

Abstract
Lymphocyte activation leads to changes in chemokine receptor expression. There are limited data, however, on how lymphocyte activators can alter chemokine signaling by affecting downstream pathways. We hypothesized that B cell-activating agents might alter chemokine responses by affecting downstream signal transducers, and that such effects might differ depending on the activator. We found that activating mouse B cells using either anti-IgM or lipopolysaccharide (LPS) increased the surface expression of CCR6 and CCR7 with large increases in chemotaxis to their cognate ligands. By contrast, while anti-IgM also led to enhanced calcium responses, LPS-treated cells showed only small changes in calcium signaling as compared with cells that were freshly isolated. Of particular interest, we found that LPS caused a reduction in the level of B-cell phospholipase C (PLC)-β2 mRNA and protein. Data obtained using PLC-β2−/− mice showed that the β2 isoform mediates close to one-half the chemokine-induced calcium signal in resting and anti-IgM-activated B cells, and we found that calcium signals in the LPS-treated cells were boosted by increasing the level of PLC-β2 using transfection, consistent with a functional effect of downregulating PLC-β2. Together, our results show activator-specific effects on responses through B-cell chemokine receptors that are mediated by quantitative changes in a downstream signal-transducing protein, revealing an activity for LPS as a downregulator of PLC-β2, and a novel mechanism for controlling chemokine-induced signals in lymphocytes.
Keywords: B cells, calcium, chemokine, chemotaxis, GPCR
Introduction
The chemokine system is critical for B-cell development, homeostasis, activation and effector function. Among the chemokine receptors with roles in B-cell biology are: CXCR4, important for B-cell lymphopoiesis,1 proper localization of plasma cells2 and the organization of germinal centers;3 CXCR5, required for the localization of B cells in B-cell follicles4 and germinal centers;3 CCR7, important for the movement of activated follicular B cells toward the T-cell zone;5 CCR6, CCR9 and CCR10, important for localization of IgA-secreting cells to intestinal and/or mammary epithelium;6, 7, 8, 9 and CXCR3, involved in optimal antibody responses to some pathogens10 and in plasmablast migration.11 Although B-cell responses to chemokines can be regulated in a straightforward fashion by changes in chemokine receptor expression, a number of reports from our laboratory and others have described alterations in responsiveness independent of changes in receptor levels.12, 13, 14, 15, 16 These studies have not, however, identified specific changes in downstream signaling elements responsible for affecting the responses to chemokines.
Chemokine receptors are members of the superfamily of G protein-coupled receptors (GPCRs). The pathways that link chemokine receptors with cellular adhesion, polarization and directional migration begin with G-protein activation and go through multiple downstream signaling proteins that include, among others, phosphoinositide 3-kinase (PI3K), phospholipase C (PLC), tyrosine kinases and small GTPases.17 The contributions of these pathways to cell migration have been studied primarily in model organisms such as Dictyostelium, transformed cell lines and neutrophils.18 Information on the roles for downstream pathways in chemoattractant responses in lymphocytes is more limited, and recent data suggest that there may be significant differences in chemoattractant-induced signaling in lymphocytes versus neutrophils.17, 19
The β-isoforms of phosphoinositide-specific PLC are activated by the βγ-dimers liberated from heterotrimeric G proteins, as well as by the α-subunits of the Gq family, and are important ‘effector' elements of GPCR signaling. PLCs produce inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG) from phosphatidylinositol 4,5-bisphosphate, leading to many downstream events, including increases in intracellular calcium and activation of protein kinase C.20 Of the PLC-β isoforms, PLC-β1 and PLC-β3 have wide tissue distribution, while PLC-β2 is expressed principally in hematopoietic cells20, 21 and is the isoform best-characterized in mediating leukocyte responses to chemoattractants.22 PLC-βs have important roles in chemoattractant-induced integrin activation, affecting cell–substrate adhesion and migration.19, 22, 23, 24, 25, 26
Through our work on CCR6 on human cells,14 we became interested in chemokine receptor signaling in B cells, and in particular how such signaling might be affected by non-GPCR-mediated B-cell activation. In the current work, we investigated the expression and activity of the chemokine receptors CCR6, CCR7, CXCR4 and CXCR5 on mouse splenic B cells, either freshly isolated or after in vitro activation with anti-IgM or lipopolysaccharide (LPS). Our principal finding was that LPS led to a significant decrease in the level of B-cell PLC-β2, which was reflected in a muted increase in intracellular calcium in response to chemokines. These data reveal activator-specific effects on responses through B-cell chemokine receptors and a novel mechanism for controlling chemokine-induced signals in lymphocytes.
Material and methods
Antibodies, LPS and chemokines
For B-cell isolation and flow cytometry, monoclonal antibodies against mouse CD16/CD32 (Fc Block), CD19, CD43, B220 and CXCR5 were from BD Biosciences (San Jose, CA, USA), anti-mouse CCR7 was from BioLegend (San Diego, CA, USA) and phycoerythrin (PE)-conjugated F(ab')2 goat anti-rabbit IgG (H+L) was from Caltag Laboratories (now Invitrogen, Carlsbad, CA, USA). Anti-FITC and CD43 MicroBeads were from Miltenyi Biotec (Auburn, CA, USA). For activation of B cells, goat anti-mouse IgM F(ab')2 was from Cappel (MP Biomedicals, Aurora, OH, USA), and for B-cell receptor (BCR) crosslinking in analysis of calcium flux, biotinylated goat anti-mouse IgM F(ab')2 was from Caltag Laboratories. Antibodies against PLC-β1, PLC-β2, PLC-β3 and PLC-γ2, horseradish peroxidase (HRP)-conjugated anti-rabbit IgG, HRP-conjugated polyclonal goat anti-actin and cell lysates for controls for western blotting or immunoprecipitation were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). LPS from Escherichia coli 0111:B4 and bovine serum albumin were from Sigma-Aldrich (St Louis, MO, USA). Normal rabbit IgG, NeutrAvidin and ECL western blotting substrate were from Thermo Scientific (Rockford, IL, USA). CXCL12, CCL20 and CCL21 were from Pepro Tech (Rocky Hill, NJ, USA), and CXCL13 was from R&D systems (Minneapolis, MN, USA).
Mice
TCRβ−/−δ−/− mice (B6.129P2-Tcrbtm1MomTcrdtm1Mom/J) were from The Jackson Laboratory (Bar Harbor, ME, USA) and C57BL/6 mice were from the National Cancer Institute (Frederick, MD, USA). PLC-β2−/− mice in the C57BL/6 background were produced as described.22 All mice were bred at the National Institutes of Health. Animal protocols were approved by the Animal Care and Use Committee of the National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Isolation and activation of B cells
For most experiments, splenic B cells were isolated from 2- to 3-month-old TCRβ−/−δ−/− mice as described.10 For purifying B cells from C56BL/6 wild-type and PLC-β2−/− mice, we used negative selection with FITC-conjugated anti-mouse CD43 and anti-FITC MicroBeads or anti-mouse CD43 directly linked to MicroBeads according to the manufacturer's protocol. B-cell purity was generally >90% as determined by staining for B220 or CD19. B cells were cultured in RPMI-1640/10% fetal bovine serum (FBS) supplemented with 1 mM sodium pyruvate, 100 µM non-essential amino acids, 100 units/ml penicillin-streptomycin and 55 µM 2-mercaptoethanol. For activation, cells were typically cultured at 1×106 cells/ml in Costar 24- or 6-well plates (Corning, Lowell, MA, USA) with 10 µg/ml anti-IgM or 50 µg/ml LPS.
Production of antibodies against mouse CCR6
A peptide containing N-terminal residues of mouse CCR6 and a carboxy-terminal cysteine, NH2-MNSTESYFGTDDYDNTEYYSIPPDHGPCSLEEVRNFTKVC-COOH, was conjugated through the cysteine residue to keyhole limpet hemocyanin and used to immunize rabbits. Affinity purification of antibodies was performed using the immunizing peptide coupled to Affi-Gel 15 (Bio-Rad, Hercules, CA, USA) according to the manufacturer's protocol. Antibody specificity was validated using transfected HEK 293 cells expressing mouse CCR6 (data not shown).
Flow cytometry
For staining CCR6, 0.5×106–1×106 cells were incubated in 100 µl Dulbecco's phosphate-buffered saline containing 2% FBS and 0.05% NaN3 with 1 µg of anti-mouse CD16/CD32 (Fc Block) for 15 min followed by 1 µg of anti-mouse CCR6 antibody or normal rabbit IgG for 45 min, washed and stained with PE-conjugated F(ab')2 goat anti-rabbit IgG for 30 min, all at 4 °C. Staining using other antibodies were done similarly, except that direct conjugates were used and incubation with a secondary antibody was omitted. Cells were analyzed using a FACScalibur or LSR II flow cytometer (BD Biosciences).
Assaying for calcium flux
For most experiments, calcium flux was measured as described.27 B cells were loaded with Fura-2 acetoxymethyl ester, washed and resuspended at 2×106 cells/ml in HBSS with 1.3 mM CaCl2, 1% FBS and 20 mM HEPES, pH 7.3. Chemokines were added at final concentrations of 500–1000 ng/ml. Emission at 510 nm was measured from cells continuously stirred at room temperature and excited alternately at 340 and 380 nm using a ratio fluorescence spectrometer (Photon Technology International, Monmouth Junction, NJ, USA). For some later experiments, calcium flux was measured using a Benchtop Scanning Fluorometer and Integrated Fluid Transfer Workstation (FlexStation; Molecular Devices, Sunnyvale, CA, USA). For these assays, B cells were suspended at 5×106 cells/ml in HBSS with calcium and magnesium, 1% FBS and 20 mM HEPES, pH 7.2, and 100 µl of the cell suspension together with 100 µl of fluorescent dye (FLIPR Calcium 3 Assay Kit; Molecular Devices) was added to each well. Cells were incubated for 30 min at 37 °C, and intracellular calcium measurements were performed in a FlexStation.
