

Abstract
Adenosine triphosphate (ATP)-binding cassette (ABC) transporters, ubiquitous proteins found in all kingdoms of life, catalyze substrates translocation across biological membranes using the free energy of ATP hydrolysis. Cystic fibrosis transmembrane conductance regulator (CFTR) is a unique member of this superfamily in that it functions as an ATP-gated chloride channel. Despite difference in function, recent studies suggest that the CFTR chloride channel and the exporter members of the ABC protein family may share an evolutionary origin. Although ABC exporters harness the free energy of ATP hydrolysis to fuel a transport cycle, for CFTR, ATP-induced dimerization of its nucleotide-binding domains (NBDs) and subsequent hydrolysis-triggered dimer separation are proposed to be coupled, respectively, to the opening and closing of the gate in its transmembrane domains. In this study, by using nonhydrolyzable ATP analogues, such as pyrophosphate or adenylyl-imidodiphosphate as baits, we captured a short-lived state (state X), which distinguishes itself from the previously identified long-lived C2 closed state by its fast response to these nonhydrolyzable ligands. As state X is caught during the decay phase of channel closing upon washout of the ligand ATP but before the channel sojourns to the C2 closed state, it likely emerges after the bound ATP in the catalysis-competent site has been hydrolyzed and the hydrolytic products have been released. Thus, this newly identified post-hydrolytic state may share a similar conformation of NBDs as the C2 closed state (i.e., a partially separated NBD and a vacated ATP-binding pocket). The significance of this novel state in understanding the structural basis of CFTR gating is discussed.
INTRODUCTION
CFTR, the culprit behind the fatal genetic disease cystic fibrosis (Riordan et al., 1989), is a member of the ATP-binding cassette (ABC) transporter superfamily, whose members exist throughout the biological universe. This family of integral membrane proteins is characterized by an evolutionarily conserved topology consisting of two transmembrane domains (TMDs) that form the cargo translocation pathway and two cytosolic nucleotide-binding domains (NBDs) serving as engines to drive the transport process. Recent structural and functional studies of ABC proteins have led to a hypothesis that the formation and separation of an NBD dimer are coupled to the conformational changes in TMDs to complete a transport cycle (Vergani et al., 2005; Dawson and Locher, 2006; Hollenstein et al., 2007; Ward et al., 2007; Khare et al., 2009).
Although nearly all ABC proteins assume the function of active transport, CFTR, a bona fide member of this superfamily, is unique in that it is an ATP-gated chloride channel (Bear et al., 1992). Nonetheless, numerous studies have indicated that CFTR shares similar architecture and mechanism of action with other ABC transporters. For example, the crystal structures of CFTR’s two NBDs are virtually indistinguishable from those of other ABC transporters (Lewis et al., 2004 and Protein Data Bank accession no. 3GD7). It has also been shown for CFTR that ATP binding induces the formation of a canonical NBD dimer seen in ABC transporters (Vergani et al., 2005; Mense et al., 2006). More recent studies (Bai et al., 2010, 2011) of CFTR’s gating conformational changes in its TMDs provided evidence supporting the notion that CFTR evolves from a primordial ABC exporter by simply removing its cytoplasmic gate (i.e., degraded transporter hypothesis). Therefore, mechanistic studies of how CFTR’s NBDs control gating transitions bear a broad implication, as the insights gained may help decipher the complex transport mechanism of all ABC proteins (Csanády, 2010).
For all ABC proteins, ATP is the source of the free energy that drives the transport cycle. Recent crystallographic studies provide snapshots of these proteins in different conformations: an “inward-facing” (Ward et al., 2007; Aller et al., 2009) configuration with separated NBDs, and an “outward-facing” (Dawson and Locher, 2006; Ward et al., 2007) configuration with dimerized NBDs. However, how they shuttle between the two states remains unclear. One major impediment to directly scrutinizing such conformational changes is the lack of techniques with high temporal resolutions. On the other hand, CFTR, as an ion channel, fills the void. It is generally accepted that the ATP-dependent NBD dimerization and separation control the gate of CFTR. Because ATP interacts exclusively with the NBDs, the gating signal upon ATP binding and subsequent NBD dimerization must be allosterically transmitted to the TMDs to initiate the gating cycle. The current model for CFTR gating (Fig. 1) dictates a strict coupling between the gating cycle and ATP hydrolysis cycle: opening of the channel into a burst and subsequent termination of the burst are synchronized to the dimerization and separation of the NBD dimer. This model, derived mostly from single-channel kinetic studies—albeit with its simplicity and elegance—is susceptible to potential technical faults. As the transitions between different conformational states could be rapid and transient in nature, classical kinetic analysis may not be sensitive enough to capture all relevant states and thus fail to detect slight deviation from the proposed scheme.
Figure 1.

A hypothetic model of CFTR gated by ATP. (A) A scheme illustrating ATP-dependent gating mechanism of CFTR. This scheme is synthesized based on several of the latest publications on CFTR gating (Csanády et al., 2010; Tsai et al., 2010b; Szollosi et al., 2011). For channel opening, two NBDs dimerize upon ATP binding to the catalysis-competent site (ABP2) in the C2 state, which harbors a partially dimerized NBD with an ATP molecule bound to the catalysis-incompetent site (ABP1). For channel closure, first the ATP in ABP2 is hydrolyzed to convert the prehydrolytic open state (O) to a post-hydrolytic open state (O′). Then, it is the separation of the NBD dimer and the dissociation of hydrolytic products that coincide with gate closure. In the continuous presence of millimolar ATP, CFTR rarely shuttles back to the C1 state, which bears completely separated NBDs. (B) A cartoon depicting how the proposed CFTR gating scheme in A correlates an experimentally observed opening and closing of a single CFTR channel to the molecular events in its NBDs and TMDs. The red box encompasses the open-channel conformations.
In the current study, by using the rapid ligand-exchange protocols developed in our laboratory (Tsai et al., 2009, 2010b), we were able to capture a transient state by locking the post-hydrolytic channel into a prolonged bursting state (i.e., locked-open state) with nonhydrolyzable ATP analogues, such as pyrophosphate (PPi) or adenylyl-imidodiphosphate (AMP-PNP) (Gunderson and Kopito, 1994; Hwang et al., 1994). In patches yielding macroscopic CFTR currents, when switching the ligand directly from ATP to PPi or AMP-PNP, we observed an unusual biphasic response to this experimental maneuver, suggesting the existence of two distinct functional states with different responsiveness to these nonhydrolyzable ligands: one C2 closed state characterized previously in Tsai et al. (2009), and the other, state X, with a faster response to PPi or AMP-PNP. These two states can be further differentiated by using mutations or the high-affinity ATP analogue, N6-phenylethyl-ATP (P-ATP). In patches containing one single CFTR channel, switching the ligand from ATP to a very brief (1-s) application of PPi in the open but not the closed state resulted in a direct transition into a locked-open state without transiting to a long interburst closure. These results suggest that state X, like the previously characterized C2 closed state, bears a partially separated NBD dimer and a vacated ATP-binding pocket (ABP)2. Although more studies are needed to determine whether this newly identified state is an open state or a short-lived closed state, our results could provide novel insight into the structural basis of CFTR gating and potentially impact our understanding of the functional mechanism for other ABC proteins.
