

Abstract
Synaptic transmission depends critically on the Sec1p/Munc18 protein Munc18-1, but it is unclear whether Munc18-1 primarily operates as a integral part of the fusion machinery or has a more upstream role in fusion complex assembly. Here, we show that point mutations in Munc18-1 that interfere with binding to the free Syntaxin1a N-terminus and strongly impair binding to assembled SNARE complexes all support normal docking, priming and fusion of synaptic vesicles, and normal synaptic plasticity in munc18-1 null mutant neurons. These data support a prevailing role of Munc18-1 before/during SNARE-complex assembly, while its continued association to assembled SNARE complexes is dispensable for synaptic transmission.
Keywords: exocytosis, Munc18-1, SM proteins, SNARE complex, Syntaxin1a
Introduction
Synaptic vesicle exocytosis is executed by a multi-subunit protein machinery between vesicle and target membrane. The central components of the machinery are the SNARE proteins Syntaxin1, SNAP25 and Synaptobrevin2/VAMP2. In preparation for the final fusion event, the exocytotic machinery goes through several molecular steps involving conformational changes and association/dissociation of additional factors. Due to the fact that individual components are integral parts of the machinery, it has proven difficult to assign a precise role for each component and to determine the precise course of events.
One key factor for which it has been difficult to define the precise role in neuronal secretion is the Sec1p/Munc18 (SM) protein Munc18-1 (Toonen and Verhage, 2003; Toonen and Verhage, 2007; Burgoyne et al, 2009; Sudhof and Rothman, 2009; Carr and Rizo, 2010; Han et al, 2010; Smyth et al, 2010). Deletion of Munc18-1 in mice leads to the complete loss of neurotransmitter secretion (Verhage et al, 2000), and similar results have been found in Caenorhabditis elegans and Drosophila melanogaster (Harrison et al, 1994; Weimer et al, 2003). Based on in vivo studies, Munc18-1 is thought to be involved in the initial docking step (Voets et al, 2001; Weimer et al, 2003; Toonen et al, 2006a; de Wit et al, 2009) but also has a post-docking role (Gulyas-Kovacs et al, 2007; de Wit et al, 2009), and is proposed to promote/regulate the final fusion reaction (Fisher et al, 2001; Graham et al, 2004; Jorgacevski et al, 2011). However, other studies suggest that Munc18-1 is no longer required as an integral part of the fusion machinery once the SNARE complex is formed (Zilly et al, 2006; Rathore et al, 2010).
Comparably, biochemical investigations on the interaction of Munc18-1 with the core SNARE machinery have provided data that are difficult to reconcile. Indisputable, however, is that Munc18-1 binds tightly to monomer Syntaxin1. Interestingly, this interaction involves two sites on opposing surfaces of Munc18-1, which bind to the far N-terminal end of Syntaxin (N-peptide) and the remainder of the protein in closed conformation (Misura et al, 2000; Burkhardt et al, 2008). In this configuration, Munc18-1 prevents Syntaxin1 from participating in SNARE complex formation (Pevsner et al, 1994a; Yang et al, 2000; Rickman et al, 2007; Brandie et al, 2008; Burkhardt et al, 2008). At first sight, this inhibitory activity appears to be inconsistent with the null mutant phenotype (Verhage et al, 2000). However, when the N-peptide of Syntaxin cannot bind, Munc18-1-bound Syntaxin1 can readily assemble into SNARE complexes (Burkhardt et al, 2008). This indicates that Munc18-1 is able to guide Syntaxin towards productive SNARE complex formation and reveals that both binding sites act together. Remarkably, a similar structure with similar functionality has recently been uncovered for the homologous Munc18/Syntaxin 1 complex of the choanoflagellate Monosiga brevicollis (Burkhardt et al, 2011). Choanoflagellates are the closest living unicellular relatives of animals, suggesting that the neuronal Munc18-1/Syntaxin1 complex is evolutionary highly conserved.
On the other hand, based mainly on insights gained upon studying the yeast ortholog Sec1p, it was suggested that SM proteins might function as part of the core vesicle fusion machinery, promoting fusion when bound to the assembled SNARE complex (Carr et al, 1999; Scott et al, 2004; Togneri et al, 2006). Whether or not Munc18-1 behaves like Sec1p and primarily acts as part of the assembled SNARE complexes has been a matter of intense debate. Indeed, this mode of action of Munc18 appears to be supported by biochemical experiments that show that Munc18-1 binds to assembled SNARE complexes via the N-peptide of Syntaxin, although with much lower affinity compared to monomeric Syntaxin (Hata et al, 1993; Pevsner et al, 1994b; Dulubova et al, 1999; Misura et al, 2000; Dulubova et al, 2007; Khvotchev et al, 2007; Rickman et al, 2007; Shen et al, 2007; Burkhardt et al, 2008; Guan et al, 2008; Rodkey et al, 2008; Weninger et al, 2008), and with even lower affinity (although still in the sub-micromolar range) to vesicle SNAREs or the core SNARE complex (Xu et al, 2010). Moreover, a number of recent studies reported an accelerating effect of Munc18-1 in a liposome fusion assay (Shen et al, 2007; Rodkey et al, 2008; Diao et al, 2010; Shen et al, 2010; Schollmeier et al, 2011).
One strategy to discriminate between action of Munc18-1 on assembled SNARE complexes or a role in more upstream events has been to introduce point mutations that interfere with binding to assembled SNARE complexes while preserving affinity for Syntaxin1 in its closed conformation (Misura et al, 2000). Several mutations in Munc18 have been described, designed to interfere either with the N-peptide or the N-terminal three-helix-bundle of Syntaxin (the Habc domain). Since the interaction of Munc18-1 with assembled SNARE complexes strongly depends on the N-peptide, at least in vitro (Dulubova et al, 2007; Shen et al, 2007; Burkhardt et al, 2008), such mutations might be instrumental to address the biological relevance of SNARE-complex binding. Mutations interfering with N-peptide binding were indeed not able to rescue the function of the nematode ortholog, UNC-18 (McEwen and Kaplan, 2008; Johnson et al, 2009), although similar mutations in Munc18-1 had a moderate effect upon overexpression in PC12 cells (Malintan et al, 2009). Moreover, mutations in the N-peptide of Syntaxin produced strong inhibition of secretion in nematodes and disrupted the accelerating effect of Munc18-1 on liposome fusion (Shen et al, 2007; Johnson et al, 2009; Rathore et al, 2010; Schollmeier et al, 2011).
Here, we combine quantitative biochemistry with synapse physiology and electron microscopy in mammalian neurons on a null mutant background to dissect Munc18-1’s function on assembled SNARE complexes and in more upstream functions. We generated two Munc18-1 mutations that inhibit binding to the Syntaxin N-peptide and a third, previously published mutation (Deak et al, 2009), which affects SNARE-complex interaction by disrupting the interaction with the Syntaxin Habc domain. As a result, these mutants have no detectable affinity for assembled SNARE complexes. Surprisingly, these mutants support synaptic transmission equally well as wild-type Munc18-1. Our data are inconsistent with the proposed role of Munc18-1 in promoting fusion via binding to assembled SNARE complexes. Instead, we propose a model in which Munc18-1 assists primarily in setting up SNARE complexes, while downstream association with fusion complexes is dispensable.
Results
Munc18-1 mutations in the N-peptide-binding pocket reduce affinity for SNARE complexes
To study the relevance for synaptic transmission of Munc18-1 binding to the N-peptide of Syntaxin and thereby to assembled SNARE complexes, we generated a new mutant inspired by Sly1pL140K (Peng and Gallwitz, 2004), which interferes with the hydrophobic nature of the N-peptide-binding pocket (M18L130K, Figure 1A). Using isothermal titration calorimetry (ITC), we previously showed that Munc18-1 binds assembled SNARE complexes with relatively low affinity, and that there is no detectable interaction if the SNARE complex contains a N-terminally truncated Syntaxin1a (Burkhardt et al, 2008). As expected, for M18L130K no binding to assembled SNARE complexes could be detected by our ITC measurement (Figure 1B; Table I). Since the N-peptide attributes to the affinity of Munc18-1 for monomeric Syntaxin1a, M18L130K showed also reduced affinity and enthalpy for Syx1a1–262 (Figure 1C; Table I). Truncating the N-terminus of Syntaxin1a did not reduce the affinity further, supporting the notion that the reduced affinity is caused by disrupted N-peptide binding (Figure 1D; Table I). The reduced affinity for monomeric Syntaxin1a can be mainly attributed to an increased off-rate of the M18-1L130K/Syx1a complex (Figure 1E), which was about 10 times higher than for M18WT (M18WT≈0.0011 s−1, M18L130K≈0.010 s−1).