Assaying chemotaxis
Chemotaxis assays were performed as described14 using Transwells with 5 µm pores (Corning). Purified B cells were resuspended at 1×107 cells/ml in RPMI-1640 containing 0.5% bovine serum albumin and 10 mM HEPES, pH 8.0 (chemotaxis medium). Inserts containing 100 µl of cell suspension were preincubated in wells containing 600 µl of chemotaxis medium for 30 min before being transferred to wells containing either chemotaxis medium alone or chemotaxis medium containing various concentrations of chemokines, and cells migrating to the lower wells after 2 h were collected and counted. All incubations were done at 37 °C in 5% CO2.
Analysis of RNA
RNA was prepared using the TRIzol reagent according to the manufacturer's protocol (Invitrogen) and Northern analysis, including detecting the 18S rRNA, was done as described.28 32P-labeled probes were prepared from fragments using the Megaprime Kit (GE Healthcare, Piscataway, NJ, USA). The mouse CXCR4 fragment was cDNA containing the entire CXCR4 open reading frame, and was a kind gift from Dr Martin Dorf (Harvard Medical School, Boston, MA, USA).29 The mouse CXCR5 fragment (used for data not shown) was cDNA containing 572–1350 bp in GenBank entry GI:2598563, and the mouse PLC-β2 fragment was cDNA containing 375–1063 bp in GeneBank entry GI:61676178. All cDNA clones were prepared by PCR with reverse transcription from B cell or spleen RNA, and their identities were verified by sequencing.
Immunoblotting
Washed cells were resuspended in lysis buffer (1% Triton X-100, 1% sodium deoxycholate, 150 mM NaCl, 20 mM HEPES, 1 mM EDTA, 20 mM NaF, 1 mM sodium orthovanadate, 10 µg/ml leupeptin, 20 µg/ml aprotinin, 1 mM PMSF and 10 µg/ml pepstatin) at 1×107 cells per 100 µl and incubated for 45 min on ice before removing insoluble material by centrifugation at 13 000 g for 25 min. Total protein was measured using Bio-Rad Protein Assay (Bio-Rad). For analysis by western blot, aliquots of 15–75 µg protein were boiled in sample buffer, separated under reducing conditions on 6% (Invitrogen) or 4–15% (Bio-Rad) precast SDS–polyacrylamide gels, and transferred to polyvinylidene fluoride membranes (Millipore, Billerica, MA, USA). Protein molecular weight standards were Kaleidoscope Prestained Standards or Precision Plus Proteins WesternC Standards from Bio-Rad. For immunoprecipitation (IP), aliquots of 100–130 µg protein were incubated first with anti-PLC-β2 for 1.5–2 h, followed by protein G agarose (Invitrogen) for 1.5 h, both at 4 °C. IP samples were washed three times with lysis buffer at 4 °C, boiled in sample buffer, and separated by SDS–polyacrylamide gel electrophoresis and transferred as above. Membranes were preincubated with 5% milk/Tris-buffered saline with 0.1% Tween 20, and then incubated with anti-PLC antibodies followed by HRP-conjugated anti-rabbit IgG. Immunoreactive proteins were visualized using ECL Western Blotting Substrate according to the manufacturer's protocol.
Expression of PLC-β2 in LPS-activated B cells by transfection
B cells were purified from spleens of C57BL/6 mice and activated as described above. At 48 h after activation, following Ficoll centrifugation, B cells were transfected with a vector control (pCEP4; Invitrogen) or DNAs encoding PLC-β2 in pMT2-PLC-β2 (Ref. 21) (a kind gift from Dr Sue Goo Rhee, Ewha Womans University, Seoul, Korea) using Amaxa Nucleofector Kit (Lonza, Walkersville, MD, USA) according to the manufacturer's instructions. At 24 h post-transfection, following Ficoll centrifugation, cells were collected for analysis.
Results
B-cell activation by anti-IgM or LPS differentially affects responses to chemokines and expression of chemokine receptors
In order to investigate how lymphocyte activation might alter chemokine receptor signaling, we analyzed the effects of two B-cell activators, anti-IgM F(ab')2 (shortened to ‘anti-IgM' in the text below) or LPS, on chemotaxis and calcium flux in mouse splenic B cells in response to chemokines. As measured by incorporation of tritiated thymidine, both anti-IgM- and LPS-treated cells were proliferating comparably at 2 days, and as expected for proliferating cells, both anti-IgM- and LPS-treated cells were larger than the freshly isolated cells, LPS-treated>anti-IgM-treated>freshly isolated. Viable cells were approximately 85 and 95% of the anti-IgM and LPS cultures, respectively, and anti-IgM and LPS cultures contained approximately 70 and 120%, respectively, of starting cell numbers (data not shown). We did not extensively analyze B cells that had been left in culture for 2 days without activation, since approximately 95% of the cells cultured in this fashion died (data not shown), leaving a selected population that we felt could not, therefore, be meaningfully compared with fresh or activated cells. As shown in Figure 1a, after 2 days of treatment with either anti-IgM or LPS, the B cells showed enhanced chemotaxis to CCL20, CCL21 and CXCL13, ligands for CCR6, CCR7 and CXCR5, respectively, and no change in response to CXCL12, the ligand for CXCR4.
Figure 1.
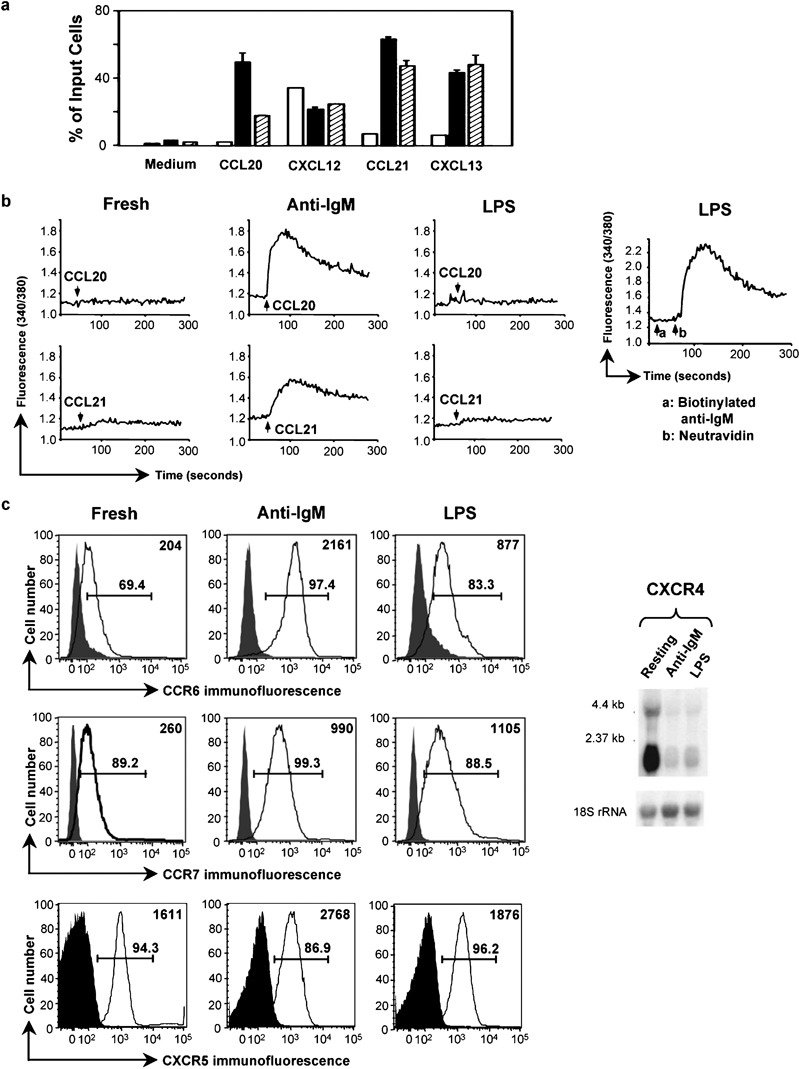
Activation by LPS produces selective effects on chemokine-induced signals in B cells. (a) Migration of mouse splenic B cells, either freshly isolated (□), or following 2-day incubations with 10 µg/ml anti-IgM F(ab')2 (▪) or 50 µg/ml LPS (┐), was measured in the absence of chemokine (‘medium') or in response to 250 ng/ml CXCL12 or 1 µg/ml of the other chemokines. Concentrations were chosen that gave maximal chemotaxis based on dose response experiments not shown. The data are shown as mean±SD of the percentage of input cells migrating into lower wells done in duplicate for each chemokine. One representative experiment is shown out of two performed with these doses of chemokines and two in which all chemokines were used at 1000 ng/ml, which gave similar results. (b) Calcium flux in mouse B cells treated with anti-IgM F(ab')2 or LPS as in (a) was measured in response to additions of CCL20 or CCL21 (indicated by the arrows) or, in the right-most panel, 5 µg/ml biotinylated anti-IgM (arrow a) followed by 10 µg/ml neutravidin (arrow b). Ratio fluorescence was recorded versus time using B cells loaded with Fura-2 AM. One representative experiment is shown out of three performed. (c) Chemokine receptor expression was analyzed either by flow cytometry (left panels) or northern blotting (right panel) for mouse B cells treated with anti-IgM F(ab')2 or LPS as in (a). Shaded histograms are of cells incubated with normal rabbit IgG or PE-conjugated isotype control and open histograms are of cells stained with anti-mouse CCR6 or PE-conjugated CCR7 or CXCR5 antibodies. CCR6 was detected using PE-conjugated F(ab')2 goat antirabbit IgG. Analysis was done on an LSR II flow cytometer (BD Biosciences). Gates show percentages of positive-staining cells as compared to controls. Mean fluorescent intensities of cells in the open histograms are shown in the upper right corner of each plot. One representative experiment is shown out of two performed for CCR7, three performed for CXCR5, and multiple experiments performed for CCR6. For Northern analysis, the blot was hybridized first to a radiolabeled mouse CXCR4 cDNA probe, and subsequently, for determining loading, to a radiolabeled 18S rRNA oligonucleotide probe. Positions of markers are noted. One representative experiment is shown out of two performed. AM, acetoxymethyl ester; LPS, lipopolysaccharide; PE, phycoerythrin.
