condensation increase the efficiency of renaturation of melted DNA (40). NC also has been shown to facilitate renaturation of complementary DNA strands (1, 41). Thus, the electrostatic attraction between the tRNA primer and the PBS-containing RNA genome caused by the presence of NC or SSHS NC greatly enhances the efficiency of annealing tRNA to the PBS. Because tRNA annealing only requires the melting of the tertiary core and 12 bp in the acceptor-TΨC stem, the enhanced electrostatic attraction of SSHS is sufficient to overcome the energy barrier to melting the tRNA.
The fact that both SSHS and wild-type NC stimulate faster minus-strand transfer than in the absence of either of these proteins (14) indicates the importance of electrostatic attraction in nucleic acid chaperone activity, regardless of the structures to be annealed. However, wild-type NC is more efficient in the case of minus-strand transfer due to its ability to destabilize complex RNA and DNA structures, as indicated by the data shown in Table 1. This finding is consistent with a recent NMR study that demonstrated significant destabilization of the 18-nt minus-strand PBS DNA hairpin by NC (42). In contrast, SSHS NC stabilizes the double-stranded form of DNA (Fig. 5).
We have shown that NC also significantly reduces the cooperativity of the helix-coil transition. Although this property may not be required for the annealing of relatively simple structures such as short DNA or RNA hairpins, the reduction of the cooperativity will be important for any rearrangement of complex nucleic acid structures that requires the melting of base pairs in the middle of a double-stranded structure. For example, if the helix-coil transition in the presence of NC were completely noncooperative (σ = 1), it would be just as easy to melt base pairs in the middle of a DNA or RNA hairpin as it is to melt a base pair on the end of the structure.
We have demonstrated three important aspects of the nucleic acid chaperone activity of HIV-1 NC. First, NC facilitates annealing of complementary strands through electrostatic attraction based on its high positive charge. Second, NC significantly destabilizes dsDNA due to its preferential binding to ssDNA. Third, NC greatly reduces the cooperativity of the helix-coil transition of dsDNA structures. In contrast, SSHS NC has only the first of these three properties, which indicates that the zinc fingers of NC are responsible for the preferential binding to ssDNA and for its capability to alter the helix-coil transition. These properties are essential for the rearrangement of complex nucleic acid structures by NC, such as those required for minus-strand transfer during retroviral DNA synthesis.
Acknowledgments
We thank Prof. Matthew Tirrell and the University of Minnesota Center for Interfacial Engineering for funding and assistance in starting the optical tweezers project. We thank Donald G. Johnson, Bradley P. Kane, and Dr. Anil Mangla for purification of wild-type and SSHS NC, as well as Dr. Michael Summers for the kind gift of the wild-type NC clone. We are grateful to Drs. Steve Smith and Christoph Baumann for help with protocols and instrument-building advice and thank Dori Henderson for taking the time to make a number of glass micropipettes for use in our experiments. We also thank Drs. Judith Levin and Jianhui Guo for helpful discussions and critical reading of the manuscript. Funding for this project was provided by National Institutes of Health Grants GM28093 (V.B.) and GM49928 (K.M.F.), as well as National Science Foundation Grant MCB9728165 (V.B.). This work was also supported in part by the National Cancer Institute, Department of Health and Human Services, under Contract NO1-CO-56000 with Science Applications International Corporation Frederick (R.J.G.).