Figure 1.

Mutation in the N-peptide-binding pocket that disrupts SNARE-complex binding supports normal synaptic transmission. (A) Crystal structure of the M18-1/Syx1a complex represented as ribbon (left), with Munc18-1 in blue–green–yellow and Syx1a in purple–orange–red (adapted from Misura et al, 2000; Burkhardt et al, 2008). Arrow is pointing to the N-peptide-binding pocket. Close-up of the N-peptide-binding pocket with the residue targeted for mutagenesis in purple and the key residue of the Syntaxin1a N-peptide in orange (right). (B) Titration of the neuronal SNARE complex containing Syx1a1–262 into M18WT (black curve) or M18L130K (blue curve). Shown are the integrated areas normalized to the amount of SNARE complex injected (kcal/mol) versus its molar ratio to Munc18-1. (C) Calorimetric titrations of monomer Syx1a1–262 into Munc18-1. (D) Calorimetric titrations of monomer Syx1a25–262 into Munc18-1. (E) Determination of the off-rate of the Munc18-1/Syntaxin1a complex by competitive dissociation (M18WT≈0.0011 s−1 (from Burkhardt et al, 2008), black curve; M18L130K≈0.010 s−1, blue curve). (F) Ternary SNARE-complex formation was followed in the absence (grey curve) or presence of Munc18-1 (M18WT, black; M18L130K, blue). (G) Electron micrographs of typical asymmetrical glutamatergic synapses from neurons rescued with M18-1WT or M18-1L130K. Scale bar=100 nm. (H) Neurons rescued with M18-1L130K have a normal SV docking and a normal ultrastructure. N=4–5 islands, n=98–102 synapses. (I) Autaptic neurons rescued with M18L130K have similar EPSC sizes as neurons rescued with M18WT. Upper panel shows typical EPSC traces (artifacts removed). (J) No differences were found in PPRs (second EPSC/first EPSC). (K) Example traces of spontaneous release in autaptic neurons. (L) Spontaneous release amplitude and decay time are comparable. (M) Cumulative frequency plot of spontaneous release events. Insert shows average frequency. (N) Depression of EPSC size during repetitive stimulation at 5 Hz is normal. Inset shows the first four pulses. (O) An estimate of RRP size was made by back-extrapolation from the cumulative synchronous charge from the last 40 pulses within a 100-pulse train at 40 Hz. No differences in RRP size were found.
Table 1. Thermodynamic parameters of the interaction of Syntaxin1a and Munc18-1 measured by ITC.
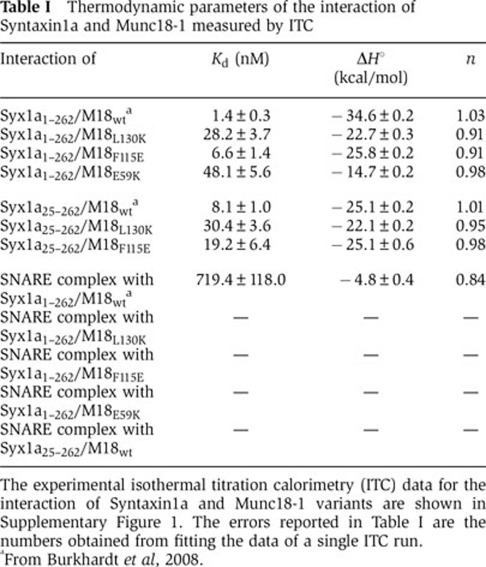
The experimental isothermal titration calorimetry (ITC) data for the interaction of Syntaxin1a and Munc18-1 variants are shown in Supplementary Figure 1. The errors reported in Table I are the numbers obtained from fitting the data of a single ITC run.
aFrom Burkhardt et al, 2008.
SNARE complex formation is blocked when the Syntaxin1a N-peptide is bound to Munc18-1 (Burkhardt et al, 2008). To test if M18L130K was still able to block ternary SNARE-complex formation, we used a fluorescence-based SNARE-binding assay. When the labelled cytosolic domain of Syb21-96 was mixed with Syx1a1–262 and SNAP25, the fluorescence anisotropy increased, indicating SNARE-complex formation (Figure 1F). M18L130K still inhibited, or at least slowed down considerably, SNARE-complex formation, but inhibition was not as complete as for M18WT (Figure 1F). Taken together, these biochemical assays confirm that interfering with N-peptide binding strongly impairs binding of Munc18-1 to the SNARE complex. In addition, the results reveal that M18L130K is much less efficient in inhibiting SNARE-complex formation than M18WT, highlighting again the crucial role of the N-peptide-binding site for the inhibitory role of Munc18 on SNARE-complex formation.
Neurons expressing an N-peptide-binding mutant show normal survival and vesicle docking
As the first step in analysing the relevance of N-peptide and SNARE-complex binding for synaptic transmission, we studied docking in autaptic munc18-1 null mutant neurons rescued with either M18WT or M18L130K. Neurons rescued with M18WT or M18L130K survived and developed with similar morphology and synapse density (Supplementary Figure S2A–D). As Munc18-1 expression level controls the number of docked vesicles and presynaptic output (Toonen et al, 2006b), we analysed synaptic Munc18-1 expression using semiquantitative immuno-fluorescence detection in VAMP2-positive synapses using an automated image analysis routine (Schmitz et al, 2011). Synaptic M18L130K levels were equal to M18WT (Supplementary Figure S2E). Docking of synaptic vesicles at the active zone and other ultrastructural features of the synapse were analysed by electron microscopy (Figure 1G and H). The number of docked vesicles in neurons rescued with M18L130K was similar to M18WT (Figure 1H). We used classical chemical fixation of these neurons, which might, together with the definition of ‘docking’ (see discussion in (Verhage and Sorensen, 2008)), produce a different absolute number of docked vesicles than when using rapid freeze fixation and/or other docking definitions, but this is unlikely to affect relative differences between experimental groups. The total number of vesicles and the amount of docked vesicles per nm active zone, as well as the size of the active zone and postsynaptic density did not differ between groups, confirming unchanged synaptic morphology (Figure 1H and Supplementary Figure S3A–B).
Synaptic transmission is unaffected by disrupted N-peptide and SNARE binding
As the second step, we studied priming and fusion in null mutant neurons rescued with either M18WT or M18L130K. Neurons rescued with M18L130K produced excitatory postsynaptic currents (EPSCs) upon depolarization that were similar to neurons rescued with M18WT (Figure 1I). The ratio between synaptic responses to two subsequent stimuli given at various time intervals (paired-pulse ratio; PPR) is a measure for release probability (Pr). The PPRs were also similar between neurons rescued with M18WT and M18L130K (Figure 1J). Furthermore, spontaneous release of single vesicles (miniature EPSCs) occurred at similar rate, and quantal size and mEPSC kinetics were similar (Figure 1K–M).
Changes in release efficiency or refilling might become evident when stimulation is continued for longer periods. However, neurons rescued with M18L130K displayed normal depression of EPSC size during stimulation trains at 5, 10 or 40 Hz (Figure 1N and Supplementary Figure S4A–B). In addition, RRP estimates derived by back-extrapolation from the cumulative synchronous charge released during a 40-Hz train indicated that the RRP was unaffected (Figure 1O) (Schneggenburger et al, 1999). Since synaptic release shifts from synchronous to asynchronous release during and after intense stimulation, a 40-Hz train can be used to monitor changes in synchronicity. Neurons rescued with M18L130K showed a similar pattern (synchronous charge/total charge) to neurons rescued with M18WT (Supplementary Figure S4C). To conclude, our data show that interfering with binding of Munc18-1 to the N-peptide of Syntaxin1 does not have a detectable effect on synaptic transmission in autaptic hippocampal neurons using our assays.
A second N-peptide mutant behaves like L130K in biochemical, electron microscopic and electrophysiological analyses
For comparison, we studied a second N-peptide-binding pocket mutant, M18F115E (Figure 2A). This mutant was inspired by published mutations in Sly1p and Munc18c, which severely impaired binding to the N-terminus of their cognate Syntaxins, Syntaxin4 and Sed5p, respectively (Peng and Gallwitz, 2004; Latham et al, 2006), and was recently also generated in Munc18-1 (Malintan et al, 2009). This mutation provides a second, independent approach for analysing the relevance of N-peptide and SNARE-complex binding within the N-peptide-binding pocket. Like for M18L130K, we found that M18F115E does not bind to the assembled SNARE complex using ITC (Figure 2B; Table I), consistent with previous co-precipitation data (Malintan et al, 2009). In addition, M18F115E showed reduced enthalpy and affinity for monomeric Syntaxin1a, and the enthalpy was not lowered further by additional removal of the N-peptide (Figure 2C and D). M18F115E behaved also very similar to M18L130K in Syx1a dissociation and SNARE-formation assays as it showed a faster dissociation rate constant (M18F115E≈0.0085 s−1) and a reduced ability to inhibit SNARE-complex formation (Figure 2E and F).