We next analyzed chemokine receptor-mediated increases in intracellular calcium. As shown in Figure 1b, freshly isolated cells showed minimal (although usually detectable when viewed with an expanded scale (see below) calcium signals, while cells that had been treated with anti-IgM showed much enhanced calcium responses to CCL20 and CCL21, similar to findings for chemotaxis in these cells. For CXCL12, as was the case for chemotaxis, anti-IgM activation did not lead to significantly enhanced calcium responses, and for CXCL13 good calcium responses could not be obtained reproducibly in any of the B-cell populations (data not shown). By contrast, responses of the LPS-treated cells to any of the chemokines tested were either not significantly different from (Figure 1b) or only slightly greater than (see figures below) those in cells assayed immediately after isolation. Nonetheless, LPS-treated cells were fully able to increase cytosolic calcium after BCR crosslinking (Figure 1b, right-most panel).
To help understand the cells' responses to chemokines, we analyzed CCR6, CCR7 and CXCR5 expression by flow cytometry using antibodies that we produced or that were commercially available. For CXCR4, the antibodies available were not sensitive enough to detect expression on B cells (data not shown), so we used northern blotting. As shown in Figure 1c, for CCR6, anti-IgM treatment caused a significant increase in expression, with a smaller increase in response to LPS. For CCR7, both anti-IgM and LPS led to increases, and for CXCR5, the activators resulted in relatively small changes in expression. For CXCR4, both agents resulted in diminished mRNA.
The effects of anti-IgM and LPS on augmenting chemotaxis (excluding the results for CXCL12/CXCR4) were mediated through receptors whose expression either increased or stayed the same. In contrast to the uniform effect of the activators on chemotaxis, only anti-IgM, but not LPS, led to dramatic increases in chemokine-induced calcium fluxes as compared with freshly isolated cells. The muted calcium responses in the LPS-treated cells were evident not only for signals through CXCR4, which had been downregulated, but also for CCR6 and CCR7, whose expression had, if anything, been increased versus the levels on freshly isolated cells. These findings led us to investigate the mechanisms underlying chemokine-mediated calcium responses in the freshly isolated and activated B cells, in order to understand, in particular, why in spite of increases in expression of some receptors and enhanced chemotaxis in the LPS-treated cells, calcium signals in these cells remained poor.
Chemokine-induced calcium flux in B cells depends on PLC and extracellular calcium, and is inhibited by protein kinase A (PKA)
We were interested in identifying the critical components in the signaling pathway leading to chemokine-induced calcium flux in B cells, since we presumed that one or more of these components would be affected specifically by LPS treatment. Using anti-IgM-treated cells and CCL20, the combination giving the largest calcium flux, we found, not unexpectedly, that the PLC inhibitors U73122 (Figure 2a) and ET-18-OCH3 (data not shown) completely blocked the calcium response. In addition, U73122 completely eliminated the small signal in the LPS-treated cells (data not shown). The PI3K inhibitors wortmannin and LY294002 had no effect.
Figure 2.
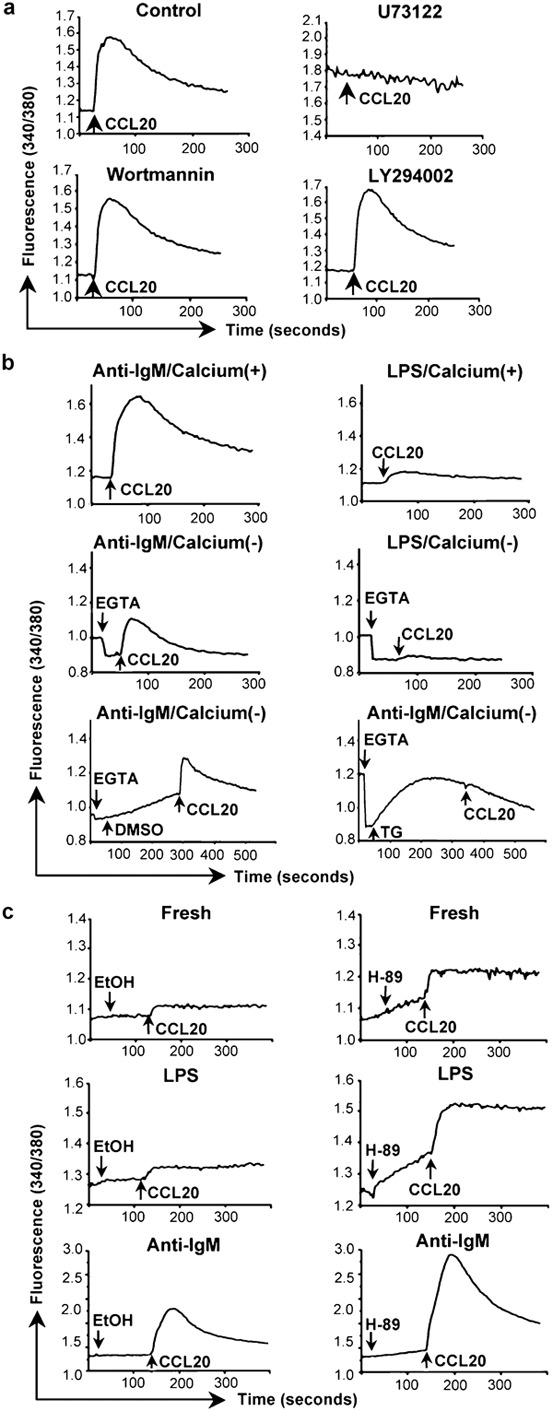
Mediators and sources of chemokine-induced calcium signals in B cells. (a) Calcium flux in mouse splenic B cells that had been incubated for 2 days with 10 µg/ml anti-IgM F(ab')2 was measured as in Figure 1b in response to additions of CCL20. Cells were pretreated for 10 min at 37 °C with the concentrations of inhibitors as noted, or with 0.2% of DMSO as a vehicle control, the cells were washed, and chemokine was added (indicated by the arrows). One representative experiment is shown out of two performed using all inhibitors. Additional experiments were done using some of these inhibitors with similar results. (b) Calcium flux in mouse B cells treated with anti-IgM F(ab')2 or LPS as in Figure 1a was measured as in Figure 1b in response to additions of CCL20 (indicated by the arrows). The top panels show cells in HBSS containing 1.3 mM calcium. The middle panels show cells in HBSS without calcium to which 1.6 mM EGTA was added (indicated by the arrows) before addition of CCL20. The bottom panels show cells in HBSS without calcium to which was added 1.6 mM EGTA and either 0.04% DMSO as a vehicle control or 1 µM TG (indicated by the arrows) before addition of CCL20. One representative experiment is shown out of two performed. (c) Calcium flux in mouse splenic B cells that had been either freshly isolated (top panels), or incubated for 2 days with 50 µg/ml LPS (middle panels) or 10 µg/ml anti-IgM F(ab')2 (bottom panels) was measured as in Figure 1b in response to additions of CCL20 following additions of either 0.625% ethanol as a vehicle control or 25 µM H-89 (indicated by the arrows). Addition of H-89 led to rising baselines before addition of CCL20. One representative experiment is shown out of two performed for all conditions. Additional experiments were done for some conditions with similar results. LPS, lipopolysaccharide; TG, thapsigargin.
We next analyzed the contributions of intracellular stores and extracellular calcium to CCL20-induced calcium signals in anti-IgM- versus LPS-treated cells. As shown in Figure 2b, chelating extracellular calcium by the addition of EGTA inhibited the majority of the strong calcium signal in the anti-IgM-treated cells and, as best could be determined, had a similar effect on the signal in the LPS-treated cells. The EGTA-resistant calcium signal in the anti-IgM-treated cells could be eliminated by pre-treatment with thapsigargin, which depletes intracellular stores. Because of the low EGTA-resistant signal in the LPS-treated cells, effects of thapsigargin on these cells could not be assessed. These data demonstrate that both intracellular stores and extracellular calcium contribute to the CCL20-induced rise in cytosolic calcium, but do not suggest that anti-IgM and LPS affect the contributions from the intracellular and extracellular pools differently.