MATERIALS AND METHODS
Cell culture and transient expression system
PolyFect transfection reagent (QIAGEN) was used to cotransfect CFTR cDNA and pEGFP-C3 (Takara Bio Inc.), encoding the green fluorescence protein into Chinese hamster ovary (CHO) cells. CHO cells were grown at 37°C in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum. 1 d before transfection, cells were trypsinized and cultured in 35-mm tissue culture dishes. After transfection, cells were cultured at 27°C for at least 2 d before electrophysiological experiments were performed.
Mutagenesis
All mutations were constructed using the QuickChange XL kit (Agilent Technologies) according to the manufacturer’s protocols, and then sequenced to confirm the mutation (DNA core; University of Missouri) on cDNA.
Electrophysiological recordings
Glass chips carrying the transfected cells were transferred to a chamber located on the stage of an inverted microscope (IX51; Olympus). Membrane patches were excised into an inside-out mode after the seal resistance was >40 GΩ. After excision, the pipette was perfused by 25 IU PKA and 2.75 mM ATP until the CFTR current reached a steady state; all other solutions containing ATP applied thereafter contained 10 IU PKA to maintain the phosphorylation level. An amplifier (EPC10; HEKA) was used to record electrophysiological data at room temperature at a −60-mV holding potential. The data were filtered online at 100 Hz with an eight-pole Bessel filter (LPF-8; Warner Instruments) and digitized to a computer at a sampling rate of 500 Hz. The inward current was inverted for clear data presentation. The resistance of pipettes for patch-clamp experiments was 2–4 MΩ in the bath solution. The pipettes were prepared from borosilicate capillary glass using a Flaming/Brown-type micropipette puller (P97; Sutter Instrument), and then polished with a homemade microforge. All inside-out patch experiments were performed with a fast solution exchange perfusion system (SF-77B; Warner Instruments). The dead time of solution change is ∼30 ms (Tsai et al., 2009).
Chemicals and composition solutions
The pipette solution contained (in mM): 140 NMDG chloride (NMDG-Cl), 2 MgCl2, 5 CaCl2, and 10 HEPES, pH 7.4 with NMDG. Cells were perfused with a bath solution containing (in mM): 145 NaCl, 5 KCl, 2 MgCl2, 1 CaCl2, 5 glucose, 5 HEPES, and 20 sucrose, pH 7.4 with NaOH. For inside-out configuration, the perfusion solution contained (in mM): 150 NMDG-Cl, 2 MgCl2, 10 EGTA, and 8 Tris, pH 7.4 with NMDG. MgATP, PPi, and PKA were purchased from Sigma-Aldrich. P-ATP was purchased from Biolog Life Science Institute. AMP-PNP was purchased from Roche. PPi and MgATP were stored in 200- and 250-mM stock solutions, respectively, at −20°C. P-ATP was stored in 10 mM of stock at −70°C. AMP-PNP was stored at −70°C and prepared before use. All chemicals were diluted to the concentration indicated in each figure using perfusion solution, and the pH was adjusted to 7.4 with NMDG. For solutions containing AMP-PNP or PPi, an equal concentration of MgCl2 was added to the solution.
Data analysis and statistics
The Igor Pro program (WaveMetrics) was used to calculate the steady-state mean current amplitude and the current relaxation time constant. Current relaxation was fitted with a single-exponential function using a Levenberg–Marquardt-based algorithm within the Igor Pro program. Channel kinetics was analyzed with a program developed by Csanády (2000) on traces that contain three or fewer channels. Kinetic modeling and computation simulations were described in Kopeikin et al. (2010).
Online supplemental material
Fig. S1 shows a representative single-channel ligand-exchange experiment for wild-type (WT)-CFTR. Fig. S2 shows a representative macroscopic trace of WT-CFTR when applying PPi 5 s after ATP washout. Fig. S3 shows the open dwell-time histograms of WT- and W401F-CFTR. Fig. S4 shows the kinetic model and parameters used for computer simulation. Figs. S1–S4 are available at http://www.jgp.org/cgi/content/full/jgp.201210789/DC1.
Results are shown as mean ± SEM. Student’s t test was performed for statistical analysis using Excel (Microsoft). P < 0.05 was considered statistically significant.
RESULTS
Identification of a new post-hydrolytic state
Once phosphorylated by PKA, CFTR channels are opened by ATP; upon ATP removal, CFTR will enter a relatively stable (dwell time of ∼25 s) closed state (so-called C2 state) that possesses a partial NBD dimer wherein the head of NBD2 and the tail of NBD1 (i.e., ABP2) are separated, but the opposite side of the NBD dimer (ABP1) remains attached (Tsai et al., 2009, 2010b; Szollosi et al., 2011). Channels in the C2 state are characterized by their capability to be locked open by nonhydrolyzable ATP analogues such as PPi or AMP-PNP (Tsai et al., 2009, 2010b) (Fig. 2, A and B); however, the lock-opening rate is very slow (time constant of current rising, τ = 4.8 ± 0.4 s; n = 8; Fig. 2 B, inset). Because the closing rate of the CFTR channel estimated from the current relaxation upon ATP removal (τrel = 380 ± 40 ms; n = 8; Fig. 2 B) is much faster, it is expected that if one switches the ligand directly from ATP to PPi, very few channel can be locked open during the current decay phase. Intuitively speaking, this is because the very short time (∼1 s) of current decay upon removing ATP is insufficient for a significant number of the closed channels to respond to PPi. Thus, nearly all channels are expected to close first upon ligand switches before they start to be opened by PPi. Indeed, when we performed computer simulations based on the model of scenario 1 (Fig. 2 A; detailed kinetic model and parameters are shown in Fig. S4), wherein the C2 state is the only state in the gating cycle that can be locked open by PPi, the simulated trace showed a decay of the macroscopic current to near the baseline (to the C2 state) before a slowly rising current was seen (Fig. 2 C). In contrast, when we performed such an experiment, the result turned out aberrant from this prediction. As shown in Fig. 2 D, upon ligand switching, the currents plummeted immediately but stopped in the middle rather than decay to the baseline, followed by a slow rising phase with a time constant (τ = 4.5 ± 0.4 s; n = 11) indistinguishable from that of channels locked open by PPi from the C2 state (Fig. 2 D, inset). Upon removal of PPi, all the currents decreased mono-exponentially with a time constant of 27.7 ± 2.9 s (n = 7), which is almost identical to the lifetime of locked-open channels from the C2 closed state (24.2 ± 2.7 s; n = 6). This result suggests that there are at least two post-hydrolytic states that can be distinguished by their responsiveness to PPi; one, named state X, can be more readily locked open within our solution exchange time and thus prevent the current from dropping to the baseline, and the other, the previously identified C2 state, responds to PPi much more sluggishly (Fig. 2 E, Scenario 2; see Discussion for alternative scheme). Because the lifetime of the locked-open state is constant in that no matter from which state, C2 or X, the channels become locked open, we conclude that a single locked-open conformation is attained, i.e., a channel with a dimerized NBD wherein ABP1 is occupied by ATP while ABP2 is taken by PPi (Tsai et al., 2009).