Figure 2.

A second mutation in the N-peptide-binding pocket that disrupts SNARE-complex binding also supports normal synaptic transmission. (A) Crystal structure of the M18-1/Syx1a complex represented as ribbon (left), with Munc18-1 in blue–green–yellow and Syx1a in purple–orange–red (adapted from Misura et al, 2000; Burkhardt et al, 2008). Arrow is pointing to the N-peptide-binding pocket. A close-up of the N-peptide-binding pocket with the residue targeted for mutagenesis in purple and the key residue of the Syntaxin1a N-peptide in orange (right). (B) Titration of the neuronal SNARE complex containing Syx1a1–262 into M18WT (black curve) or M18F115E (red curve). Shown are the integrated areas normalized to the amount of SNARE complex injected (kcal/mol) versus its molar ratio to Munc18-1. (C) Calorimetric titrations of monomer Syx1a1–262 into Munc18-1. (D) Calorimetric titrations of Syx1a25–262 into Munc18-1. (E) Determination of the off-rate of the Munc18-1/Syntaxin1a complex by competitive dissociation (M18WT≈0.0011 s−1 (from Burkhardt et al, 2008), black curve; M18F115E≈0.0085 s−1, red curve). (F) Ternary SNARE-complex formation was followed in the absence (grey curve) or presence of Munc18-1 (M18WT, black; M18F115E, red). (G) Electron micrographs of typical asymmetrical glutamatergic synapses from neurons rescued with M18-1WT or M18-1F115E. Scale bar=100 nm. (H) Neurons rescued with M18-1F115E have normal numbers of vesicles docked at the active zone and in total. Active zone size is normal. N=4–6 islands, n=98–100 synapses. (I) Autaptic neurons rescued with M18F115E have similar EPSC sizes as neurons rescued with M18WT. Upper panel shows typical EPSC traces (artifacts removed). (J) No differences were found in PPRs (second EPSC/first EPSC). (K) Example traces of spontaneous release in autaptic neurons. (L) Spontaneous release amplitude and decay time are comparable. (M) Cumulative frequency plot of spontaneous release events. Insert shows average frequency. (N) Depression of EPSC size during repetitive stimulation at 5 Hz is normal. Inset shows first four pulses. (O) An estimate of RRP size was made by back-extrapolation from the cumulative synchronous charge from the last 40 pulses within a 100-pulse train at 40 Hz. Inset shows average RRP sizes. No differences in RRP size were found.
Munc18-1 null neurons could also be rescued with M18F115E in autaptic culture. Again, overall morphology and synapse density of rescued neurons was not different between neurons rescued with M18WT and M18F115E (Supplementary Figure S2A–D). Unexpectedly, synaptic Munc18-1 levels were higher than in neurons rescued with M18WT or M18L130K (Supplementary Figure S2E). Reducing infection titres could not reduce cellular levels of M18F115E without compromising rescue efficiency/survival. Hence, M18F115E, unlike M18L130K (and M18E59K, see below), can be considered as an over-expression model and allows answering the question whether higher levels of interfering mutations might inhibit SNARE-complex formation, alter synaptic transmission and produce different phenotypes.
Despite higher synaptic levels, the number of docked vesicles was similar between neurons rescued with M18F115E or M18WT (Figure 2G and H) and therefore also similar to M18-1L130K (Figure 1G and H). The total number of vesicles, size of the active zone and other morphometric parameters all did not differ between null mutant neurons rescued with M18WT or M18F115E (Figure 2G and H and Supplementary Figure S3). Overexpression (Supplementary Figure S2E) is not as high as with Semliki Forest viral infection that we previously used to produce the highest cellular Munc18-1 levels (Toonen et al, 2006b), and the lack of ultrastructural effects of higher cellular Munc18-1 levels are consistent with earlier findings (Toonen et al, 2006b). EPSC sizes were also identical between neurons rescued with M18WT and M18F115E as were PPRs, indicating a normal Pr (Figure 2I and J). Spontaneous release was also normal in event frequency, size and kinetics (Figure 2K–M). EPSCs obtained by a train of stimulation at 5, 10 or 40 Hz followed a normal decline of amplitude and shift from synchronous to asynchronous release (Figure 2N and Supplementary Figure S4D–F). In addition, RRP estimates derived by back-extrapolation from the cumulative synchronous charge released during a 40-Hz train indicated a normal RRP size in neurons rescued with M18F115E (Figure 2O). Together, these results show that another point mutation within the N-peptide-binding pocket, M18F115E, behaves identically to M18L130K in biochemical assays and electrophysiological measurements. This further supports our hypothesis that N-peptide and SNARE-complex binding are not required for normal synaptic transmission.
A mutant that disrupts SNARE binding also supports normal synaptic transmission
Munc18-1 has been reported to control synaptic vesicle priming via interactions with the assembled SNARE complex. A mutation, M18E59K, has been introduced in the N-terminal domain of Munc18-1, which contacts the Syntaxin Habc domain (Figure 3A) and was previously characterized to disrupt binding to the SNARE complex while leaving the N-peptide-binding pocket intact. Compound EPSCs and spontaneous release frequency were severely reduced in network cultures of neurons expressing this mutant (Deak et al, 2009). Since our findings do not support a critical role for Munc18-1 at the assembled SNARE complex in synaptic transmission, we also tested M18E59K in our autaptic cultures. Our ITC measurements confirmed that M18E59K lowered the affinity for SNARE complexes beyond detection (Figure 3B; Table I). M18E59K also exhibited a lower affinity than M18WT for monomer Syntaxin1a (Kd≈48.1 nM, Figure 3C; Table I). Notably, the affinity determined by us is somewhat lower than what has been reported originally (12 nM, (Deak et al, 2009)). In this study, the affinity for the wild-type interaction was determined to be ≈7.5 nM, which suggested that the E59K mutation only slightly affected the interaction with the closed conformation of Syntaxin1a. By contrast, our data, in agreement with a recent study (Han et al, 2011), reveal that the affinity is more strongly affected by the point mutation. The reduced overall affinity for Syntaxin1a is probably caused by a faster rate of dissociation (k off≈0.072 s−1) (Figure 3D), a feature not previously documented. Also, we found that M18E59K completely lost the ability to inhibit SNARE-complex formation, which occurred at about similar speed as in the absence of Munc18-1 (Figure 3E).
Figure 3.

A subpopulation of M18E59K expressing neurons support neurotransmission. (A) Crystal structure of the M18-1/Syx1a complex represented as ribbon (left), with Munc18-1 in blue–green–yellow and Syx1a in purple–orange–red (adapted from Misura et al, 2000; Burkhardt et al, 2008). Arrow is pointing to the site of mutagenesis. Close-up of the E59 residue in grey (right). (B) Titration of the neuronal SNARE complex containing Syx1a1–262 into Munc18-1. (C) Titration of monomer Syx1a1-262 into Munc18-1. (D) Determination of the off-rate of the Munc18-1/Syntaxin1a complex by competitive dissociation (M18WT≈0.0011 s−1 (from Burkhardt et al, 2008), black curve; M18E59K≈0.072 s−1, green curve). (E) Ternary SNARE-complex formation was followed in the absence (grey curve) or presence of Munc18-1 (M18WT, black; M18E59K, green). (F) Electron micrographs of typical asymmetrical glutamatergic synapses from neurons rescued with M18-1WT or M18-1E59K. Scale bar=100 nm. (G) Neurons rescued with M18-1E59K are capable of docking synaptic vesicles. Active zone size is normal. N=3–4 islands, n=85–98 synapses. (H) Autaptic neurons rescued with M18E59K have similar EPSC sizes as neurons rescued with M18WT. Left panel shows typical EPSC traces (artifacts removed). (I) PPR was increased at the 20-ms pulse interval in neurons rescued with M18E59K (unpaired t-test, P=0.047). (J) Example traces of spontaneous release. (K) Spontaneous release amplitude is decreased in neurons rescued with M18E59K (unpaired t-test with Welch correction, P=0.016). Decay time is comparable to control mEPSCs. (L) Cumulative frequency plot of spontaneous release events. Insert shows average frequency. (M) Neurons rescued with M18E59K show normal depression of EPSC size during repetitive stimulation at 10 Hz. Insets show the first three pulses and the average absolute amplitude of the first pulse. (N) An estimate of RRP size was made by back-extrapolation from the cumulative synchronous charge from the last 40 pulses within a 100-pulse train at 40 Hz. Inset shows average RRP sizes. No difference in RRP size was found.