Abbreviations
- NC
nucleocapsid protein
- dsDNA
double-stranded DNA
- ssDNA
single-stranded DNA
- PBS
primer binding site
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Tsuchihashi Z, Brown P O. J Virol. 1994;68:5863–5870. doi: 10.1128/jvi.68.9.5863-5870.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rein A, Henderson L E, Levin J G. Trends Biochem Sci. 1998;23:297–301. doi: 10.1016/s0968-0004(98)01256-0. [DOI] [PubMed] [Google Scholar]
- 3.Berg J M. Science. 1986;232:485–487. doi: 10.1126/science.2421409. [DOI] [PubMed] [Google Scholar]
- 4.Covey S N. Nucleic Acids Res. 1986;14:623–633. doi: 10.1093/nar/14.2.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vuilleumier C, Bombarda E, Morellet N, Ge'rard D, Roques B P, Me'ly Y. Biochemistry. 1999;38:16816–16825. doi: 10.1021/bi991145p. [DOI] [PubMed] [Google Scholar]
- 6.De Guzman R N, Wu Z R, Stalling C C, Pappalardo L, Borer P N, Summers M F. Science. 1998;279:384–388. doi: 10.1126/science.279.5349.384. [DOI] [PubMed] [Google Scholar]
- 7.Amarasinghe G K, Guzman R N D, Turner R B, Chancellor K J, Wu Z R, Summers M F. J Mol Biol. 2000;301:491–511. doi: 10.1006/jmbi.2000.3979. [DOI] [PubMed] [Google Scholar]
- 8.Gorelick R J, Nigida S M, Jr, Bess J W, Jr, Arthur L O, Henderson L E, Rein A. J Virol. 1990;64:3207–3211. doi: 10.1128/jvi.64.7.3207-3211.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dorfman T, Luban J, Goff S P, Haseltine W A, Gottlinger H G. J Virol. 1993;67:6159–6169. doi: 10.1128/jvi.67.10.6159-6169.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Y, Barklis E. J Virol. 1995;69:5716–5722. doi: 10.1128/jvi.69.9.5716-5722.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berkowitz R, Fisher J, Goff S P. Curr Top Microbiol Immunol. 1996;214:177–218. doi: 10.1007/978-3-642-80145-7_6. [DOI] [PubMed] [Google Scholar]
- 12.Tanchou V, Decimo D, Pechoux C, Lener D, Rogemond V, Berthoux L, Ottmann M, Darlix J L. J Virol. 1998;72:4442–4447. doi: 10.1128/jvi.72.5.4442-4447.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gorelick R, Gagliardi T, Bosche W, Wiltrout T, Coren L, Chabot D, Lifson J, Henderson L, Arthur A. Virology. 1999;256:92–104. doi: 10.1006/viro.1999.9629. [DOI] [PubMed] [Google Scholar]
- 14.Guo J, Wu T, Anderson J, Kane B F, Johnson D G, Gorelick R J, Henderson L E, Levin J G. J Virol. 2000;74:8980–8988. doi: 10.1128/jvi.74.19.8980-8988.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Williams M C, Wenner J R, Rouzina I, Bloomfield V A. Biophys J. 2001;80:1932–1939. doi: 10.1016/S0006-3495(01)76163-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Williams M C, Wenner J R, Rouzina I, Bloomfield V A. Biophys J. 2001;80:874–881. doi: 10.1016/S0006-3495(01)76066-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee B M, De Guzman R N, Turner B G, Tjandra N, Summers M F. J Mol Biol. 1998;279:633–649. doi: 10.1006/jmbi.1998.1766. [DOI] [PubMed] [Google Scholar]
- 18.Cluzel P, Lebrun A, Heller C, Lavery R, Viovy J L, Chatenay D, Caron F. Science. 1996;271:792–794. doi: 10.1126/science.271.5250.792. [DOI] [PubMed] [Google Scholar]
- 19.Smith S B, Cui Y J, Bustamante C. Science. 1996;271:795–799. doi: 10.1126/science.271.5250.795. [DOI] [PubMed] [Google Scholar]
- 20.Rouzina I, Bloomfield V A. Biophys J. 2001;80:882–893. doi: 10.1016/S0006-3495(01)76067-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rouzina I, Bloomfield V A. Biophys J. 2001;80:894–900. doi: 10.1016/S0006-3495(01)76068-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rouzina I, Bloomfield V A. Biophys Chem. 1997;64:139–155. doi: 10.1016/s0301-4622(96)02231-4. [DOI] [PubMed] [Google Scholar]