Figure 2.

PPi captures a short-lived, post-hydrolytic state. (A) A cartoon depicting the mechanism based in Fig. 1 A by which PPi locks open CFTR. O, open state with a dimerized NBD; C2, closed state with a partially separated NBD; LO, locked-open state with PPi occupying the second ABP, ABP2. (B, D, F, and G) Macroscopic current of WT-CFTR channels was activated by ATP to a steady state before carrying out different ligand-switch protocols: (B) washout for 10 s before applying PPi; (D) direct switch from ATP to PPi; (F) direct switched from ATP to a 1-s PPi pulse; and (G) washout for 3 s before applying a 1-s PPi pulse. (C) Computer simulation of macroscopic currents based on scenario 1. (E) A cartoon depicting a revised CFTR gating model, wherein state X can respond to PPi rapidly. Insets in B and D show the current rising phase upon PPi application; τ represents the relaxation time constant obtained by fitting the current rise with a single-exponential function (mean ± SEM was specified in Results). Bars above each trace mark the perfused ligand denoted on the very left (applied to every figure).
This newly identified state can be further probed by applying PPi for a brief duration that is deemed too short for channels in the C2 state to respond. Fig. 2 F shows such an experiment where ATP was switched to 10 mM PPi for just 1 s. This maneuver indeed effectively prevented a complete current decay seen upon removal of ATP and at the same time eliminated the slow rising phase seen in Fig. 2 D. In contrast, the application of the same 1-s pulse of PPi 3 s after ATP washout resulted in only minuscule current (Fig. 2 G), indicating that a 1-s pulse application of PPi is indeed too short for the C2 state to respond to a measurable extent, and that this newly identified state X is short-lived. Similar results were obtained with AMP-PNP instead of PPi as a nonhydrolyzable ligand (Fig. 3).
Figure 3.

Identification of a post-hydrolytic state by AMP-PNP. Macroscopic current of WT-CFTR channels was activated by ATP to a steady state before carrying out similar ligand-exchange protocols as shown in Fig. 2: (A) washout for 10 s and before applying AMP-PNP; (B) directly switched to a 1-s AMP-PNP pulse; and (C) washout for 3 s and before applying a 1-s AMP-PNP pulse.
The results in Figs. 2 and 3 not only demonstrate a novel state with a distinct responsiveness to PPi or AMP-PNP. The fact that this state vanishes within seconds after ATP washout (Figs. 2 G and 3 C) also suggests that it is less stable compared with the C2 state. The time when state X emerges is also intriguing in that it is not present before opening of the channel with ATP, and it ceases to exist just 3 s after closing of the channel after ATP hydrolysis. If we accept the idea that the open channel at least initially harbors an NBD dimer with both ABPs occupied (Vergani et al., 2005), this unique timing of state X surfacing leads to a conclusion that state X represents a post-hydrolytic state appearing before the C2 closed state as depicted in Fig. 2 E (see supplemental Discussion for details). More important, the fact that PPi or AMP-PNP can occupy ABP2 while the channel resides in state X indicates that the NBD dimer must have separated to the extent that it can accommodate a large ligand like AMP-PNP.
Competition between ADP and PPi for ABP2 of state X
The proposition that PPi or AMP-PNP enters ABP2 in state X to lock open CFTR suggests that the hydrolytic products, ADP and Pi, have been released. Thus, one expects that when applying ADP together with PPi, the effectiveness of PPi should be decreased, as ADP may compete with PPi for the binding site, namely ABP2. Using the protocol shown in Fig. 2 F, we compared the fraction of locked-open channels by a 1-s pulse of 10 mM PPi in the presence of three different concentrations of ADP (1, 2, and 10 mM). Indeed, ADP inhibits the effect of PPi in a dose-dependent manner (Fig. 4). These results again support the notion that in state X, the ABP2 is exposed and ready to accommodate another ligand. As our previous studies have demonstrated that once the channel is opened by ATP, the ABP1 remains inaccessible for ∼30 s even after channel closure (Vergani et al., 2005; Tsai et al., 2010b), we conclude that the NBDs in state X most likely assume a partial dimeric conformation, where the head of NBD2 and the tail of NBD1 are disengaged to expose the ATP-binding site in ABP2, similar to the configuration of the previously identified C2 state (Tsai et al., 2010b).
Figure 4.

ADP competes with PPi for state X. (A–C) Macroscopic current of WT-CFTR was activated by ATP to a steady state before switching the ligand to a 1-s pulse of 10 mM PPi in the presence of 1 mM (A), 2 mM (B), or 10 mM (C) ADP. (D) Summary of the proportion of locked-open currents (I LO) relative to ATP-activated currents (I ATP) at different ADP concentrations (mean ± SEM). *, P < 0.05 compared with 0 ADP.