We then expressed M18E59K in autaptic hippocampal munc18-1 null neurons. Neurons rescued with M18E59K had a similar morphology and synapse density as neurons rescued with M18WT (Supplementary Figure S2A–D). However, synaptic Munc18-1 levels were lower (Supplementary Figure S2E–F). This could not be normalized by increasing viral load and might be due to decreased protein stability upon introduction of the mutation (as also noticed previously (Deak et al, 2009)). Under these conditions, ultrastructural analyses on rescued neurons showed that neurons rescued with M18E59K were capable of docking synaptic vesicles and have a total number of vesicles and ultrastructural features comparable to neurons expressing M18WT (Figure 3F and G and Supplementary Figure S3).
Nonetheless, neurons rescued with M18E59K showed normal EPSC sizes (Figure 3H), confirming that the interaction between Munc18-1 and the SNARE complex is not essential for synaptic transmission. However, we observed that a smaller number of neurons responded to stimulation, resulting in a higher number of cells being excluded from analysis (see Materials and methods). The average synaptic M18E59K levels per neuron varied widely, and despite the lower average M18E59K level, some neurons did express higher levels (Supplementary Figure S2F). It is conceivable that neurotransmission in these neurons is normal, while in low-expressing neurons neurotransmission is abolished. Responsive neurons expressing M18E59K displayed an enhanced PPR at the 20-ms inter-pulse interval (Figure 3I), suggesting a decreased initial Pr. Spontaneous release frequency was unchanged in neurons rescued with M18E59K but the average mEPSC amplitude was slightly lower (Figure 3J–L). Stimulation at 10 and 40 Hz resulted in normal rundown of EPSCs (Figure 3M and Supplementary Figure S4G). RRP estimates derived from 40-Hz stimulation trains are not significantly different from neurons rescued with M18WT (Figure 3N), nor is the shift in synchronicity in these trains (Supplementary Figure S4H).
Since the initial hypothesis that Munc18-1 controls synaptic vesicle priming via interactions with the SNARE complex originated from data obtained in network cultures (Deak et al, 2009), we also tested all three mutants in network cultures (Supplementary Figure S5). Networks of cortical neurons rescued with M18E59K responded with smaller peak amplitudes to local field stimulation (Supplementary Figure S5G), consistent with previous data (Deak et al, 2009). The high proportion of nonresponding neurons as found in autaptic cultures (>60%) provides a likely explanation for this reduced average response in mass culture. As in autaptic cultures, network cultures rescued with the N-peptide mutants M18L130K and M18F115E supported synaptic transmission and responded to field stimulation with similar peak amplitudes as cultures rescued with M18WT (Supplementary Figure S5A–F). However, interpretation of network cultures is more complex compared to autaptic cultures due to confounding factors like cell density and homoeostatic plasticity, especially when cell survival depends on efficient protein expression as is the case here.
Discussion
In this study, we tested the concept that binding of Munc18-1 to assembled SNARE complexes via the N-terminus is an important aspect of Munc18-1’s role in synaptic transmission. We tested this in situ, by analysing synaptic transmission in living neurons from munc18-1 null mutant mice. In these ‘rescued’ neurons, synapses depend solely on Munc18-1 carrying a mutation that interferes with binding to the N-peptide of Syntaxin and strongly impairs binding to the assembled SNARE complex. Using this system, we found no support for the concept that Munc18-1 promotes fusion efficiency by binding to SNARE complexes. The Munc18-1 mutations that lost affinity for assembled SNARE complexes support synaptic transmission equally well as M18WT. Hence, while binding assembled SNARE complexes and thereby promoting fusion may be a central aspect of some SM proteins (Carr et al, 1999; Scott et al, 2004; Togneri et al, 2006; McEwen and Kaplan, 2008; Hashizume et al, 2009; Johnson et al, 2009; Pieren et al, 2010), this is probably not a universal feature of all SM proteins.
The idea that SM proteins primarily act on assembled SNARE complexes was originally proposed on the basis of studies on the yeast SM protein involved in plasma membrane fusion, Sec1p. Sec1p has been shown to be unable to bind its cognate Syntaxin, Sso1p, but is thought to interact primarily with the assembled ternary SNARE complex (Carr et al, 1999; Scott et al, 2004; Togneri et al, 2006; Hashizume et al, 2009). The recently determined structure of the primordial Munc18/Syntaxin 1 complex of the choanoflagellate M. brevicollis suggests, however, that not the neuronal Munc18-1/Syntaxin complex but rather yeast Sec1p is deviated. In fact, the secretory Syntaxin of yeast, Sso1p, in contrast to most other eukaryotic lineages, seems to have lost a canonical N-terminal peptide sequence, suggesting that the mode of its interaction with the SNARE machinery diverged during fungal evolution. Moreover, a recent study shows that Sec1p contains a unique C-terminal tail, which preferentially binds SNARE complexes and is absent in other SM proteins (Weber-Boyvat et al, 2011). Furthermore, several additional actions of Sec1p have been characterized, suggesting that Sec1p also has more upstream actions potentially similar to those of Munc18-1 (Hashizume et al, 2009).
The fact that SNARE-complex binding is not crucial for synaptic transmission in mammalian neurons might be considered an evolutionary adaptation. However, disrupting the N-peptide interaction between two other SM proteins, Sly1p or Vsp45p, and their cognate Syntaxins also had no functional impact (Peng and Gallwitz, 2004; Carpp et al, 2006), suggesting that actions on assembled SNARE complexes might already vary among different SM proteins. The lack of effect of N-peptide mutations on synaptic transmission (and other fusion events in the cell) might in principle also be an additive effect of two separate effects, one being inhibited and one being stimulated by loss of N-peptide binding. However, the different functional assays used in this study are expected to unmask such dual, opposite effects, since these assays probe different aspects of Munc18-1 function to a different extent. For example, morphological docking analysis and RRP estimates probe specifically the more upstream actions of Munc18-1 and should report changes if the overall lack of effect on synaptic transmission would result from an additive effect of two underlying opposite effects of N-peptide mutations.
While in agreement with studies on yeast Sly1p and Vsp45p listed above, our findings contrast somewhat with several others. First, C. elegans UNC-18 binding to the N-peptide of Syntaxin/UNC-64 is required for synaptic function (McEwen and Kaplan, 2008; Johnson et al, 2009). Second, Syntaxin N-peptide injected or overexpressed in different systems inhibits fusion (Yamaguchi et al, 2002; Dulubova et al, 2003; Williams et al, 2004) including mammalian neurotransmitter secretion (Khvotchev et al, 2007). It is conceivable that the N-peptide fragment interferes with fusion in these systems irrespective of SM protein binding to the SNARE complex, or that the N-peptide of Syntaxin has a role beyond Munc18. Furthermore, the contribution of N-peptide binding can be different for different systems. For example, disrupting the N-peptide binding of UNC-18, unlike Munc18-1 (Burkhardt et al, 2008), also disrupted binding to monomeric Syntaxin (Johnson et al, 2009). It seems thus possible that in C. elegans the two binding sites cooperate much more strongly than in Munc18-1. Nevertheless, it needs to be tested by more rigorous biophysical methods like ITC whether the loss of the N-peptide entirely abolishes the binding of Syntaxin.