Phosphorylation by PKA, whose activity depends on cyclic AMP (cAMP), has been reported to inhibit PLCs.30, 31, 32 We therefore tested the possibility that differences in PKA-mediated inhibition might be responsible for differences in calcium signaling among the variously treated B cells. As shown in Figure 2c, pre-incubating cells with the PKA inhibitor H-89 led to increases in the calcium responses to CCL20 as compared with untreated controls in all three groups of cells—freshly isolated, anti-IgM- and LPS-treated. Consistent with these findings, we also detected significantly lower calcium signals after treating the B cells with 8-(4-chlorophenylthio)-cAMP, a cell-permeable analog of cAMP (data not shown). Although these data suggest an inhibitory role for PKA-mediated phosphorylation of PLC-βs in limiting the chemokine-induced calcium responses in B cells, we did not find consistent differences among the three groups of cells in the fold increase induced by H-89 (data not shown). Therefore, the poor calcium signals specific to the LPS-treated cells cannot be explained by a greater level of PKA-mediated inhibition.
LPS downregulates PLC-β2 in B cells
The central role for PLCs in the chemokine receptor-mediated calcium signals led us to analyze the expression of PLCs in the mouse B cells. Initially, we analyzed the levels of PLC-β2, which is expressed preferentially in hematopoietic cells.21 As shown in Figure 3a, the PLC-β2 mRNA is much reduced in the LPS-treated cells as compared with the fresh B cells and the cells activated with anti-IgM. Analysis of PLC-β2 protein by western blot of total cell extracts or by immunoprecipitation, as shown in Figure 3b, gave analogous results. Treatment with anti-IgM-treated cells also tended to reduce levels of PLC-β2 versus the fresh B cells, but these effects were small as compared with the cells treated with LPS and were not investigated further. LPS treatment downregulated PLC-β2 not only in cells treated immediately out of the animal, but also in cells that had been pre-treated with anti-IgM. As shown in Figure 4a, by 24 h after adding LPS to cells that had been activated for 1 day with anti-IgM, the level of PLC-β2 had fallen significantly. At the same time, the CCL20-induced calcium flux was much diminished in the cells treated with anti-IgM and LPS versus those treated with anti-IgM alone (Figure 4b), although adding LPS had resulted in only a small decrease in the surface expression of CCR6 on the cells that had been pre-treated with anti-IgM (Figure 4c). Together, these data demonstrate that LPS downregulates PLC-β2 mRNA and protein in B cells and suggest that such downregulation may contribute to diminished chemokine-induced calcium signals in the LPS-treated cells.
Figure 3.
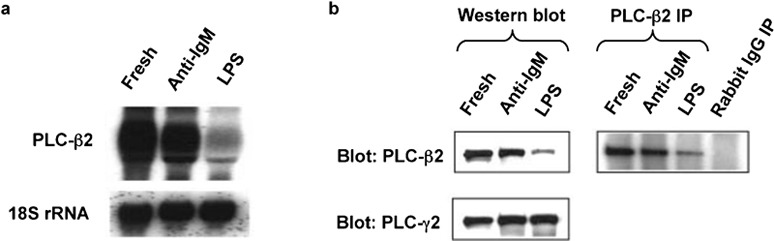
Activation of B cells with LPS decreases levels of PLC-β2 mRNA and protein. (a) Total RNA was prepared from mouse B cells treated with anti-IgM F(ab')2 or LPS as in Figure 1a. The blot was hybridized first to a radiolabeled mouse PLC-β2 cDNA probe, and subsequently, for determining loading, to a radiolabeled 18S rRNA oligonucleotide probe. One representative experiment is shown out of two performed. (b) For the left panels, lysates of mouse B cells treated with anti-IgM F(ab')2 or LPS as in Figure 1a were analyzed by western blot with sequential probing using antibodies against mouse PLC-β2 and PLC-γ2, and HRP-conjugated goat antirabbit IgG for detection. Probing for PLC-γ2 was for determining loading. Loading was also determined by probing for the clathrin heavy chain, which gave results that matched those for PLC-γ2 (not shown). For the right panel, the lysates were incubated with polyclonal anti-PLC-β2 or rabbit IgG, immune complexes were precipitated (IP) using protein G agarose, and the precipitates were analyzed by western blot for PLC-β2 as for the left panel. The PLC-β2 band was positioned between protein markers of 210 kDa (myosin) and 131 kDa (β-galactosidase), with an apparent molecular mass of 140 kDa, as reported.21 One representative experiment is shown out of many (western blotting) or two (IP) performed. IP, immunoprecipitation; LPS, lipopolysaccharide; PLC, phospholipase C.
Figure 4.
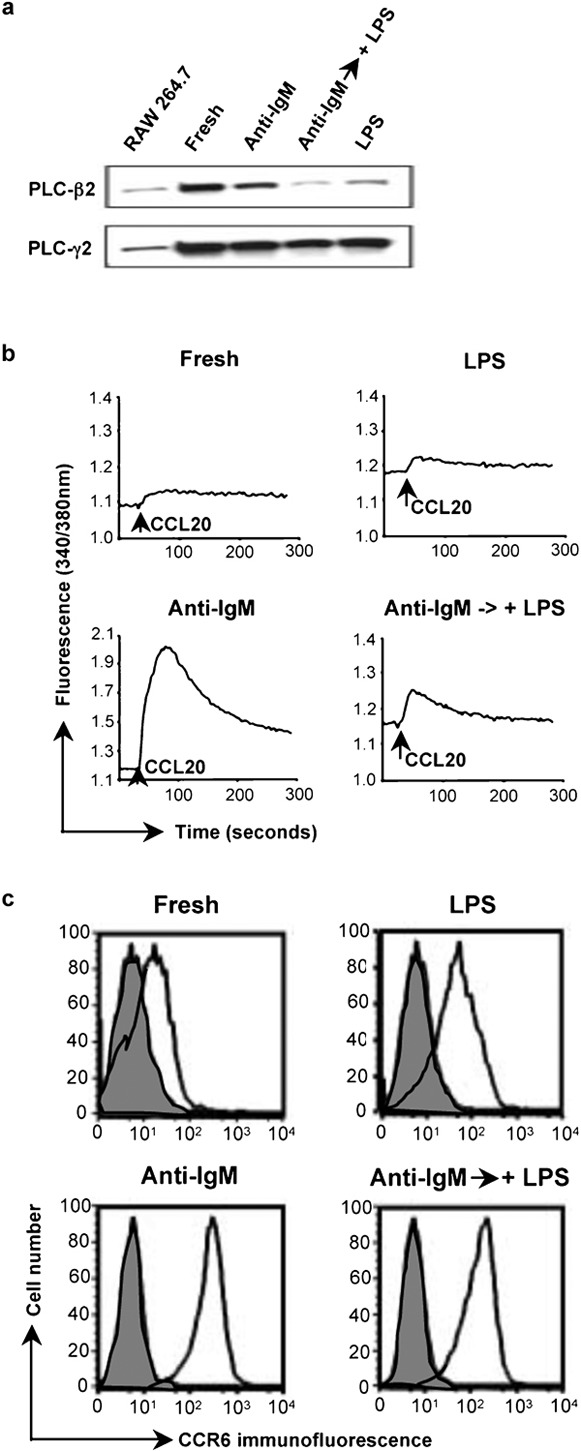
LPS downregulates the level of PLC-β2 and chemokine-induced calcium signals in B cells that had been activated with anti-IgM. (a) Lysates of mouse B cells treated with anti-IgM F(ab')2 or LPS as in Figure 1a, or after a 1-day incubation with 10 µg/ml anti-IgM F(ab')2 followed by a 1-day incubation with 50 µg/ml LPS were analyzed by western blot as in Figure 3b. A lysate of the RAW 264.7 mouse macrophage cell line was included as a positive control/marker for detecting PLC-β2. The amounts of protein loaded for the RAW 264.7 cells were not matched to the B-cell samples. Probing for PLC-γ2 was for determining loading of B-cell samples. The PLC-β2 band was positioned between protein markers of 210 kDa (myosin) and 131 kDa (β-galactosidase), with an apparent molecular mass, on this gel, of 150 kDa. (b) Calcium flux for the same B cells as in (a) was measured as in Figure 1b in response to additions of CCL20 (indicated by the arrows). (c) Surface expression of CCR6 is shown for the same B cells as in (a). Shaded histograms are of cells incubated with normal rabbit IgG and open histograms are of cells stained with antimouse CCR6. CCR6 was detected using PE-conjugated F(ab')2 goat anti-rabbit IgG. Analysis was done on a FACSCalibur flow cytometer (BD Biosciences). One representative experiment is shown out of two performed. LPS, lipopolysaccharide; PE, phycoerythrin; PLC, phospholipase C.
Downregulation of PLC-β2 contributes to the low calcium signals in LPS-treated B cells
To further investigate the functional significance of downregulating PLC-β2 in LPS-treated B cells, we analyzed the limiting condition represented by cells from PLC-β2−/− mice. We first characterized the expression of the additional, potentially relevant isoforms of PLC-β in freshly isolated and activated B cells from wild-type and PLC-β2−/− mice. As shown in Figure 5a, there were no differences in levels of PLC-β1 or PLC-β3 in the wild-type versus PLC-β2−/− mice or in B cells treated with LPS versus anti-IgM. Again, LPS treatment led to downregulation of PLC-β2 in cells from the wild-type mice, and, as expected, PLC-β2 was undetectable in PLC-β2−/− cells.