Differential modulation of state X and the C2 state
Although both state X and the C2 state may harbor conformationally similar NBDs (i.e., a partial NBD dimer) as revealed by their responsiveness to AMP-PNP or PPi, they must differ in the overall conformation because the rate of their response to these nonhydrolyzable analogues is not the same, and their respective lifetime is very different. Here, we provide evidence that these two states can be differentially modulated by different experimental maneuvers. First, we used P-ATP, a high-affinity hydrolyzable ATP analogue (Zhou et al., 2005), as the initial ligand and performed the protocols shown in Fig. 2 (B and F) to isolate the fast and slow PPi-responsive states, respectively. Interestingly, the proportion of the fast locked-open current relative to the current before washout remained unchanged when we directly switched the ligand from P-ATP to a 1-s PPi pulse (46 ± 3% and n = 8 for P-ATP vs. 49 ± 2% and n = 13 for ATP), whereas the slow locked-open current relative to the ATP-activated current was increased when we applied PPi after a 10-s washout (83 ± 11% and n = 7 for P-ATP vs. 32 ± 2% and n = 6 for ATP; Fig. 5 A). Thus, opening CFTR channels with P-ATP improves the responsiveness of the C2 state (comparing to Fig. 2 B), but not state X, to PPi. On the other hand, mutating the residue Trp401 (W401), which forms ring–ring stacking interactions with ATP in ABP1 (Lewis et al., 2004; Zhou et al., 2006), to phenylalanine significantly increased the propensity of state X in response to PPi (i.e., a higher percentage of channels locked open from state X; WT, 49 ± 2% and n = 13; W401F, 59 ± 2% and n = 13; P < 0.05). But this same mutation dramatically reduced the lock-open current from the C2 state (0.08 ± 0.01% and n = 6; Fig. 5 B). Therefore, opposite to P-ATP, the W401F mutation enhances the responsiveness of state X to PPi but may destabilize the C2 state. These differential modulations of the properties of state X and the C2 state reinforce our interpretation that these two states cannot be conformationally identical.
Figure 5.

Differential modulation of the C2 state and state X. (A) WT-CFTR channels were first activated by ATP, and then the ligand was switched to P-ATP until the current reached a steady state. Next, the ligand was switched directly to a 1-s PPi pulse (top) to assess the responsiveness of state X to PPi. P-ATP was washed out for 10 s before applying PPi (bottom) to assess the responsiveness of the C2 state to PPi. (B) A similar protocol was conducted with W401F-CFTR channels. The percentage of locked-open current relative to ATP- (IATP) or P-ATP–induced (IP-ATP) current is indicated.
State X is an open state or a very brief closed state
To further investigate the nature of state X, we performed single-channel ligand-exchange experiments, which allow close monitoring of the channel gating upon ligand exchange. After witnessing the channel entering an open state, we switched the perfusate from ATP to PPi or AMP-PNP for just 1 s before washout. In this way, we could be certain that the channel was exposed to PPi or AMP-PNP in the open state. Such protocol could be technically difficult to perform with the WT channel because its short mean open time (∼300 ms) provides a limited operational window. However, the conserved W401F mutation (see below), which significantly prolongs the open time (Tsai et al., 2010a), makes such experiments more feasible.
In the presence of ATP, the W401F-CFTR channel exhibits similar behavior as WT-CFTR: the channel opens into bursts that last for hundreds of milliseconds to seconds, and each opening burst is separated by a long interburst that also closes in the range of hundreds of milliseconds (Fig. 6). Upon ligand switches from ATP to a 1-s pulse of PPi or AMP-PNP (Fig. 6, A and C), the channel can sojourn into a long-lasting locked-open burst without entering into a long interburst closure (see Discussion for details). For control, we performed the same ligand-exchange protocol when the channel was closed after washout of ATP and did not see any locked-open event (Fig. 6, B and D). These results suggest that state X is either an open state or a short-lived closed state that is indistinguishable from the flickers within each opening burst. We measured the duration of each closed event within that 1-s time window upon which PPi or AMP-PNP was applied. The maximal duration among 68 events is 86 ms with an average value of 25.1 ± 2.4 ms, which is similar to the mean lifetime of flickering closures characterized previously for hydrolysis-deficient mutants (Bompadre et al., 2005b) but is indeed much shorter than the interburst closed time (≥∼400 ms). It is noteworthy that the success rate of this line of experiments is lower for WT channels (see supplemental Discussion for more details), but similar observations were made for WT-CFTR (Fig. S1), indicating that such open X–locked-open transitions are not just idiosyncratic for W401F mutant channels. Furthermore, this measurement also established an upper limit of tens of milliseconds for the lifetime of state X if state X is a closed state.
Figure 6.

Single-channel ligand exchange for W401F-CFTR. (A and C) A single W401F-CFTR was activated by ATP, and the ligand was switched to a 1-s PPi (A) or AMP-PNP (C) pulse in the open state. The channel was locked open after the short application of PPi or AMP-PNP. The current trace was expanded in the red box to better discern events during ligand switch. (B and D) The same 1-s PPi (B) or AMP-PNP (D) pulse was applied when the channel resided in the closed state; no locked-open event was seen. Channels can be reactivated by ATP after long washout.
The lifetime of opening bursts forW401F-CFTR is [ATP] dependent
So far, our results have laid out a picture in which state X, like the C2 closed state, is a state with a vacant ABP2 that is readily accessible to sizable ligands such as AMP-PNP or PPi. But unlike the C2 state, which can be very stable, state X is likely short-lived (Figs. 1 and 6). Because both the shape and size between AMP-PNP and ATP are very similar, we reasoned that ATP is also capable of binding to state X, redimerizing the NBD, and bringing the channel to the initial, prehydrolytic open state (X→X′→O in Fig. 8 below). This hypothesis would predict that the mean open burst time should increase by increasing [ATP] as more reentry events (X→X′→O in Fig. 8) are likely to occur at higher [ATP]. This prediction is self-explanatory if state X is an open state. Even if state X is a short-lived closed state, this phenomenon should be observed, as the analytic method used for microscopic kinetic analysis of CFTR discards short-lived closings to extract “ATP-dependent” events (see Carson et al., 1995; Csanády and Gadsby, 1999; Csanády et al., 2005a,b, 2006, 2010; Vergani et al., 2005; Cai et al., 2006; Tsai et al., 2010a, and Discussion for details).
Figure 8.

A revised CFTR gating scheme showing hypothetical conformational transitions that take place during an opening burst. C1, closed state with two separated NBDs; C2, partial NBD dimeric closed state; O1, prehydrolytic open state; O1′, post-hydrolytic open state with a full NBD dimer; X, the newly identified state with a partially separated NBD dimer; X′, one ATP bound to the vacant ABP2 in state X. *, the ATP-independent, short-lived flickery closed events that are discounted in data analysis and interpretation (see Discussion for details). The red box encompasses the reentry pathway that may occur within each opening burst.