A number of recent studies using liposome fusion assays conclude that Munc18-1, like Sec1, promotes SNARE-dependent fusion and that binding the N-peptide is crucial in this respect (Shen et al, 2007; Rathore et al, 2010; Shen et al, 2010), probably by bringing Munc18-1 in close contact with the SNARE bundle (Diao et al, 2010; Rathore et al, 2010). In vivo, proximity between Munc18-1 and SNARE complexes could be achieved by additional factors or by Munc18-1 binding to closed Syntaxin, a step that most liposome fusion assays bypass by using liposomes with preassembled t-SNARE complexes. Indeed, a recent study revealed that Munc18-1 inhibits SNARE-mediated liposome fusion when the t-SNARE complex is not preassembled (Schollmeier et al, 2011), consistent with the well-established inhibitory effect of Munc18-1 on SNARE-complex formation in vitro (Pevsner et al, 1994b; Yang et al, 2000; Rickman et al, 2007; Burkhardt et al, 2008). Furthermore, stimulation of Munc18-1 in liposome fusion assays can only be observed after pre-incubation on ice with t-SNARE- and v-SNARE-containing liposomes and is limited to the beginning of the reaction. Therefore, it is conceivable that Munc18-1 promotes fusion by promoting SNARE assembly more than by acting on already assembled SNARE complexes. Furthermore, it was recently shown that purified squid Munc18-1 tends to aggregate in solution (Xu et al, 2011). This is consistent with our own observations on rat Munc18-1, which slowly aggregates and precipitates at higher concentrations (and also forced us to conduct binding experiments at concentrations lower than 20 μM in a previous study (Burkhardt et al, 2008). This tendency of Munc18 complicates standard liposome fusion assays due to potential light scattering artifacts and liposome clustering (Xu et al, 2011).
It has been known for a long time that Munc18-1 inhibits SNARE-complex formation in vitro (Pevsner et al, 1994a; Yang et al, 2000; Rickman et al, 2007; Burkhardt et al, 2008). Surprisingly, however, this aspect of Munc18’s activity has not been studied much in the last years as it is often considered to be a peculiarity of the neuronal isoform that does not play a direct role in exocytosis. Nevertheless, the ability of choanoflagellate Munc18 to block SNARE assembly reveals that this feature of Munc18 is conserved and thus very likely must play an important role. Interestingly, although all mutants tested still bound with relatively high affinity to individual Syntaxin, the ability to control SNARE assembly was lost to a variable extent among the mutations described here, including one mutation, M18E59K, which showed no inhibition at all (Figure 3E). Still all mutants supported transmission and no excess or aberrant fusion was observed. If anything, neurons rescued with M18E59K showed a slightly decreased Pr (Figure 3I). The reason that M18E59K is much more affected in its capacity for SNARE inhibition than the N-peptide mutants could be caused by the fast dissociation rate from monomeric Syntaxin1a (Figure 3D), a feature that could also be responsible for the reduced capacity of M18E59K to stimulate liposome fusion (Rodkey et al, 2008; Shen et al, 2010). Alternatively, binding of Munc18-1 to the SNARE complex might still have a subtle role on synaptic transmission, which for example might be reflected in the decreased Pr (increased PPR) of neurons rescued with M18E59K. A similar trend can be observed in the N-terminal mutants, although we must stress that this cannot be seen as a true difference since statistical significance is not reached.
Taken together, recently published work and our own findings here suggest a revised model for the actions of Munc18-1 in exocytosis (Figure 4). As an initial step in the secretory pathway, Munc18 binds tightly to monomeric Syntaxin en route to the target membrane and probably sequesters individual Syntaxin molecules from larger multimers in the plasma membrane. As suggested by the crystal structure of both rat and choanoflagellate, Munc18-1 readily makes use of two binding sites in Syntaxin, the N-peptide and the remainder of the molecule in closed conformation. It is conceivable that in this complex, Munc18 establishes the right conformation and localization of Syntaxin to assemble into the acceptor complex (Syntaxin plus SNAP25). This might allow synaptotagmin to bind to this acceptor complex and stably dock vesicles (de Wit et al, 2009). The Munc18/Syntaxin complex probably undergoes a conformational change during this process, which might be induced/controlled by other factors such as Munc13 and/or CAPS (Varoqueaux et al, 2002; Jockusch et al, 2007; Ma et al, 2011). Finally, the N-terminus of synaptobrevin associates to the Munc18/Syntaxin/SNAP25 complex to form a partially zippered, trans-SNARE complex, and might be supervised by other molecules such as complexin and synaptotagmin (see for a review (Walter et al, 2011)). In this situation, the vesicle is ready for Ca2+-triggered fusion, which, according to our data, no longer requires Munc18–Syntaxin interactions. Hence, in this model, Munc18-1 does not need to dissociate from Syntaxin during SNARE assembly, but instead assists Syntaxin in forming SNARE complexes, but its association is dispensable once the N-terminus of synaptobrevin associates. Indeed, one study has shown that Munc18-1 dissociates from SNARE complexes in exchange for synaptobrevin association (Zilly et al, 2006). Alternatively, Munc18-1 stays attached and performs a role that is too subtle to be detected by the tools used in this study.
Figure 4.

Schematic representation of the involvement of Munc18-1 in synaptic vesicle release. As an initial step in the secretory pathway, Munc18-1 (shown in cyan) binds tightly to monomeric Syntaxin (shown in red) via the N-peptide and the remainder of Syntaxin in closed conformation, probably sequestering individual Syntaxin molecules from larger multimers. Munc18 then establishes the right conformation and localization of Syntaxin to assemble into the acceptor complex (Syntaxin plus SNAP25 (shown in green)), which allows Synaptotagmin (shown in yellow) to bind to this acceptor complex and stably dock vesicles. This represents the established role of Munc18-1 in docking. Munc18-1 then assists Syntaxin in forming SNARE complexes, transferring vesicles from the docked state to the primed state. However, once the N-terminus of Synaptobrevin (shown in blue) associates to the Munc18/Syntaxin/SNAP25 complex to form a partially zippered, trans-SNARE complex and the vesicle is ready for Ca2+-triggered fusion, Munc18–Syntaxin interactions are no longer required.
Notably, the docking defect in Munc18-1 knockout neurons can be rescued by SNAP25 overexpression, although secretion remains arrested (de Wit et al, 2009). This observation suggests that Munc18, in addition to its established role in docking, also has a role in the following steps. The data in our current study position this post-docking role at the step(s) before the SNARE complex is fully assembled, that is, during association of the N-terminus of synaptobrevin with the Syntaxin:SNAP25 acceptor complex. At this stage, SM proteins may no longer be required and their continued association to assembled SNARE complexes is dispensable.
Materials and methods
Munc18-1-deficient mice
Munc18-1-deficient mice were generated as described previously (Verhage et al, 2000). Munc18-1 null mutant mice are stillborn and can be easily distinguished from wild-type or heterozygous littermates. E18 embryos were obtained by caesarian section of pregnant females from timed heterozygous mating. Animals were housed and bred according to the Institutional, Dutch and US governmental guidelines.
Constructs
We substituted leucine L130 for lysine or phenylalanine F115 for glutamic acid by subcloning the fragments in PCRblunt (Invitrogen) to create M18L130K and M18F115E, respectively. M18WT, M18L130K and M18F115E were cloned into pIRES2EGFP (Clontech). M18E59K was created from M18WTIRES2EGFP by substituting glutamic acid E59 for lysine using Quickchange (Stratagene). All constructs were subcloned into pLenti vectors, and viral particles were produced as described (Naldini et al, 1996). Transduction efficiencies were assessed on HEK cells and taken into account when applied to rescue neuronal cultures.
Protein expression and purification
The bacterial expression constructs for SNARE proteins, cysteine-free SNAP-25A (1–206), the soluble portion of Syntaxin1a (1–262) and the soluble portion of Synaptobrevin 2 (1–96) have been described before. Likewise, the single-cysteine SNARE protein variants used for labelling Syb2 Cys79 has been published (Burkhardt et al, 2008). Full-length Munc18-1 and the three Munc18-1 mutants L130K, F115E and E59K were constructed by site-directed mutagenesis. All constructs were cloned into a pET28a vector and expressed in E. coli. Proteins and SNARE complexes assembled from purified monomers were purified by Ni2+-NTA affinity chromatography followed by ion-exchange chromatography essentially as described (Burkhardt et al, 2008).
Isothermal titration calorimetry
ITC was performed on a VP-ITC instrument (GE Healthcare) at 25 °C. Samples were dialyzed against degassed PBS buffer (20 mM sodium phosphate, pH 7.4, 150 mM NaCl and 1 mM DTT). Titrations were carried out by 20 μl injections. The measured heat released upon binding was integrated and analysed with Microcal Origin 7.0 using a single-site binding model, yielding the equilibrium association constant Ka, the enthalpy of binding ΔH and the stoichiometry N. Experimental data are shown in Supplementary Figure S1.