Figure 5.
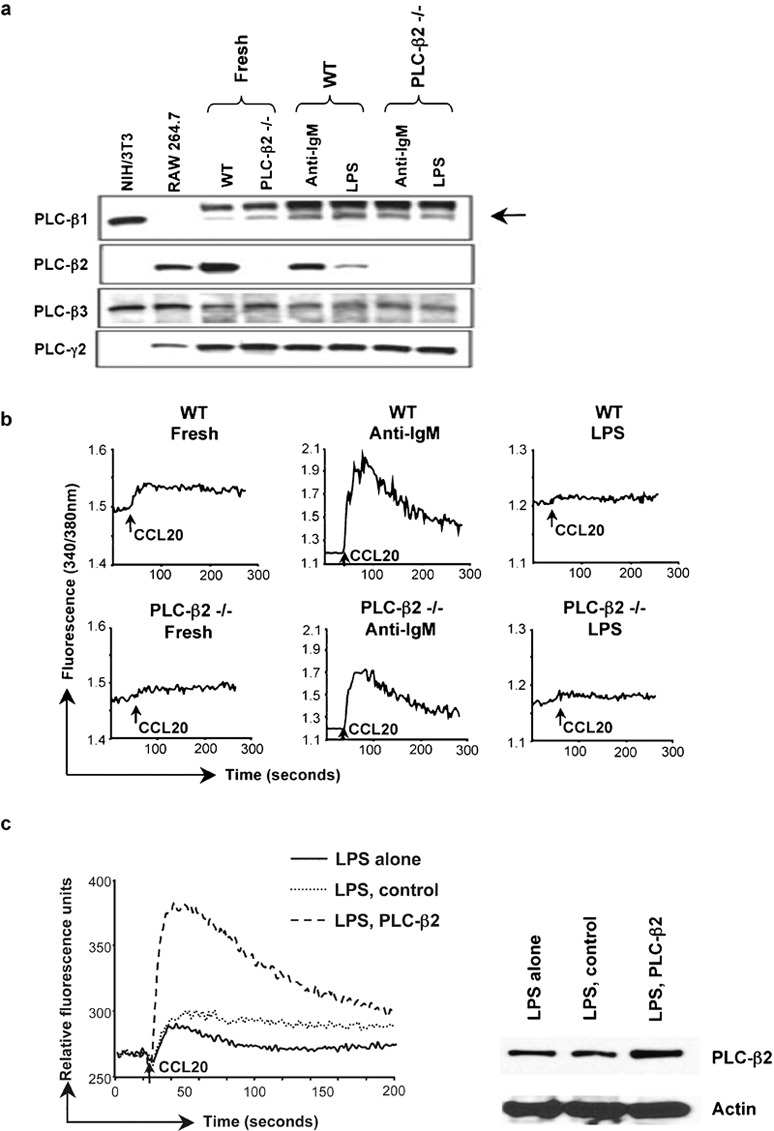
Levels of PLC-β2 affect calcium signaling in LPS-treated B cells. (a) Lysates of B cells from C57BL/6 wild-type or PLC-β2−/− mice treated with anti-IgM F(ab')2 or LPS as in Figure 1a were analyzed by western blot with sequential probing using antibodies against mouse PLC-β1, PLC-β2, PLC-β3 and PLC-γ2, and HRP-conjugated goat antirabbit IgG for detection. Lysates of cell lines were included as positive controls/markers for PLC-β1 and PLC-β3 (NIH/3T3), and PLC-β2 (RAW 264.7). Probing for PLC-γ2 was for determining loading of B-cell samples. The arrow indicates the weak band for PLC-β1. The amounts of protein loaded for the RAW 264.7 and NIH/3T3 cells were not matched to the B-cell samples. The PLC bands were positioned between protein markers of 210 kDa (myosin) and 131 kDa (β-galactosidase), with apparent molecular masses of 150, 140, 155 and 155 kDa for PLC-β1, PLC-β2, PLC-β3 and PLC-γ2, respectively. One representative experiment is shown out of two performed. (b) Calcium flux in B cells from C57BL/6 wild-type or PLC-β2−/− mice that had been treated with anti-IgM F(ab')2 or LPS as in Figure 1a was measured as in Figure 1b in response to additions of CCL20 (indicated by the arrows). One representative experiment is shown out of four performed. (c) B cells from C57BL/6 mice were treated with LPS as in Figure 1a and at 2 days, some cells were transfected either with control DNA or with DNA-encoding PLC-β2. In the left panel, calcium flux was measured 1 day after transfections in non-transfected (LPS alone), control-transfected (LPS, control) and PLC-β2-transfected (LPS, PLC-β2) cells in response to CCL20 using a FlexStation (see the section on ‘Material and methods'). Baselines of each tracing were overlaid for purposes of comparison. In the right panel, cell lysates were analyzed by western blot with sequential probing using antibodies against PLC-β2 and actin. The PLC-β2 band was positioned between protein markers of 150 and 100 kDa, with an apparent molecular mass of 140 kDa. One experiment is shown out of seven where calcium signals were measured. All seven experiments showed an increase in calcium flux in the PLC-β2-transfected cells. In three of the seven experiments, like the one shown, samples were also analyzed by western blot. HRP, horseradish peroxidase; LPS, lipopolysaccharide; PLC, phospholipase C; WT, wild-type.
We next analyzed calcium signals in response to CCL20 and CCL21 in B cells from the wild-type and the PLC-β2−/− mice in order to establish the contribution of PLC-β2 to chemokine-induced signals. As shown in Figure 5b, freshly isolated and anti-IgM-activated B cells isolated from PLC-β2−/− mice showed a little more than one-half the increase in cytosolic calcium as compared with the wild-type cells in response to CCL20 (ratio for PLC-β2−/−/wild-type cells of approximately 0.6). The low signals in the freshly isolated and LPS-treated cells limited the value of the quantitative comparisons in these cells. Analyzing calcium signals after treating cells with CCL21 gave similar results (data not shown).
In order to provide additional evidence that diminished levels of PLC-β2 in the LPS-treated cells could affect signaling, we tested whether or not by increasing levels of PLC-β2 in the LPS-treated cells we could boost the chemokine-induced signals in the calcium assay. After culturing for 2 days in LPS, cells were transfected with an expression vector encoding PLC-β2 and responses to chemokine were measured 1 day later. As shown in Figure 5c, the LPS-treated cells transfected with PLC-β2 sequences showed an enhanced CCL20-induced calcium response as compared to non-transfected or control-transfected cells. Western blotting confirmed an increase in PLC-β2 in the cells transfected with PLC-β2 sequences.
Discussion
Our initial experiments revealed that activating mouse B cells by crosslinking surface IgM led to significant enhancement of chemokine-induced migration and calcium signals (with exception of responses to CXCL12). However, whereas chemokine-induced migration was also enhanced in the LPS-treated cells, these cells showed little increase in calcium flux compared with freshly isolated cells, demonstrating a significant discordance between changes in migration versus calcium signaling. Both anti-IgM and LPS led to changes in chemokine receptor expression, ranging from profound downregulation of CXCR4 to, in the anti-IgM-treated cells, dramatic upregulation of CCR6. Typically, levels of response to chemokines correlate with levels of expression of the cognate receptors, and changes in receptor levels could at least partly explain some of our results, such as the increases in chemotaxis and calcium signals induced by CCL20 in the anti-IgM-treated versus the fresh cells. However, changes in receptor levels often did not correlate with responses to chemokines, such as in the large increases in chemotaxis to CXCL13 in both anti-IgM- and LPS-treated cells without a concomitant increase in CXCR5. Little is known either about the molecular mechanisms regulating chemokine receptor expression, or how responses to chemokines can be affected by regulating the expression of downstream signaling proteins. In this study we addressed the latter question by focusing on the anomalously low chemokine-induced calcium signal in the LPS-activated B cells.
The similar effects of depleting extracellular calcium on the signals in anti-IgM- and LPS-treated cells suggested that LPS did not have a selective effect on plasma membrane channels. The demonstration that BCR-induced calcium signaling was intact following crosslinking of IgM on the LPS-treated B cells suggested that the limiting step(s) in the chemokine-induced calcium response would be in the pathway for generating IP3. In fact, we found a selective decrease in the level of PLC-β2 and the PLC-β2 mRNA in the LPS-treated B cells. PLC-β2 was downregulated not only during LPS activation of freshly isolated B cells, but also by LPS treatment of anti-IgM-activated cells. In the latter case, the downregulation of PLC-β2 was associated with a marked reduction in the chemokine-mediated calcium signal. Using PLC-β2−/− mice, we showed that PLC-β2 accounted for approximately 40% of the chemokine-induced calcium signal in B cells; and we found that calcium signals in the LPS-treated cells were boosted by increasing the level of PLC-β2 using transient transfections. Together, these data show that the chemokine-induced signals were sensitive to levels of PLC-β2 and are consistent with a functional effect of downregulating PLC-β2 in the LPS-treated cells. Similar to previous results for mouse neutrophils33 and T cells,19 our preliminary findings using B cells from PLC-β2−/−PLC-β3−/− mice show that the remaining chemokine-induced calcium signals in the PLC-β2−/− cells are mediated by PLC-β3 (data not shown).