However, this [ATP]-dependent increase of the open burst time was not observed for WT-CFTR (Winter et al., 1994; Zeltwanger et al., 1999; Vergani et al., 2003; Bompadre et al., 2005a) probably because state X in WT channels is very unstable and/or it responds extremely poorly to ATP (see supplemental Discussion for detail). For reasons unclear at this moment, the W401F mutation could somehow improve the responsiveness of state X to PPi (Fig. 5) and therefore presumably to ATP as well. We therefore quantified the open burst time of W401F-CFTR at different [ATP]. Consistent with our idea, there is indeed a discernible [ATP]-dependent increase of the mean open burst time when the [ATP] is >1 mM (Fig. 7). Furthermore, the notion that a prolonged open burst time seen with the W401F mutation is caused by reentry of the channel from state X predicts a disparity between the mean open time estimated from microscopic analysis and the relaxation time constant obtained from macroscopic current decay upon ATP removal, which prohibits reentry to occur. Fig. 7 (B and C) shows that when fitting the current relaxation of W401F macroscopic current after washout of 10 mM ATP, the time constant is almost identical to the mean open time of W401F channels in the presence of micromolar ATP.
Figure 7.

The mean open time of W401F-CFTR is [ATP] dependent. (A) Microscopic current traces of W401F-CFTR in the presence of 10 mM (top), 1 mM (middle), or 100 µM (bottom) ATP. (B) Mean open time of W401F-CFTR at different [ATP] (closed circles). *, P < 0.05 when compared with 10 mM [ATP]. (C) Macroscopic current of W401F-CFTR was activated by 10 mM ATP to a steady state. The current relaxation upon ATP washout was fitted with a single-exponential function (red curve). τ = 389 ± 13 ms (n = 8).
DISCUSSION
Here, using nonhydrolyzable nucleotide analogues as “baits,” we were able to capture a short-lived post-hydrolytic state (state X; Fig. 8). This newly identified state distinguishes itself by its fast response to nonhydrolyzable ligands, PPi or AMP-PNP, the telltale sign for the presence of an exposed nucleotide-binding site. In light of the evolutionary relationship between CFTR and ABC transporters, our results have the potential to not only bring new insights to the coupling mechanism between ATP hydrolysis and gating transitions of CFTR, but they also bear structural/functional implications on the operational mechanism of the widespread ABC transporters.
Of note, the ligand-exchange method and subsequent interpretations of our data are based on one well-established theory—for WT-CFTR, channel opening is associated with NBD dimerization that sandwiches two ATP molecules in the two ABPs (Vergani et al., 2005)—and two simple physical rules: First, for a ligand to leave or enter an encaged space (i.e., NBD dimer interface), there must be a cleft equal to or larger than the size of the ligand. Second, for ligand A to be substituted by ligand B, ligand A must leave first. Thus, ligand exchanges between ATP and AMP-PNP during state X yield an inevitable conclusion that state X harbors a conformation that features a vacated ATP-binding site. Because state X is caught during the current decay phase (Fig. 1), which is embedded with ATP hydrolysis and release of the hydrolytic products, but before the channel transits to the previously characterized C2 closed state (Tsai et al., 2009, 2010b), we conclude that state X, similar to the C2 closed state, is very likely to bear a partially separated NBD with an exposed ABP2. However, the partial NBD dimeric conformation in state X may not be identical to the one in the C2 state. As shown in Fig. 5, the W401F mutation and the high-affinity ATP analogue P-ATP, both acting on ABP1, affect state X and the C2 state very differently. One simple scenario is that separation of the NBD dimer is incremental, so that procession of the channel from state X to the C2 state is accompanied by a stepwise increase of the separation of NBDs. This idea, albeit awaiting further experimental substantiation, could account for the observation that state X exhibits a much faster response to PPi or AMP-PNP than the C2 state (Fig. 2), as if these ligands can redimerize the NBDs of state X with ease.
However, caution should be taken when interpreting our data with such a simple model (Scheme 1), as the actual mechanism could be far more complicated and may entail more kinetic steps than what we proposed. For example, although our data shown in Fig. 2 strongly suggest that state X appears after ATP is hydrolyzed, this result by itself cannot exclude the possibility that state X and the C2 state emerge in two parallel pathways both derived from the open state and subsequently sojourn to the C1 state (Scheme 2).
| (SCHEME 1) |
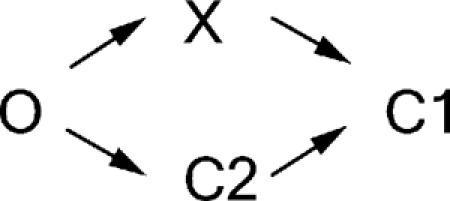 |
(SCHEME 2) |
According to Scheme 2, results in Fig. 2 F indicate that after ATP washout, about half of the open channels would travel to state X to be locked open by PPi immediately, and the other half would travel to the C2 state that does not respond to a 1-s application of PPi. As state X is a transient state with a lifetime likely in the millisecond range (see Fig. 6 above), those channels that take the O→X route would reach the C1 state within a second. This then means that during ATP-dependent gating, ∼50% of the “closed” channels will be in the C1 state with two NBDs separated and ATP-binding sites vacated. If half of the channels rapidly dissipate into the C1 state, it is expected that when conducting single-channel ligand-exchange experiments reported in Tsai et al. (2010b), about half of the experiments would show that both the opening rate and the open time increase concurrently. That is clearly not the case.
Another implication derived from Scheme 2 is that channels in state X and the C2 state are not interchangeable in the absence of ATP. In other words, once the channel departs from the O state upon ATP washout, its destiny is determined. Depleting the population of channels in one particular state will not affect the amount of channels in the other state. On the other hand, Scheme 1 would predict that the number of channels in the C2 state will increase initially when channels in state X dissipate. Because the slow responsive component (labeled in blue in Fig. 2 D) upon ligand switch from ATP to PPi reflects the portion of the channels in the C2 state, we can further differentiate these two schemes by comparing this component with different ATP washout times, as state X is expected to disappear after just seconds of ATP removal. Fig. 2 D shows that the slow responsive component is 0.24 ± 0.05 (n = 12; Fig. 2 D, the blue portion, I LO/I ATP) when PPi was applied immediately upon ATP washout. This is significantly lower (P < 0.05) than the same ratio measured when PPi is applied after 5 s of ATP washout (0.38 ± 0.04 and n = 7; Fig. S2), when all channels in state X have disappeared from the system. These results again contradict Scheme 2 but are consistent with Scheme 1.
Perhaps the most important question pertaining to the newly identified state is the functional status of its gate. As described above, state X is either an open state or a brief closed state. In the next section, we will discuss in detail the structure/function implications as well as the technical consequences of these two equally possible scenarios.