Fluorescence spectroscopy
Fluorescence measurements were carried out in a Fluorolog 3 spectrometer in T-configuration equipped for polarization (Horiba Scientific). Single-cysteine variants were labelled with Texas Red C5 bromoacetamide or Oregon Green 488 iodoacetamide according to the manufacturer’s instructions (Invitrogen). All experiments were performed at 25 °C in 1-cm quartz cuvettes in PBS buffer. Fluorescence anisotropy, which is used to indicate the local flexibility of the labelled residue, and which increases upon complex formation and decreases upon dissociation, was measured essentially as described (Burkhardt et al, 2008). The G factor was calculated according to G=I HV/I HH, where I is the fluorescence intensity, and the first subscript letter indicates the direction of the exciting light and the second subscript letter the direction of emitted light. The intensities of the vertically (V) and horizontally (H) polarized emission light after excitation by vertically polarized light were measured. The anisotropy (r) was determined according to r=(I VV−GI VH)/(I VV+2GI VH). During competitive dissociation experiments, an excess of unlabelled Syx1a1–262 (5 μM) was added to a premix of 100-nM Oregon Green-labelled Syx1a1–262 and 200 nM Munc18, and the decrease in fluorescence anisotropy measured. The dissociation was fitted by a single exponential fit. Ternary SNARE-complex formation was followed by the increase in fluorescence anisotropy of labelled Syb21-96 (40 nM) upon mixing with 500 nM Syntaxin1a and 750 nM SNAP25 in the absence or presence of 750 nM Munc18-1.
Neuronal cultures
Hippocampi and cortices were separately collected in ice-cold Hanks Buffered Salt Solution (HBSS; Sigma) buffered with 7 mM HEPES (Invitrogen). After removal of the meninges, neurons were incubated in Hanks-HEPES with 0.25% trypsin (Invitrogen) for 20 min at 37 °C. After washing, neurons were triturated and counted in a Fuchs–Rosenthal chamber. Neurons were plated in prewarmed Neurobasal medium (Invitrogen) supplemented with 2% B-27 (Invitrogen), 1.8% HEPES, 0.25% glutamax (Invitrogen) and 0.1% Pen/Strep (Invitrogen). To achieve autaptic cultures, hippocampal neurons were plated at a density of 6 K/well of a 12-well plate on microislands of rat glia. These microislands were generated by plating 8 K/well rat glia on UV-sterilized agarose-coated etched glass coverslips stamped with a 0.1 mg/ml poly-D-lysine (Sigma) and 0.2 mg/ml rat tail collagen (BD Biosciences) solution. Network cultures were generated by plating cortical neurons (100–500 K/well on a 12-well plate) on a confluent layer of rat glia grown on etched glass coverslips sprayed with a 0.1 mg/ml poly-D-lysine and 0.2 mg/ml rat tail collagen (BD Biosciences) solution. Neurons were infected at DIV0 with lentiviral particles encoding M18WT, M18L130K, M18F115E and M18E59K, and were allowed to develop for 13–18 days before measuring. For protein level measurements, neurons were plated at 100 K/well in a 12-well plate containing etched glass coverslips coated with 0.5 milli-percent poly-L-ornithine (Sigma) and 2 μg/ml laminin (Sigma), and were grown for 10–11 days before fixation.
Electrophysiological recordings
Whole-cell voltage-clamp recordings (Vm=−70 mV) were performed on autaptic or network cultures on DIV 13–18 at room temperature. External solution contained the following (in mM): 10 HEPES, 10 glucose, 140 NaCl, 2.4 KCl, 4 MgCl2 and 4 CaCl2 (pH=7.30, 300 mOsmol). Gabazine (Sigma) was added to the external solution at a concentration of 20 μM to isolate excitatory currents. Patch-pipette solution contained the following (in mM): 125 K+–gluconic acid, 10 NaCl, 4.6 MgCl2, 15 creatine phosphate, 10 U/ml phosphocreatine kinase and 1 EGTA (pH 7.30). Recordings were performed with an Axopatch 200A amplifier (Molecular Devices). Digidata 1322A and Clampex 9.0 (Molecular Devices) were used for signal acquisition. After whole-cell mode was established, only cells with an access resistance of <12 MΩ and leak current of <500 pA were accepted for analysis. In autaptic neurons, EPSCs were elicited by a 0.5 ms depolarization to 30 mV. Responses below 300 pA were excluded from analysis. The percentage of excluded cells was similar for all groups except for M18E59K rescued neurons (M18WT, 24%; M18L130K, 29%; M18F115E, 28%; M18E59K, 48% of cells excluded). Local field stimulation (1 mA, 1 ms) was applied with a bipolar concentric electrode placed in the vicinity of the patched neuron. Spontaneous release in networks was measured in the presence of 1 μM tetrodotoxin (Ascent) to block Na+ currents. Offline analysis of electrophysiology was performed using Clampfit v9.0 (Axon Instruments), Mini Analysis Program v6.0 (synaptosoft) and custom-written software routines in Matlab 7.1 or R2009b (Mathworks).
Immunocytochemistry
Cultures were fixed with 3.7% formaldehyde (Electron Microscopy Sciences). After washing with PBS, cells were permeated with 0.1% Triton X-100 for 5 min and incubated in 2% normal goat serum for 20 min to block aspecific binding. Cells were incubated for 1 h at room temperature in a mixture of monoclonal mouse anti-VAMP (1:1000, SySy), polyclonal chicken anti-MAP2 (1:10 000, Abcam) and polyclonal rabbit anti-Munc18-1 (1:500, SySy) antibodies. After washing, cells were incubated for 1 h at room temperature with second antibodies conjugated to Alexa dyes (1:1000, Molecular Probes) and washed again. Coverslips were mounted with Mowiol-Dabco and imaged with a confocal LSM510 microscope (Carl Zeiss) using a ×40 oil immersion objective with ×0.7 zoom at 1024×1024 pixels. Neuronal morphology and protein levels were analysed using a published automated image analysis routine (Schmitz et al, 2011).
Electron microscopy
Autaptic hippocampal cultures of munc18-1 null mutant mice (E18) obtained from two different litters were fixed at DIV14-16 for 45 min at room temperature with 2.5% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.4) (de Wit et al, 2006; Wierda et al, 2007). As for electrophysiology, only glia islands containing a single neuron were used for analysis. After fixation, cells were washed three times for 5 min with 0.1 M cacodylate buffer (pH 7.4), post-fixed for 2 h at room temperature with 1% Osmium tetroxide/1% potassium ferrocyanide in bidest, washed and stained with 1% uranyl acetate for 40 min in the dark. Following dehydration through a series of increasing ethanol concentrations, cells were embedded in Epon and polymerized for 24 h at 60 °C. After polymerization of the Epon, the coverslip was removed by alternately dipping it in liquid nitrogen and hot water. Cells of interest were selected by observing the flat Epon-embedded cell monolayer under the light microscope, and mounted on prepolymerized Epon blocks for thin sectioning. Ultrathin sections (∼90 nm) were cut parallel to the cell monolayer and collected on single-slot, formvar-coated copper grids, and stained in uranyl acetate and lead citrate. Autaptic synapses were selected at low magnification using a JEOL 1010 electron microscope. All analyses were performed on single ultrathin sections of randomly selected synapses. The distribution of synaptic vesicles, total synaptic vesicle number and active zone length were measured with Image J (National Institute of Health, USA) on digital images of synapses taken at ×100 000 magnification using analySIS software (Soft Imaging System, Gmbh, Germany). The observer was blinded for the genotype. For all morphological analyses, we selected only synapses with intact synaptic plasma membranes with a recognizable pre- and postsynaptic density and clear synaptic vesicle membranes. Docked synaptic vesicles had a distance of 0 nm from the synaptic vesicle membrane to the active zone membrane. The active zone membrane was recognized as a specialized part of the presynaptic plasma membrane that contained a clear presynaptic density. To get an estimate size of the total synaptic vesicle pool, distances of undocked synaptic vesicles to the active zone membrane were also included in our measurements.
Data analysis
Data are presented as mean values±s.e.m., with n referring to the number of cells from each group. Statistical analysis was performed with Instat v3.05 software (GraphPad Software). Data samples were first tested for normality with the Kolmogorov and Smirnov test, and for heterogeneity of variance with the method of Bartlett. If data allowed an unpaired t-test (Welch correction was adopted when variances were unequal) or an ANOVA was used to determine statistical significance. Alternatively, the nonparametric Mann–Whitney U-test or the Kruskal–Wallis Test (for comparing one or multiple groups, respectively) was used. If a test for multiple groups reached significance, post-testing to compare the experimental to the control group was performed using the parametric Bonferroni post-test or the nonparametric Dunn’s post-test. P-values below 0.05 are considered significant and are indicated by asterisks (*P<0.05, **P<0.01 and ***P<0.001).