Our data naturally raise the issue of the roles for PLC-β2 and its products, and their downstream effects, in the cells' physiological responses to chemokines. Firstly, it is important to note that assaying for a global rise in intracellular calcium in response to high, uniform concentrations of chemokines as is commonly done, and as done in this study, is non-physiological. Although these assays are good tools for evaluating PLC-dependent pathways and mechanisms underlying calcium flux per se, the assays do not mimic the way calcium signals are occurring in vivo where chemokines fixed on cell surfaces and/or extracellular matrix, often in concentration gradients, produce intracellular gradients and changes in calcium concentrations that are highly localized and evolving with time and cell movement.34, 35 In fact, chemoattractant-induced calcium fluxes have been implicated in a number of processes important for cell migration, including integrin activation through CalDAG-GEFI, Rap1 and RAPL17, 24, 25, 36 and de-adhesion/uropod release through effects on integrin trafficking,23 myosin light chain kinase/myosin-II activation37 and synaptotagmin-mediated lysosome fusion.38 Nonetheless, controversy remains as to whether or not calcium flux is required for chemotaxis. Mouse neutrophils lacking both PLC-β2 and PLC-β3 showed no chemoattractant-induced increase in intracellular calcium, yet migrated the same or better than wild-type cells,33 and human T cells migrated well to CCR4 agonists after calcium flux was blocked using an inhibitor of IP3-mediated calcium release.39 However, PLC-βs and calcium flux have both been shown to be important in the chemotaxis of mouse T cells.19 Contradictory findings may reflect limitations and/or differences in calcium assays, methods for blocking calcium signals, and assays of cell migration, as well as real differences among cell types.
An increase in intracellular calcium is only one of the effects of activating PLCs. The other principal result of PLC activity is DAG, which has been implicated in chemoattractant-mediated adhesion and/or migration, either directly through interactions with CalDAG-GEFI40 and the Rac1 GTPase-activating protein, β2-chimaerin,41 or indirectly through activation of protein kinase C isoforms.39, 42 Targeting the PLC-β2 gene specifically has revealed a critical function in neutrophil responses to chemoattractants in calcium flux, superoxide production and expression of the αMβ2 integrin.22 Some of the relevant signaling pathways centered on PLCs are diagrammed in Figure 6. Although we have shown in this report that LPS can downregulate the PLC-β2 isoform in B cells with effects on calcium signaling, it is clear that any resulting negative consequences for chemotaxis are more than compensated for by other changes in these cells, at least in our in vitro assays. The large differences in chemokine-induced calcium flux between the anti-IgM- and LPS-treated cells, despite only modest differences in their abilities to migrate, reflect, likely for some of the reasons discussed above, what is often a poor correlation between assays for calcium flux and chemotaxis.
Figure 6.
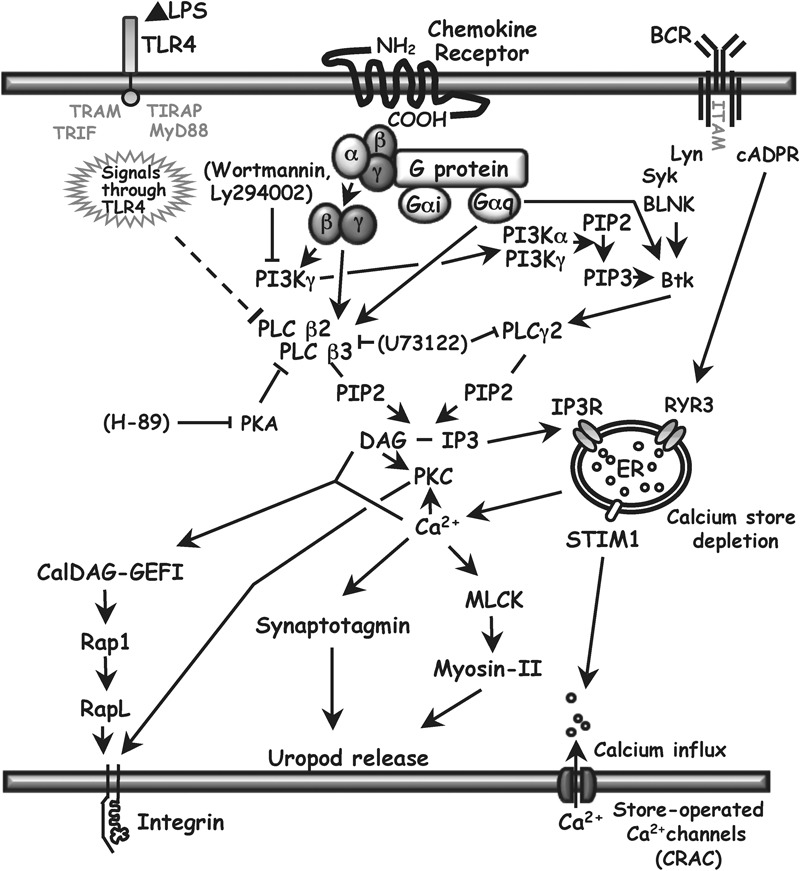
PLC signaling in B cells. The principal isoforms of PLC-β in hematopoietic cells are PLC-β2 and PLC-β3. They can be activated by βγ-dimers or Gα subunits of the Gq family of heterotrimeric G proteins that are coupled to seven transmembrane receptors. PLC-βs can be inhibited by PKA, and expression of PLC-β2 can be downregulated by LPS (this report), which signals through TLR4. PLC converts PIP2 to DAG and IP3. IP3 binding to the IP3R on the ER leads to a rise in cytoplasmic calcium by the release of calcium from ER stores and, indirectly, by opening store-operated plasma membrane calcium channels, the major ones being CRAC. DAG and IP3 can also be produced by PLC-γ2, activated primarily through signals from the BCR. There is an alternate calcium release pathway from BCR through cADPR to RYR3 in the ER. There is a possible cross-talk pathway from G protein through PI3Kγ, PIP3 and Bruton's tyrosine kinase to activate PLC-γ2, but we found that inhibiting PI3K with wortmannin or Ly294002 had no effect on chemokine-induced calcium flux (see text). DAG, together with calcium, can bind to the GEF CalDAG-GEFI, leading to integrin activation. DAG can also enhance integrin avidity for adhesion proteins through a PKC-dependent pathway. Calcium has roles in uropod release at the back of migrating cells by contributing to the activation of myosin II and the activity of synaptotagmins, which are important for lysosome fusion. Arrows show positive effects. Solid lines ending in bars show inhibitory effects. Pharmacological inhibitors used in this study are shown in brackets. Downregulation of PLC-β2 demonstrated in this study is shown using the dashed line. For relevant references, see text and Refs. 52 and 53. BCR, B-cell receptor; cADPR, cyclic ADP ribose; CRAC, calcium release-activated calcium channels; DAG, diacylglycerol; ER, endoplasmic reticulum; GEF, guanine nucleotide exchange factor; IP3, inositol 1,4,5-trisphosphate; IP3R, inositol trisphosphate receptor; LPS, lipopolysaccharide; MLCK, myosin light chain kinase; PIP2, phosphatidylinositol 4,5-bisphosphate; PIP3, phosphatidylinositol 3,4,5-trisphosphate; PI3K, phosphatidylinositol 3-kinase; PKA, protein kinase A; PKC, protein kinase C; PLC, phospholipase C; RYR3, ryanodine receptor 3; STIM1, stromal interaction molecule 1; TLR, Toll-like receptor.
Regarding previous information on factors influencing expression of PLC-βs, there are reports of changes in levels of PLC-β2 during granulocyte differentiation43 and reports of changes in levels of other PLC-β isoforms in cardiac muscle associated with hypertrophy44 or ischemia,45 and in brain46 and smooth muscle47 induced by dexamethasone. While the current manuscript was being prepared for submission, Grinberg et al., in a collaborative study with one of us (DW), reported that LPS could downregulate PLC-β1 and PLC-β2 in mouse macrophages, and that suppression of PLC-β2 was important for the effects of the adenosine A2A receptor in mediating a switch in macrophage phenotype from inflammatory to angiogenic.48 These data suggest that the effects that we observed of LPS on PLC-β2 may extend well beyond B cells. Unlike in Grinberg et al., however, we found no LPS-induced downregulation of PLC-β1 in B cells, suggesting that effects on this isoform may be cell-type specific.
Of particular relevance for our findings is a report of a human-inherited disorder in platelet function characterized by impaired GPCR-induced calcium signals due to a selective decrease in expression of PLC-β2,49 supporting our data that the level of PLC-β2 can determine the magnitude of the calcium signal mediated by GPCRs.
In comparing the LPS-treated versus freshly isolated B cells, it is plausible that the downregulation of PLC-β2 contributed to the absence of significantly enhanced CCL20- and CCL21-induced calcium signals in the LPS-treated cells. In this case, there were only little increases in calcium signaling despite increases in chemokine receptor expression. However, in comparing the 2-day LPS-treated versus anti-IgM-treated B cells for responses through CCR7, or in comparing the cells treated for 1 day with anti-IgM followed by 1 day with LPS versus cells treated for 2 days with anti-IgM for responses through CCR6, it would seem that the changes we have identified in levels of PLC-β2 are not sufficient to explain the large differences in calcium signals between anti-IgM- and LPS-treated B cells—since approximately 60% of the calcium signal in the anti-IgM-treated cells was not mediated through PLC-β2. Experiments using cDNA microarrays to compare levels of mRNAs encoding relevant signaling proteins in anti-IgM versus LPS-treated B cells did not reveal any additional, obvious differences that could explain our findings with respect to chemokine receptor signaling (data not shown), and identifying such differences awaits further investigation.