But first, it is necessary to delineate the generally adopted definition of “open” and “closed” events in CFTR gating. It has been long held that CFTR channels open in bursts; there are actually two distinct groups of closed events in CFTR’s gating: the [ATP]-dependent one (i.e., interburst closure) with a time constant of hundreds of milliseconds to seconds (Zeltwanger et al., 1999; Vergani et al., 2003), and the other one buried in an opening burst with a time constant in tens of milliseconds that was known as flickers (marked by stars in Fig. 8). These flickering closings have been long thought to be ATP independent, as they can be easily discerned in a complete absence of ATP within an opening burst of hydrolysis-deficient mutants, such as E1371S or K1250A (Carson et al., 1995; Powe et al., 2002; Bompadre et al., 2005b; Vergani et al., 2005). Although the nature and the mechanism of such flickers have not yet been rigorously studied, it was proposed that at least some of them are caused by pore blockage by anionic molecules in the solution (Zhou et al., 2001). These flickering closings have been excluded by various means in the literature upon data analysis to extract gating parameters that are associated with ATP binding and hydrolysis (Carson et al., 1995; Csanády and Gadsby, 1999; Bompadre et al., 2005a,b; Csanády et al., 2005a,b, 2006, 2010; Vergani et al., 2005; Cai et al., 2006; Tsai et al., 2010a). Therefore, the strict coupling model depicted in Fig. 1 means that each opening burst, which may contain several flickering closings, corresponds to hydrolysis of one ATP molecule.
Scenario 1: State X is a transient closed state
Because we cannot distinguish between the presumed closed state X and other ATP-independent flickers in the single-channel traces, an accurate estimation of the lifetime of state X is unattainable. However, as discussed in Results, should state X be a closed state, its dwell time is unlikely much longer than that of flickering closings (∼25 ms in Fig. 6 and in Bompadre et al., 2005b). Furthermore, the prolonged open burst time in the presence of higher [ATP] (>1 mM; P < 0.05 when comparing 1 and 10 mM) for W401F-CFTR indicates that the lifetime of state X as a closed state must be indistinguishable from that of the flickers so that closed events corresponding to state X are excluded by the analysis. In other words, the longer “observable” openings (or the open bursts), which have been considered hydrolysis-coupled events (Carson et al., 1995; Csanády and Gadsby, 1999; Bompadre et al., 2005a,b; Csanády et al., 2005a,b, 2006, 2010; Vergani et al., 2005; Cai et al., 2006; Tsai et al., 2010a), in fact could contain more than one “true” opening event separated by the closed state X. As explained in detail below, contrary to the model depicted in Fig. 1, it is this “true” opening event that is coupled to hydrolysis of one ATP under the premise that state X is a closed state.
As described above, in order for state X to be accommodating to PPi or AMP-PNP, it has to bear a partially separated NBD dimer with a vacated ABP2, to which ATP should also be able to bind. Furthermore, PPi or AMP-PNP needs to act on this presumed, barely visible closed state at an extremely fast rate; otherwise, this closed event will become more discernible in the single-channel recording. As the chemical structure of AMP-PNP is nearly identical to that of ATP (Yount, 1975), and this ATP analogue can be found comfortably nestled in the ATP-binding sites of ABC transporters (Dawson and Locher, 2007), one comes to an ineluctable implication that ATP can also readily reopen the channel from state X. Thus, contrary to the long-held idea that ATP opens CFTR solely through the long interburst closure, one has to conclude that some of the short-lived closures that are buried in an opening burst can respond to ATP to enter the open state. Thus, ATP hydrolysis is not coupled to the “observable” opening burst, but to the “true” opening that can happen more than one time within an observable burst.
Scenario 2: State X is an open state
If we interpret state X as an open state by discarding short-lived closures as adopted previously (Carson et al., 1995; Csanády and Gadsby, 1999; Bompadre et al., 2005a,b; Csanády et al., 2005a,b, 2006, 2010; Vergani et al., 2005; Cai et al., 2006; Tsai et al., 2010a), one immediate consequence is a violation of one-to-one stoichiometry between CFTR’s gating cycle and the ATP hydrolysis cycle because when the channel resides in state X, the vacated ATP-binding site is now accessible to a new ATP molecule, which subsequently redimerizes the NBDs and returns the channel to the original open state (X (open)→X′ (open)→O1 in Fig. 8).
At first glance, this conclusion based on the assumption that state X is an open state seems in disagreement with that reported in Csanády et al. (2010), but, in fact, it is not in conflict with but complementary to what they had discovered. The bimodal distribution in their open-time histograms convincingly demonstrated that closure of CFTR involves at least two steps, presumably ATP hydrolysis and NBD dimer separation. Csanády et al. (2010) concluded that in WT-CFTR, the majority of channel closings are the consequence of ATP hydrolysis rather than through a nonhydrolytic pathway. If state X is an open state, our data lead us to propose that for those channels undergoing hydrolysis-dependent closing, the hydrolysis per se does not guarantee a gate closure, and neither does the release of the hydrolytic products. It suggests a delay of the gating signal transmission from NBDs to TMDs; the NBD dimer separates before the gate closure, resulting in a post-hydrolytic open state (state X) with a partially separated NBD. Indeed, this addition of a post-hydrolytic open state (state X) and a reentry pathway to the prehydrolytic open state (O1 state) (X→X′→O1; Fig. 8) still predicts an open-time histogram with a bimodal distribution similar to that reported in Csanády et al. (2010). By comparing WT-CFTR and W401F-CFTR, the increasing reentry events (X→X′→O1) results in differences only in the mean open time and the tail of the fitted curve (Fig. S3), which is supplanted with an increased number of long opening bursts (>1,000 ms).
It is noteworthy that the idea of a post-hydrolytic open state was proposed 17 years ago. Gunderson and Kopito (1995) observed two open states with different single-channel conductances in WT-CFTR. Because the appearance of the two open states follows a preferred order (i.e., small conductance→large conductance), they proposed that by providing an input of the free energy, ATP hydrolysis per se triggered such a transition. Could the O2 state seen by Gunderson and Kopito correspond to state X here? Further studies are needed to provide the answer.