Supplementary Material
Acknowledgments
We thank R Zalm, D Schut, I Saarloos, J Hoetjes, B Beuger, H Lodder and J Wortel for technical assistance, and C van der Meer, B Tersteeg and M Boin for animal breeding and maintenance. R Dekker is acknowledged for excellent electron microscopical analyses. This work was supported by the EU (EUSynapse project 019055 to MV, EUROSPIN project HEALTH-F2-2009-241498 to MV and HEALTH-F2-2009-242167 SynSys project to MV), the Netherlands Organization for Scientific Research, NWO (Pionier/VICI 900-01-001 and ZonMW 903-42-095 to MV; and VENI 916-36-043 to HdW) and the NeuroBsik Mouse Phenomics Consortium (BSIK03053).
Author contributions: MM and PB performed most of the experiments, and analysed and interpreted the data. HdW contributed to the EM data. MV, DF and RFT conceived–designed the project and interpreted the results. MM, MV and DF wrote the manuscript with inputs from all authors.
Footnotes
The authors declare that they have no conflict of interest.
References
- Brandie FM, Aran V, Verma A, McNew JA, Bryant NJ, Gould GW (2008) Negative regulation of syntaxin4/SNAP-23/VAMP2-mediated membrane fusion by Munc18c in vitro. PLoS ONE 3: e4074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgoyne RD, Barclay JW, Ciufo LF, Graham ME, Handley MT, Morgan A (2009) The functions of Munc18-1 in regulated exocytosis. Ann N Y Acad Sci 1152: 76–86 [DOI] [PubMed] [Google Scholar]
- Burkhardt P, Hattendorf DA, Weis WI, Fasshauer D (2008) Munc18a controls SNARE assembly through its interaction with the syntaxin N-peptide. EMBO J 27: 923–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhardt P, Stegmann CM, Cooper B, Kloepper TH, Imig C, Varoqueaux F, Wahl MC, Fasshauer D (2011) Primordial neurosecretory apparatus identified in the choanoflagellate Monosiga brevicollis. Proc Natl Acad Sci USA 108: 15264–15269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpp LN, Ciufo LF, Shanks SG, Boyd A, Bryant NJ (2006) The Sec1p/Munc18 protein Vps45p binds its cognate SNARE proteins via two distinct modes. J Cell Biol 173: 927–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr CM, Grote E, Munson M, Hughson FM, Novick PJ (1999) Sec1p binds to SNARE complexes and concentrates at sites of secretion. J Cell Biol 146: 333–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr CM, Rizo J (2010) At the junction of SNARE and SM protein function. Curr Opin Cell Biol 22: 488–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit H, Cornelisse LN, Toonen RF, Verhage M (2006) Docking of secretory vesicles is syntaxin dependent. PLoS ONE 1: e126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit H, Walter AM, Milosevic I, Gulyas-Kovacs A, Riedel D, Sorensen JB, Verhage M (2009) Synaptotagmin-1 docks secretory vesicles to syntaxin-1/SNAP-25 acceptor complexes. Cell 138: 935–946 [DOI] [PubMed] [Google Scholar]
- Deak F, Xu Y, Chang WP, Dulubova I, Khvotchev M, Liu X, Sudhof TC, Rizo J (2009) Munc18-1 binding to the neuronal SNARE complex controls synaptic vesicle priming. J Cell Biol 184: 751–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diao J, Su Z, Lu X, Yoon TY, Shin YK, Ha T (2010) Single-vesicle fusion assay reveals Munc18-1 binding to the SNARE core is sufficient for stimulating membrane fusion. ACS Chem Neurosci 1: 168–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulubova I, Khvotchev M, Liu S, Huryeva I, Sudhof TC, Rizo J (2007) Munc18-1 binds directly to the neuronal SNARE complex. Proc Natl Acad Sci USA 104: 2697–2702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulubova I, Sugita S, Hill S, Hosaka M, Fernandez I, Sudhof TC, Rizo J (1999) A conformational switch in syntaxin during exocytosis: role of munc18. EMBO J 18: 4372–4382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulubova I, Yamaguchi T, Arac D, Li H, Huryeva I, Min SW, Rizo J, Sudhof TC (2003) Convergence and divergence in the mechanism of SNARE binding by Sec1/Munc18-like proteins. Proc Natl Acad Sci USA 100: 32–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher RJ, Pevsner J, Burgoyne RD (2001) Control of fusion pore dynamics during exocytosis by Munc18. Science (New York, NY 291: 875–878 [DOI] [PubMed] [Google Scholar]
- Graham ME, Barclay JW, Burgoyne RD (2004) Syntaxin/Munc18 interactions in the late events during vesicle fusion and release in exocytosis. J Biol Chem 279: 32751–32760 [DOI] [PubMed] [Google Scholar]
- Guan R, Dai H, Rizo J (2008) Binding of the Munc13-1 MUN domain to membrane-anchored SNARE complexes. Biochemistry 47: 1474–1481 [DOI] [PubMed] [Google Scholar]
- Gulyas-Kovacs A, de Wit H, Milosevic I, Kochubey O, Toonen R, Klingauf J, Verhage M, Sorensen JB (2007) Munc18-1: sequential interactions with the fusion machinery stimulate vesicle docking and priming. J Neurosci 27: 8676–8686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han GA, Malintan NT, Collins BM, Meunier FA, Sugita S (2010) Munc18-1 as a key regulator of neurosecretion. J Neurochem 115: 1–10 [DOI] [PubMed] [Google Scholar]
- Han GA, Malintan NT, Saw NM, Li L, Han L, Meunier FA, Collins BM, Sugita S (2011) Munc18-1 domain-1 controls vesicle docking and secretion by interacting with syntaxin-1 and chaperoning it to the plasma membrane. Mol Biol Cell 22: 4134–4149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison SD, Broadie K, van de Goor J, Rubin GM (1994) Mutations in the Drosophila Rop gene suggest a function in general secretion and synaptic transmission. Neuron 13: 555–566 [DOI] [PubMed] [Google Scholar]
- Hashizume K, Cheng YS, Hutton JL, Chiu CH, Carr CM (2009) Yeast Sec1p functions before and after vesicle docking. Mol Biol Cell 20: 4673–4685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata Y, Slaughter CA, Sudhof TC (1993) Synaptic vesicle fusion complex contains unc-18 homologue bound to syntaxin. Nature 366: 347–351 [DOI] [PubMed] [Google Scholar]
- Jockusch WJ, Speidel D, Sigler A, Sorensen JB, Varoqueaux F, Rhee JS, Brose N (2007) CAPS-1 and CAPS-2 are essential synaptic vesicle priming proteins. Cell 131: 796–808 [DOI] [PubMed] [Google Scholar]
- Johnson JR, Ferdek P, Lian LY, Barclay JW, Burgoyne RD, Morgan A (2009) Binding of UNC-18 to the N-terminus of syntaxin is essential for neurotransmission in caenorhabditis elegans. Biochem J 418: 73–80 [DOI] [PubMed] [Google Scholar]
- Jorgacevski J, Potokar M, Grilc S, Kreft M, Liu W, Barclay JW, Buckers J, Medda R, Hell SW, Parpura V, Burgoyne RD, Zorec R (2011) Munc18-1 tuning of vesicle merger and fusion pore properties. J Neurosci 31: 9055–9066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khvotchev M, Dulubova I, Sun J, Dai H, Rizo J, Sudhof TC (2007) Dual modes of Munc18-1/SNARE interactions are coupled by functionally critical binding to syntaxin-1 N terminus. J Neurosci 27: 12147–12155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latham CF, Lopez JA, Hu SH, Gee CL, Westbury E, Blair DH, Armishaw CJ, Alewood PF, Bryant NJ, James DE, Martin JL (2006) Molecular dissection of the Munc18c/syntaxin4 interaction: implications for regulation of membrane trafficking. Traffic (Copenhagen, Denmark) 7: 1408–1419 [DOI] [PubMed] [Google Scholar]
- Ma C, Li W, Xu Y, Rizo J (2011) Munc13 mediates the transition from the closed syntaxin-Munc18 complex to the SNARE complex. Nat Struct Mol Biol 18: 542–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malintan NT, Nguyen TH, Han L, Latham CF, Osborne SL, Wen PJ, Lim SJ, Sugita S, Collins BM, Meunier FA (2009) Abrogating Munc18-1-SNARE complex interaction has limited impact on exocytosis in PC12 cells. J Biol Chem 284: 21637–21646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen JM, Kaplan JM (2008) UNC-18 promotes both the anterograde trafficking and synaptic function of syntaxin. Mol Biol Cell 19: 3836–3846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misura KM, Scheller RH, Weis WI (2000) Three-dimensional structure of the neuronal-Sec1-syntaxin 1a complex. Nature 404: 355–362 [DOI] [PubMed] [Google Scholar]