With regard to our data on the anti-IgM-treated cells, one question is how our findings for changes in chemokine receptor activity and expression fit within the programmed movements of follicular B cells following their activation. Elegant in vivo studies5, 50 have investigated the migration of follicular B cells after encountering antigen, and the roles of chemokines in directing these movements. Within hours after the injection of antigen, antigen-binding follicular B cells show enhanced motility, a modest increase in expression of CCR7, and directed migration to and accumulation at the boundary between the follicle and the interfollicular (T cell) region (B/T boundary). Subsequently, in the presence of sufficient numbers of antigen-specific CD4+ T cells, the B cells form clusters within the interfollicular region. The enhanced response that we found to CCR7 ligands would contribute directly to migration of cells up the gradient of CCL2150 to the B/T boundary. For CXCR5, given the reasonably uniform expression of CXCL13 in the B-cell follicle and the drop-off in CXCL13 expression in the T-cell zone,3 the enhanced activity of CXCR5 would not be expected to impede B-cell movement through the follicle, but might serve to prevent movement down the CXCL13 gradient, thereby arresting cells at the B/T, i.e., the CXCL13/CCL21 boundary. In data not shown, we found that treatment with anti-IgM led to a fall in Cxcr5 mRNA, even though we did not detect a change in surface CXCR5 in the anti-IgM-treated cells. Because decreases in response to CXCL13 are presumably necessary for B cells to move subsequently to the interfollicular regions, the fall in receptor mRNA may be preparatory for the changes in receptor level and signaling which, acting in concert, will selectively downregulate CXCL13 responses.
To our knowledge, our data contain the first demonstrations that levels of a PLC-β—and GPCR-induced PLC-β-mediated signals—can be altered in lymphocytes by activating stimuli, and more generally that lymphocyte responses to chemokines can be regulated by activation-induced changes in the level of a downstream signaling ‘effector' protein. It is of interest that certain B-cell activators can also lead to induction of negative regulators of G protein signaling, namely, the RGS proteins.17 The molecular mechanisms of downregulation of PLC-β2 by LPS warrant additional study, as do the possible consequences of this downregulation during exposure to LPS in vivo. In vivo, LPS has profound effects on B cells—not only on proliferation and antibody production, but also on the integrin-mediated localization of B-cell subsets,51 which would be predicted to be affected by changes in chemoattractant-induced, PLC-β-dependent signals.
Acknowledgments
We would like to thank Martin Dorf, Harvard Medical School, for providing the cDNA for mouse CXCR4; Kaimei Song, for sharing reagents; Paul Goldsmith, for help in making antibodies; and Sue Goo Rhee for advice and for supplying pMT2-PLC-β2. The Intramural Research Program of NIAID, NIH, supported this research.
References
- Nagasawa T, Hirota S, Tachibana K, Takakura N, Nishikawa S, Kitamura Y, et al. Defects of B-cell lymphopoiesis and bone-marrow myelopoiesis in mice lacking the CXC chemokine PBSF/SDF-1. Nature. 1996;382:635–638. doi: 10.1038/382635a0. [DOI] [PubMed] [Google Scholar]
- Hargreaves DC, Hyman PL, Lu TT, Ngo VN, Bidgol A, Suzuki G, et al. A coordinated change in chemokine responsiveness guides plasma cell movements. J Exp Med. 2001;194:45–56. doi: 10.1084/jem.194.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen CD, Ansel KM, Low C, Lesley R, Tamamura H, Fujii N, et al. Germinal center dark and light zone organization is mediated by CXCR4 and CXCR5. Nat Immunol. 2004;5:943–952. doi: 10.1038/ni1100. [DOI] [PubMed] [Google Scholar]
- Forster R, Mattis AE, Kremmer E, Wolf E, Brem G, Lipp M. A putative chemokine receptor, BLR1, directs B cell migration to defined lymphoid organs and specific anatomic compartments of the spleen. Cell. 1996;87:1037–1047. doi: 10.1016/s0092-8674(00)81798-5. [DOI] [PubMed] [Google Scholar]
- Reif K, Ekland EH, Ohl L, Nakano H, Lipp M, Forster R, et al. Balanced responsiveness to chemoattractants from adjacent zones determines B-cell position. Nature. 2002;416:94–99. doi: 10.1038/416094a. [DOI] [PubMed] [Google Scholar]
- Cook DN, Prosser DM, Forster R, Zhang J, Kuklin NA, Abbondanzo SJ, et al. CCR6 mediates dendritic cell localization, lymphocyte homeostasis, and immune responses in mucosal tissue. Immunity. 2000;12:495–503. doi: 10.1016/s1074-7613(00)80201-0. [DOI] [PubMed] [Google Scholar]
- Kunkel EJ, Kim CH, Lazarus NH, Vierra MA, Soler D, Bowman EP, et al. CCR10 expression is a common feature of circulating and mucosal epithelial tissue IgA Ab-secreting cells. J Clin Invest. 2003;111:1001–1010. doi: 10.1172/JCI17244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hieshima K, Kawasaki Y, Hanamoto H, Nakayama T, Nagakubo D, Kanamaru A, et al. CC chemokine ligands 25 and 28 play essential roles in intestinal extravasation of IgA antibody-secreting cells. J Immunol. 2004;173:3668–3675. doi: 10.4049/jimmunol.173.6.3668. [DOI] [PubMed] [Google Scholar]
- Wilson E, Butcher EC. CCL28 controls immunoglobulin (Ig)A plasma cell accumulation in the lactating mammary gland and IgA antibody transfer to the neonate. J Exp Med. 2004;200:805–809. doi: 10.1084/jem.20041069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park MK, Amichay D, Love P, Wick E, Liao F, Grinberg A, et al. The CXC chemokine murine monokine induced by IFN-gamma (CXC chemokine ligand 9) is made by APCs, targets lymphocytes including activated B cells, and supports antibody responses to a bacterial pathogen in vivo. . J Immunol. 2002;169:1433–1443. doi: 10.4049/jimmunol.169.3.1433. [DOI] [PubMed] [Google Scholar]
- Hauser AE, Debes GF, Arce S, Cassese G, Hamann A, Radbruch A, et al. Chemotactic responsiveness toward ligands for CXCR3 and CXCR4 is regulated on plasma blasts during the time course of a memory immune response. J Immunol. 2002;169:1277–1282. doi: 10.4049/jimmunol.169.3.1277. [DOI] [PubMed] [Google Scholar]
- Honczarenko M, Douglas RS, Mathias C, Lee B, Ratajczak MZ, Silberstein LE. SDF-1 responsiveness does not correlate with CXCR4 expression levels of developing human bone marrow B cells. Blood. 1999;94:2990–2998. [PubMed] [Google Scholar]
- Brandes M, Legler DF, Spoerri B, Schaerli P, Moser B. Activation-dependent modulation of B lymphocyte migration to chemokines. Int Immunol. 2000;12:1285–1292. doi: 10.1093/intimm/12.9.1285. [DOI] [PubMed] [Google Scholar]
- Liao F, Shirakawa AK, Foley JF, Rabin RL, Farber JM. Human B cells become highly responsive to macrophage-inflammatory protein-3 alpha/CC chemokine ligand-20 after cellular activation without changes in CCR6 expression or ligand binding. J Immunol. 2002;168:4871–4880. doi: 10.4049/jimmunol.168.10.4871. [DOI] [PubMed] [Google Scholar]
- Casamayor-Palleja M, Mondiere P, Verschelde C, Bella C, Defrance T. BCR ligation reprograms B cells for migration to the T zone and B-cell follicle sequentially. Blood. 2002;99:1913–1921. doi: 10.1182/blood.v99.6.1913. [DOI] [PubMed] [Google Scholar]
- Badr G, Borhis G, Treton D, Richard Y. IFN{alpha} enhances human B-cell chemotaxis by modulating ligand-induced chemokine receptor signaling and internalization. Int Immunol. 2005;17:459–467. doi: 10.1093/intimm/dxh227. [DOI] [PubMed] [Google Scholar]
- Kehrl JH. Chemoattractant receptor signaling and the control of lymphocyte migration. Immunol Res. 2006;34:211–227. doi: 10.1385/IR:34:3:211. [DOI] [PubMed] [Google Scholar]
- Stephens L, Milne L, Hawkins P. Moving towards a better understanding of chemotaxis. Curr Biol. 2008;18:R485–494. doi: 10.1016/j.cub.2008.04.048. [DOI] [PubMed] [Google Scholar]
- Bach TL, Chen QM, Kerr WT, Wang Y, Lian L, Choi JK, et al. Phospholipase cbeta is critical for T cell chemotaxis. J Immunol. 2007;179:2223–2227. doi: 10.4049/jimmunol.179.4.2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee SG. Regulation of phosphoinositide-specific phospholipase C. Annu Rev Biochem. 2001;70:281–312. doi: 10.1146/annurev.biochem.70.1.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park D, Jhon DY, Kriz R, Knopf J, Rhee SG. Cloning, sequencing, expression, and Gq-independent activation of phospholipase C-beta 2. J Biol Chem. 1992;267:16048–16055. [PubMed] [Google Scholar]