In addition, if separation of NBDs could occur before closure of the gate in TMDs takes place, it would suggest that these two domains in CFTR may assume a certain degree of autonomy; movement of one domain does not have to be coupled to the motion of the other. Then, instead of physical coupling of each TMD–NBD complex as a rigid body (Ivetac et al., 2007; Khare et al., 2009), one will entertain the possibility of an energetic coupling between NBDs and TMDs. Indeed, based on thermodynamic analysis of CFTR gating, Csanády et al. (2006) proposed that the gate in TMDs opens after the two NBDs have dimerized. The delay between NBD dimerization and gate opening was posited to represent the lifetime of a high-energy transition state. In a separate study, a transition state had been reported to have a lifetime in the microsecond range for acetylcholine receptor (Chakrapani and Auerbach, 2005). Also consistent with this idea, lately, we (Bai et al., 2010) showed that chemical modifications of an introduced cysteine in the TMDs can render the CFTR channel almost completely ATP independent. As this dramatic effect was also observed in a construct devoid of the entire NBD2 (unpublished data), at least in this extreme case, opening and closing of the gate in TMDs can take place without the necessity of NBD dimerization.
In conclusion, by using nonhydrolytic ATP analogues as tools, we uncovered a novel post-hydrolytic state that has a vacant ABP2 readily accessible to ATP. Regardless of the status of the gate for this state, our results argue that each previously defined opening burst in CFTR gating may entail hydrolysis of more than one ATP. An imminent goal is to answer a crucial question: is state X an open or a closed state? Answering this question could lead to a better understanding of the coupling mechanism between TMDs and NBDs not only for CFTR but also for ABC transporters at large.
Acknowledgments
We thank Dr. Kevin Gillis for critical reading of the manuscript, Dr. Ming-Feng Tsai for useful discussion, and Cindy Chu for technical assistance.
This work was supported by National Institutes of Health (grants R01HL53445 and R01DK55835 to T.-C. Hwang), Grants-in-Aid for Scientific Research (KAKENHI; JSPS22590212 and MEXT23118714), and Keio Gijuku Academic Development Funds (to Y. Sohma).
Christopher Miller served as editor.
Footnotes
Abbreviations used in this paper:
- ABC
- ATP-binding cassette
- ABP
- ATP-binding pocket
- AMP-PNP
- adenylyl-imidodiphosphate
- NBD
- nucleotide-binding domain
- P-ATP
- N6-phenylethyl-ATP
- PPi
- pyrophosphate
- TMD
- transmembrane domain
- WT
- wild type
References
- Aller S.G., Yu J., Ward A., Weng Y., Chittaboina S., Zhuo R., Harrell P.M., Trinh Y.T., Zhang Q., Urbatsch I.L., Chang G. 2009. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science. 323:1718–1722 10.1126/science.1168750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y., Li M., Hwang T.C. 2010. Dual roles of the sixth transmembrane segment of the CFTR chloride channel in gating and permeation. J. Gen. Physiol. 136:293–309 10.1085/jgp.201010480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y., Li M., Hwang T.C. 2011. Structural basis for the channel function of a degraded ABC transporter, CFTR (ABCC7). J. Gen. Physiol. 138:495–507 10.1085/jgp.201110705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear C.E., Li C.H., Kartner N., Bridges R.J., Jensen T.J., Ramjeesingh M., Riordan J.R. 1992. Purification and functional reconstitution of the cystic fibrosis transmembrane conductance regulator (CFTR). Cell. 68:809–818 10.1016/0092-8674(92)90155-6 [DOI] [PubMed] [Google Scholar]
- Bompadre S.G., Ai T., Cho J.H., Wang X., Sohma Y., Li M., Hwang T.C. 2005a. CFTR gating I: Characterization of the ATP-dependent gating of a phosphorylation-independent CFTR channel (ΔR-CFTR). J. Gen. Physiol. 125:361–375 10.1085/jgp.200409227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bompadre S.G., Cho J.H., Wang X., Zou X., Sohma Y., Li M., Hwang T.C. 2005b. CFTR gating II: Effects of nucleotide binding on the stability of open states. J. Gen. Physiol. 125:377–394 10.1085/jgp.200409228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Z., Taddei A., Sheppard D.N. 2006. Differential sensitivity of the cystic fibrosis (CF)-associated mutants G551D and G1349D to potentiators of the cystic fibrosis transmembrane conductance regulator (CFTR) Cl- channel. J. Biol. Chem. 281:1970–1977 10.1074/jbc.M510576200 [DOI] [PubMed] [Google Scholar]
- Carson M.R., Travis S.M., Welsh M.J. 1995. The two nucleotide-binding domains of cystic fibrosis transmembrane conductance regulator (CFTR) have distinct functions in controlling channel activity. J. Biol. Chem. 270:1711–1717 10.1074/jbc.270.4.1711 [DOI] [PubMed] [Google Scholar]
- Chakrapani S., Auerbach A. 2005. A speed limit for conformational change of an allosteric membrane protein. Proc. Natl. Acad. Sci. USA. 102:87–92 10.1073/pnas.0406777102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csanády L. 2000. Rapid kinetic analysis of multichannel records by a simultaneous fit to all dwell-time histograms. Biophys. J. 78:785–799 10.1016/S0006-3495(00)76636-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csanády L. 2010. Degenerate ABC composite site is stably glued together by trapped ATP. J. Gen. Physiol. 135:395–398 10.1085/jgp.201010443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csanády L., Gadsby D.C. 1999. CFTR channel gating: Incremental progress in irreversible steps. J. Gen. Physiol. 114:49–53 10.1085/jgp.114.1.49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csanády L., Chan K.W., Nairn A.C., Gadsby D.C. 2005a. Functional roles of nonconserved structural segments in CFTR’s NH2-terminal nucleotide binding domain. J. Gen. Physiol. 125:43–55 10.1085/jgp.200409174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csanády L., Seto-Young D., Chan K.W., Cenciarelli C., Angel B.B., Qin J., McLachlin D.T., Krutchinsky A.N., Chait B.T., Nairn A.C., Gadsby D.C. 2005b. Preferential phosphorylation of R-domain serine 768 dampens activation of CFTR channels by PKA. J. Gen. Physiol. 125:171–186 10.1085/jgp.200409076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csanády L., Nairn A.C., Gadsby D.C. 2006. Thermodynamics of CFTR channel gating: A spreading conformational change initiates an irreversible gating cycle. J. Gen. Physiol. 128:523–533 10.1085/jgp.200609558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csanády L., Vergani P., Gadsby D.C. 2010. Strict coupling between CFTR’s catalytic cycle and gating of its Cl- ion pore revealed by distributions of open channel burst durations. Proc. Natl. Acad. Sci. USA. 107:1241–1246 10.1073/pnas.0911061107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson R.J., Locher K.P. 2006. Structure of a bacterial multidrug ABC transporter. Nature. 443:180–185 10.1038/nature05155 [DOI] [PubMed] [Google Scholar]