- Naldini L, Blomer U, Gallay P, Ory D, Mulligan R, Gage FH, Verma IM, Trono D (1996) In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science (New York, NY 272: 263–267 [DOI] [PubMed] [Google Scholar]
- Peng R, Gallwitz D (2004) Multiple SNARE interactions of an SM protein: Sed5p/Sly1p binding is dispensable for transport. EMBO J 23: 3939–3949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pevsner J, Hsu SC, Braun JE, Calakos N, Ting AE, Bennett MK, Scheller RH (1994a) Specificity and regulation of a synaptic vesicle docking complex. Neuron 13: 353–361 [DOI] [PubMed] [Google Scholar]
- Pevsner J, Hsu SC, Scheller RH (1994b) n-Sec1: a neural-specific syntaxin-binding protein. Proc Natl Acad Sci USA 91: 1445–1449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieren M, Schmidt A, Mayer A (2010) The SM protein Vps33 and the t-SNARE H(abc) domain promote fusion pore opening. Nat Struct Mol Biol 17: 710–717 [DOI] [PubMed] [Google Scholar]
- Rathore SS, Bend EG, Yu H, Hammarlund M, Jorgensen EM, Shen J (2010) Syntaxin N-terminal peptide motif is an initiation factor for the assembly of the SNARE-Sec1/Munc18 membrane fusion complex. Proc Natl Acad Sci USA 107: 22399–22406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickman C, Medine CN, Bergmann A, Duncan RR (2007) Functionally and spatially distinct modes of munc18-syntaxin 1 interaction. J Biol Chem 282: 12097–12103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodkey TL, Liu S, Barry M, McNew JA (2008) Munc18a scaffolds SNARE assembly to promote membrane fusion. Mol Biol Cell 19: 5422–5434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz SK, Hjorth JJ, Joemai RM, Wijntjes R, Eijgenraam S, de Bruijn P, Georgiou C, de Jong AP, van Ooyen A, Verhage M, Cornelisse LN, Toonen RF, Veldkamp WJ (2011) Automated analysis of neuronal morphology, synapse number and synaptic recruitment. J Neurosci Methods 195: 185–193 [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Meyer AC, Neher E (1999) Released fraction and total size of a pool of immediately available transmitter quanta at a calyx synapse. Neuron 23: 399–409 [DOI] [PubMed] [Google Scholar]
- Schollmeier Y, Krause JM, Kreye S, Malsam J, Sollner T (2011) Resolving the function of distinct Munc18-1-SNARE interaction modes in a reconstituted membrane fusion assay. J Biol Chem 286: 30582–30590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott BL, Van Komen JS, Irshad H, Liu S, Wilson KA, McNew JA (2004) Sec1p directly stimulates SNARE-mediated membrane fusion in vitro. J Cell Biol 167: 75–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Rathore SS, Khandan L, Rothman JE (2010) SNARE bundle and syntaxin N-peptide constitute a minimal complement for Munc18-1 activation of membrane fusion. J Cell Biol 190: 55–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Tareste DC, Paumet F, Rothman JE, Melia TJ (2007) Selective activation of cognate SNAREpins by Sec1/Munc18 proteins. Cell 128: 183–195 [DOI] [PubMed] [Google Scholar]
- Smyth AM, Duncan RR, Rickman C (2010) Munc18-1 and syntaxin1: unraveling the interactions between the dynamic duo. Cellular Mol Neurobiol 30: 1309–1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC, Rothman JE (2009) Membrane fusion: grappling with SNARE and SM proteins. Science (New York, NY 323: 474–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Togneri J, Cheng YS, Munson M, Hughson FM, Carr CM (2006) Specific SNARE complex binding mode of the Sec1/Munc-18 protein, Sec1p. Proc Natl Acad Sci USA 103: 17730–17735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toonen RF, Kochubey O, de Wit H, Gulyas-Kovacs A, Konijnenburg B, Sorensen JB, Klingauf J, Verhage M (2006a) Dissecting docking and tethering of secretory vesicles at the target membrane. EMBO J 25: 3725–3737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toonen RF, Verhage M (2003) Vesicle trafficking: pleasure and pain from SM genes. Trends Cell Biol 13: 177–186 [DOI] [PubMed] [Google Scholar]
- Toonen RF, Verhage M (2007) Munc18-1 in secretion: lonely Munc joins SNARE team and takes control. Trends Neurosci 30: 564–572 [DOI] [PubMed] [Google Scholar]
- Toonen RF, Wierda K, Sons MS, de Wit H, Cornelisse LN, Brussaard A, Plomp JJ, Verhage M (2006b) Munc18-1 expression levels control synapse recovery by regulating readily releasable pool size. Proc Natl Acad Sci USA 103: 18332–18337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varoqueaux F, Sigler A, Rhee JS, Brose N, Enk C, Reim K, Rosenmund C (2002) Total arrest of spontaneous and evoked synaptic transmission but normal synaptogenesis in the absence of Munc13-mediated vesicle priming. Proc Natl Acad Sci USA 99: 9037–9042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhage M, Maia AS, Plomp JJ, Brussaard AB, Heeroma JH, Vermeer H, Toonen RF, Hammer RE, van den Berg TK, Missler M, Geuze HJ, Sudhof TC (2000) Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science (New York, NY 287: 864–869 [DOI] [PubMed] [Google Scholar]
- Verhage M, Sorensen JB (2008) Vesicle docking in regulated exocytosis. Traffic (Copenhagen, Denmark) 9: 1414–1424 [DOI] [PubMed] [Google Scholar]
- Voets T, Toonen RF, Brian EC, de Wit H, Moser T, Rettig J, Sudhof TC, Neher E, Verhage M (2001) Munc18-1 promotes large dense-core vesicle docking. Neuron 31: 581–591 [DOI] [PubMed] [Google Scholar]
- Walter AM, Groffen AJ, Sorensen JB, Verhage M (2011) Multiple Ca(2+) sensors in secretion: teammates, competitors or autocrats? Trends Neurosci 34: 487–497 [DOI] [PubMed] [Google Scholar]
- Weber-Boyvat M, Aro N, Chernov KG, Nyman T, Jantti J (2011) Sec1p and Mso1p C-terminal tails cooperate with the SNAREs and Sec4p in polarized exocytosis. Mol Biol Cell 22: 230–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weimer RM, Richmond JE, Davis WS, Hadwiger G, Nonet ML, Jorgensen EM (2003) Defects in synaptic vesicle docking in unc-18 mutants. Nat Neurosci 6: 1023–1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weninger K, Bowen ME, Choi UB, Chu S, Brunger AT (2008) Accessory proteins stabilize the acceptor complex for synaptobrevin, the 1:1 syntaxin/SNAP-25 complex. Structure 16: 308–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierda KD, Toonen RF, de Wit H, Brussaard AB, Verhage M (2007) Interdependence of PKC-dependent and PKC-independent pathways for presynaptic plasticity. Neuron 54: 275–290 [DOI] [PubMed] [Google Scholar]
- Williams AL, Ehm S, Jacobson NC, Xu D, Hay JC (2004) rsly1 binding to syntaxin 5 is required for endoplasmic reticulum-to-Golgi transport but does not promote SNARE motif accessibility. Mol Biol Cell 15: 162–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Seven AB, Su L, Jiang QX, Rizo J (2011) Membrane bridging and hemifusion by denaturated munc18. PLoS ONE 6: e22012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Su L, Rizo J (2010) Binding of Munc18-1 to synaptobrevin and to the SNARE four-helix bundle. Biochemistry 49: 1568–1576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi T, Dulubova I, Min SW, Chen X, Rizo J, Sudhof TC (2002) Sly1 binds to Golgi and ER syntaxins via a conserved N-terminal peptide motif. Dev Cell 2: 295–305 [DOI] [PubMed] [Google Scholar]
- Yang B, Steegmaier M, Gonzalez LC Jr., Scheller RH (2000) nSec1 binds a closed conformation of syntaxin1A. J Cell Biol 148: 247–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilly FE, Sorensen JB, Jahn R, Lang T (2006) Munc18-bound syntaxin readily forms SNARE complexes with synaptobrevin in native plasma membranes. PLoS Biol 4: e330. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.