- 23.Urbaneja M A, Kane B P, Johnson D G, Gorelick R J, Henderson L E, Casas-Finet J R. J Mol Biol. 1999;287:59–75. doi: 10.1006/jmbi.1998.2521. [DOI] [PubMed] [Google Scholar]
- 24.Stoylov S P, Vuilleumier C, Stoylova E, Rocquigny H D, Roques B P, Gérard D, Mély Y. Biopolymers. 1997;41:301–312. doi: 10.1002/(SICI)1097-0282(199703)41:3<301::AID-BIP5>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 25.Le Cam E, Coulaud D, Delain E, Petitjean P, Roques B P, Gérard D, Stoylova E, Vuilleumier C, Stoylov S P, Mély Y. Biopolymers. 1998;45:217–229. doi: 10.1002/(SICI)1097-0282(199803)45:3<217::AID-BIP4>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 26.Hegner M, Smith S B, Bustamante C. Proc Natl Acad Sci USA. 1999;96:10109–10114. doi: 10.1073/pnas.96.18.10109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baumann C G, Smith S B, Bloomfield V A, Bustamante C. Proc Natl Acad Sci USA. 1997;94:6185–6190. doi: 10.1073/pnas.94.12.6185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bloomfield V A. Biopolymers. 1997;44:269–282. doi: 10.1002/(SICI)1097-0282(1997)44:3<269::AID-BIP6>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 29.Baumann C G, Bloomfield V A, Smith S B, Bustamante C, Wang M D, Block S M. Biophys J. 2000;78:1965–1978. doi: 10.1016/S0006-3495(00)76744-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zimm B H. J Chem Phys. 1960;33:1349–1356. [Google Scholar]
- 31.Cantor C R, Schimmel P R. Biophysical Chemistry. New York: Freeman; 1980. [Google Scholar]
- 32.Fisher R J, Rein A, Fivash M, Urbaneja M A, Casas-Finet J R, Medaglia M, Henderson L E. J Virol. 1998;72:1902–1909. doi: 10.1128/jvi.72.3.1902-1909.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.De Rocquigny H, Gabus C, Vincent A, Fournie-Zaluski M C, Roques B, Darlix J L. Proc Natl Acad Sci USA. 1992;89:6472–6476. doi: 10.1073/pnas.89.14.6472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li X, Quan Y, Arts E J, Li Z, Preston B D, de Rocquigny H, Roques B P, Darlix J L, Kleiman L, Parniak M A, Wainberg M A. J Virol. 1996;70:4996–5004. doi: 10.1128/jvi.70.8.4996-5004.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chan B, Musier-Forsyth K. Proc Natl Acad Sci USA. 1997;94:13530–13535. doi: 10.1073/pnas.94.25.13530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith S B, Finzi L, Bustamante C. Science. 1992;258:1122–1126. doi: 10.1126/science.1439819. [DOI] [PubMed] [Google Scholar]
- 37.Huang Y, Khorchid A, Gabor J, Wang J, Li X G, Darlix J L, Wainberg M A, Kleiman L. J Virol. 1998;72:3907–3915. doi: 10.1128/jvi.72.5.3907-3915.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rong L W, Liang C, Hsu M L, Kleiman L, Petitjean P, Derocquigny H, Roques B P, Wainberg M A. J Virol. 1998;72:9353–9358. doi: 10.1128/jvi.72.11.9353-9358.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guo J H, Henderson L E, Bess J, Kane B, Levin J G. J Virol. 1997;71:5178–5188. doi: 10.1128/jvi.71.7.5178-5188.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sikorav J L, Church G M. J Mol Biol. 1991;222:1085–1108. doi: 10.1016/0022-2836(91)90595-w. [DOI] [PubMed] [Google Scholar]
- 41.Dib-Hajj F, Khan R, Giedroc D P. Protein Sci. 1993;2:231–243. doi: 10.1002/pro.5560020212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Johnson P E, Turner R B, Wu Z R, Hairston L, Guo J, Levin J G, Summers M F. Biochemistry. 2000;39:9084–9091. doi: 10.1021/bi000841i. [DOI] [PubMed] [Google Scholar]
- 43.Odijk T. Macromolecules. 1995;28:7016–7018. [Google Scholar]