- Dawson R.J., Locher K.P. 2007. Structure of the multidrug ABC transporter Sav1866 from Staphylococcus aureus in complex with AMP-PNP. FEBS Lett. 581:935–938 10.1016/j.febslet.2007.01.073 [DOI] [PubMed] [Google Scholar]
- Gunderson K.L., Kopito R.R. 1994. Effects of pyrophosphate and nucleotide analogs suggest a role for ATP hydrolysis in cystic fibrosis transmembrane regulator channel gating. J. Biol. Chem. 269:19349–19353 [PubMed] [Google Scholar]
- Gunderson K.L., Kopito R.R. 1995. Conformational states of CFTR associated with channel gating: the role ATP binding and hydrolysis. Cell. 82:231–239 10.1016/0092-8674(95)90310-0 [DOI] [PubMed] [Google Scholar]
- Hollenstein K., Frei D.C., Locher K.P. 2007. Structure of an ABC transporter in complex with its binding protein. Nature. 446:213–216 10.1038/nature05626 [DOI] [PubMed] [Google Scholar]
- Hwang T.C., Nagel G., Nairn A.C., Gadsby D.C. 1994. Regulation of the gating of cystic fibrosis transmembrane conductance regulator C1 channels by phosphorylation and ATP hydrolysis. Proc. Natl. Acad. Sci. USA. 91:4698–4702 10.1073/pnas.91.11.4698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivetac A., Campbell J.D., Sansom M.S. 2007. Dynamics and function in a bacterial ABC transporter: simulation studies of the BtuCDF system and its components. Biochemistry. 46:2767–2778 10.1021/bi0622571 [DOI] [PubMed] [Google Scholar]
- Khare D., Oldham M.L., Orelle C., Davidson A.L., Chen J. 2009. Alternating access in maltose transporter mediated by rigid-body rotations. Mol. Cell. 33:528–536 10.1016/j.molcel.2009.01.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopeikin Z., Sohma Y., Li M., Hwang T.C. 2010. On the mechanism of CFTR inhibition by a thiazolidinone derivative. J. Gen. Physiol. 136:659–671 10.1085/jgp.201010518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis H.A., Buchanan S.G., Burley S.K., Conners K., Dickey M., Dorwart M., Fowler R., Gao X., Guggino W.B., Hendrickson W.A., et al. 2004. Structure of nucleotide-binding domain 1 of the cystic fibrosis transmembrane conductance regulator. EMBO J. 23:282–293 10.1038/sj.emboj.7600040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mense M., Vergani P., White D.M., Altberg G., Nairn A.C., Gadsby D.C. 2006. In vivo phosphorylation of CFTR promotes formation of a nucleotide-binding domain heterodimer. EMBO J. 25:4728–4739 10.1038/sj.emboj.7601373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powe A.C., Jr, Al-Nakkash L., Li M., Hwang T.C. 2002. Mutation of Walker-A lysine 464 in cystic fibrosis transmembrane conductance regulator reveals functional interaction between its nucleotide-binding domains. J. Physiol. 539:333–346 10.1113/jphysiol.2001.013162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riordan J.R., Rommens J.M., Kerem B., Alon N., Rozmahel R., Grzelczak Z., Zielenski J., Lok S., Plavsic N., Chou J.L., et al. 1989. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 245:1066–1073 10.1126/science.2475911 [DOI] [PubMed] [Google Scholar]
- Szollosi A., Muallem D.R., Csanády L., Vergani P. 2011. Mutant cycles at CFTR’s non-canonical ATP-binding site support little interface separation during gating. J. Gen. Physiol. 137:549–562 10.1085/jgp.201110608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai M.F., Shimizu H., Sohma Y., Li M., Hwang T.C. 2009. State-dependent modulation of CFTR gating by pyrophosphate. J. Gen. Physiol. 133:405–419 10.1085/jgp.200810186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai M.F., Jih K.Y., Shimizu H., Li M., Hwang T.C. 2010a. Optimization of the degenerated interfacial ATP binding site improves the function of disease-related mutant cystic fibrosis transmembrane conductance regulator (CFTR) channels. J. Biol. Chem. 285:37663–37671 10.1074/jbc.M110.172817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai M.F., Li M., Hwang T.C. 2010b. Stable ATP binding mediated by a partial NBD dimer of the CFTR chloride channel. J. Gen. Physiol. 135:399–414 10.1085/jgp.201010399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergani P., Nairn A.C., Gadsby D.C. 2003. On the mechanism of MgATP-dependent gating of CFTR Cl− channels. J. Gen. Physiol. 121:17–36 10.1085/jgp.20028673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergani P., Lockless S.W., Nairn A.C., Gadsby D.C. 2005. CFTR channel opening by ATP-driven tight dimerization of its nucleotide-binding domains. Nature. 433:876–880 10.1038/nature03313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward A., Reyes C.L., Yu J., Roth C.B., Chang G. 2007. Flexibility in the ABC transporter MsbA: Alternating access with a twist. Proc. Natl. Acad. Sci. USA. 104:19005–19010 10.1073/pnas.0709388104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter M.C., Sheppard D.N., Carson M.R., Welsh M.J. 1994. Effect of ATP concentration on CFTR Cl- channels: a kinetic analysis of channel regulation. Biophys. J. 66:1398–1403 10.1016/S0006-3495(94)80930-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yount R.G. 1975. ATP analogs. Adv. Enzymol. Relat. Areas Mol. Biol. 43:1–56 [DOI] [PubMed] [Google Scholar]
- Zeltwanger S., Wang F., Wang G.T., Gillis K.D., Hwang T.C. 1999. Gating of cystic fibrosis transmembrane conductance regulator chloride channels by adenosine triphosphate hydrolysis. Quantitative analysis of a cyclic gating scheme. J. Gen. Physiol. 113:541–554 10.1085/jgp.113.4.541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z., Hu S., Hwang T.C. 2001. Voltage-dependent flickery block of an open cystic fibrosis transmembrane conductance regulator (CFTR) channel pore. J. Physiol. 532:435–448 10.1111/j.1469-7793.2001.0435f.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z., Wang X., Li M., Sohma Y., Zou X., Hwang T.C. 2005. High affinity ATP/ADP analogues as new tools for studying CFTR gating. J. Physiol. 569:447–457 10.1113/jphysiol.2005.095083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z., Wang X., Liu H.Y., Zou X., Li M., Hwang T.C. 2006. The two ATP binding sites of cystic fibrosis transmembrane conductance regulator (CFTR) play distinct roles in gating kinetics and energetics. J. Gen. Physiol. 128:413–422 10.1085/jgp.200609622 [DOI] [PMC free article] [PubMed] [Google Scholar]