- Jiang H, Kuang Y, Wu Y, Xie W, Simon MI, Wu D. Roles of phospholipase C beta2 in chemoattractant-elicited responses. Proc Natl Acad Sci USA. 1997;94:7971–7975. doi: 10.1073/pnas.94.15.7971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson MA, Maxfield FR. Ca2+- and calcineurin-dependent recycling of an integrin to the front of migrating neutrophils. Nature. 1995;377:75–79. doi: 10.1038/377075a0. [DOI] [PubMed] [Google Scholar]
- Katagiri K, Ohnishi N, Kabashima K, Iyoda T, Takeda N, Shinkai Y, et al. Crucial functions of the Rap1 effector molecule RAPL in lymphocyte and dendritic cell trafficking. Nat Immunol. 2004;5:1045–1051. doi: 10.1038/ni1111. [DOI] [PubMed] [Google Scholar]
- McLeod SJ, Li AH, Lee RL, Burgess AE, Gold MR. The Rap GTPases regulate B cell migration toward the chemokine stromal cell-derived factor-1 (CXCL12): potential role for Rap2 in promoting B cell migration. J Immunol. 2002;169:1365–1371. doi: 10.4049/jimmunol.169.3.1365. [DOI] [PubMed] [Google Scholar]
- Bergmeier W, Goerge T, Wang HW, Crittenden JR, Baldwin AC, Cifuni SM, et al. Mice lacking the signaling molecule CalDAG-GEFI represent a model for leukocyte adhesion deficiency type III. J Clin Invest. 2007;117:1699–1707. doi: 10.1172/JCI30575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao F, Rabin RL, Yannelli JR, Koniaris LG, Vanguri P, Farber JM. Human Mig chemokine: biochemical and functional characterization. J Exp Med. 1995;182:1301–1314. doi: 10.1084/jem.182.5.1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amichay D, Gazzinelli RT, Karupiah G, Moench TR, Sher A, Farber JM. Genes for chemokines MuMig and Crg-2 are induced in protozoan and viral infections in response to IFN-gamma with patterns of tissue expression that suggest nonredundant roles in vivo. . J Immunol. 1996;157:4511–4520. [PubMed] [Google Scholar]
- Heesen M, Berman MA, Benson JD, Gerard C, Dorf ME. Cloning of the mouse fusin gene, homologue to a human HIV-1 co-factor. J Immunol. 1996;157:5455–5460. [PubMed] [Google Scholar]
- Ali H, Fisher I, Haribabu B, Richardson RM, Snyderman R. Role of phospholipase Cbeta3 phosphorylation in the desensitization of cellular responses to platelet-activating factor. J Biol Chem. 1997;272:11706–11709. doi: 10.1074/jbc.272.18.11706. [DOI] [PubMed] [Google Scholar]
- Liu M, Simon MI. Regulation by cAMP-dependent protein kinease of a G-protein-mediated phospholipase C. Nature. 1996;382:83–87. doi: 10.1038/382083a0. [DOI] [PubMed] [Google Scholar]
- Park DJ, Min HK, Rhee SG. Inhibition of CD3-linked phospholipase C by phorbol ester and by cAMP is associated with decreased phosphotyrosine and increased phosphoserine contents of PLC-gamma 1. J Biol Chem. 1992;267:1496–1501. [PubMed] [Google Scholar]
- Li Z, Jiang H, Xie W, Zhang Z, Smrcka AV, Wu D. Roles of PLC-beta2 and -beta3 and PI3Kgamma in chemoattractant-mediated signal transduction. Science. 2000;287:1046–1049. doi: 10.1126/science.287.5455.1046. [DOI] [PubMed] [Google Scholar]
- Pettit EJ, Fay FS. Cytosolic free calcium and the cytoskeleton in the control of leukocyte chemotaxis. Physiol Rev. 1998;78:949–967. doi: 10.1152/physrev.1998.78.4.949. [DOI] [PubMed] [Google Scholar]
- Wei C, Wang X, Chen M, Ouyang K, Song LS, Cheng H. Calcium flickers steer cell migration. Nature. 2009;457:901–905. doi: 10.1038/nature07577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reedquist KA, Bos JL. Costimulation through CD28 suppresses T cell receptor-dependent activation of the Ras-like small GTPase Rap1 in human T lymphocytes. J Biol Chem. 1998;273:4944–4949. doi: 10.1074/jbc.273.9.4944. [DOI] [PubMed] [Google Scholar]
- Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, et al. Cell migration: integrating signals from front to back. Science. 2003;302:1704–1709. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- Colvin RA, Means TK, Diefenbach TJ, Moita LF, Friday RP, Sever S, et al. Synaptotagmin-mediated vesicle fusion regulates cell migration. Nat Immunol. 2010;11:495–502. doi: 10.1038/ni.1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronshaw DG, Kouroumalis A, Parry R, Webb A, Brown Z, Ward SG. Evidence that phospholipase-C-dependent, calcium-independent mechanisms are required for directional migration of T-lymphocytes in response to the CCR4 ligands CCL17 and CCL22. J Leukoc Biol. 2006;79:1369–1380. doi: 10.1189/jlb.0106035. [DOI] [PubMed] [Google Scholar]
- Crittenden JR, Bergmeier W, Zhang Y, Piffath CL, Liang Y, Wagner DD, et al. CalDAG-GEFI integrates signaling for platelet aggregation and thrombus formation. Nat Med. 2004;10:982–986. doi: 10.1038/nm1098. [DOI] [PubMed] [Google Scholar]
- Siliceo M, Garcia-Bernal D, Carrasco S, Diaz-Flores E, Coluccio Leskow F, Teixido J, et al. Beta2-chimaerin provides a diacylglycerol-dependent mechanism for regulation of adhesion and chemotaxis of T cells. J Cell Sci. 2006;119:141–152. doi: 10.1242/jcs.02722. [DOI] [PubMed] [Google Scholar]
- Pasvolsky R, Grabovsky V, Giagulli C, Shulman Z, Shamri R, Feigelson SW, et al. RhoA is involved in LFA-1 extension triggered by CXCL12 but not in a novel outside-in LFA-1 activation facilitated by CXCL9. J Immunol. 2008;180:2815–2823. doi: 10.4049/jimmunol.180.5.2815. [DOI] [PubMed] [Google Scholar]
- Bertagnolo V, Marchisio M, Pierpaoli S, Colamussi ML, Brugnoli F, Visani G, et al. Selective up-regulation of phospholipase C-beta2 during granulocytic differentiation of normal and leukemic hematopoietic progenitors. J Leukoc Biol. 2002;71:957–965. [PubMed] [Google Scholar]
- Schnabel P, Mies F, Nohr T, Geisler M, Bohm M. Differential regulation of phospholipase C-beta isozymes in cardiomyocyte hypertrophy. Biochem Biophys Res Commun. 2000;275:1–6. doi: 10.1006/bbrc.2000.3255. [DOI] [PubMed] [Google Scholar]
- Asemu G, Tappia PS, Dhalla NS. Identification of the changes in phospholipase C isozymes in ischemic-reperfused rat heart. Arch Biochem Biophys. 2003;411:174–182. doi: 10.1016/s0003-9861(02)00733-6. [DOI] [PubMed] [Google Scholar]
- Dwivedi Y, Pandey GN. Repeated administration of dexamethasone increases phosphoinositide-specific phospholipase C activity and mRNA and protein expression of the phospholipase C beta 1 isozyme in rat brain. J Neurochem. 1999;73:780–790. doi: 10.1046/j.1471-4159.1999.0730780.x. [DOI] [PubMed] [Google Scholar]
- Cueille C, Frayon S, de Vernejoul MC, Garel JM. Dexamethasone decreases phospholipase C beta1 isozyme expression in human vascular smooth muscle cells. J Steroid Biochem Mol Biol. 2003;86:173–178. doi: 10.1016/s0960-0760(03)00271-1. [DOI] [PubMed] [Google Scholar]
- Grinberg S, Hasko G, Wu D, Leibovich SJ. Suppression of PLCbeta2 by endotoxin plays a role in the adenosine A(2A) receptor-mediated switch of macrophages from an inflammatory to an angiogenic phenotype. Am J Pathol. 2009;175:2439–2453. doi: 10.2353/ajpath.2009.090290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SB, Rao AK, Lee KH, Yang X, Bae YS, Rhee SG. Decreased expression of phospholipase C-beta 2 isozyme in human platelets with impaired function. Blood. 1996;88:1684–1691. [PubMed] [Google Scholar]
- Okada T, Miller MJ, Parker I, Krummel MF, Neighbors M, Hartley SB, et al. Antigen-engaged B cells undergo chemotaxis toward the T zone and form motile conjugates with helper T cells. e150; PLoS Biol. 2005;3 doi: 10.1371/journal.pbio.0030150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu TT, Cyster JG. Integrin-mediated long-term B cell retention in the splenic marginal zone. Science. 2002;297:409–412. doi: 10.1126/science.1071632. [DOI] [PubMed] [Google Scholar]
- Sambrano GR, Chandy G, Choi S, Decamp D, Hsueh R, Lin KM, et al. Unravelling the signal-transduction network in B lymphocytes. Nature. 2002;420:708–710. doi: 10.1038/nature01305. [DOI] [PubMed] [Google Scholar]
- Feske S. Calcium signalling in lymphocyte activation and disease. Nat Rev Immunol. 2007;7:690–702. doi: 10.1038/nri2152. [DOI] [PubMed] [Google Scholar]