

Abstract
Methods to induce antigen-specific immune responses in mice using insect cells infected with recombinant baculoviruses are described in this unit. Although this vaccine strategy has been used to generate both antibody and T cell responses, it has been more thoroughly characterized for the peptide-specific cytotoxic T cell responses. Nonspecific responses to the vaccine vehicle are controlled for by vaccinating with insect cells infected with baculoviruses encoding irrelevant antigens or no antigen. The baculovirus-infected insect cells alone are an effective immune adjuvant to elicit antigen-specific T cells. Overall, immune responses generated using this approach are similar to those generated by more conventional vaccine strategies.
Keywords: vaccine, immune responses, peptides, baculovirus, T cells, molecular cloning, insect cells
Unit 2.15. Vaccination of mice with baculovirus-infected insect cells expressing antigenic proteins
This unit describes how to induce antigen-specific immune responses in mice using insect cells infected with recombinant baculoviruses. This straightforward strategy stimulates peptide-MHC-specific T cell responses, which may be employed in basic immunology experiments. For example, these vaccines facilitate a comparison of cytotoxic T cell responses to peptides identified from peptide-MHC libraries (1). Although this unit describes induction of T cell responses using infected insect cells expressing specific peptide-MHC molecules, this vaccine also induces B cell responses and elicits antigen-specific antibody responses. To produce these vaccines, plasmids encoding peptide-MHC molecules are constructed (see Basic protocol 1), homologously recombined with linearized baculovirus DNA in insect cells (see Basic protocol 2), and resulting recombinant baculoviruses are cloned by limiting dilution so that uniform viruses are analyzed (see Basic protocol 3). The expression of the recombinant proteins and the DNA sequence are confirmed (see Basic Protocol 2 and Supporting Protocol 1). Finally, the insect cells are expanded, infected, and injected into mice (see Basic protocol 4). The ensuing peptide-specific T cell responses are evaluated. No additional adjuvants, other than the xenogenic insect cells and double-stranded DNA virus, are required to generate antigen specific responses. The responses are similar to those generated by more conventional vaccine strategies (see Commentary).
Strategic Planning
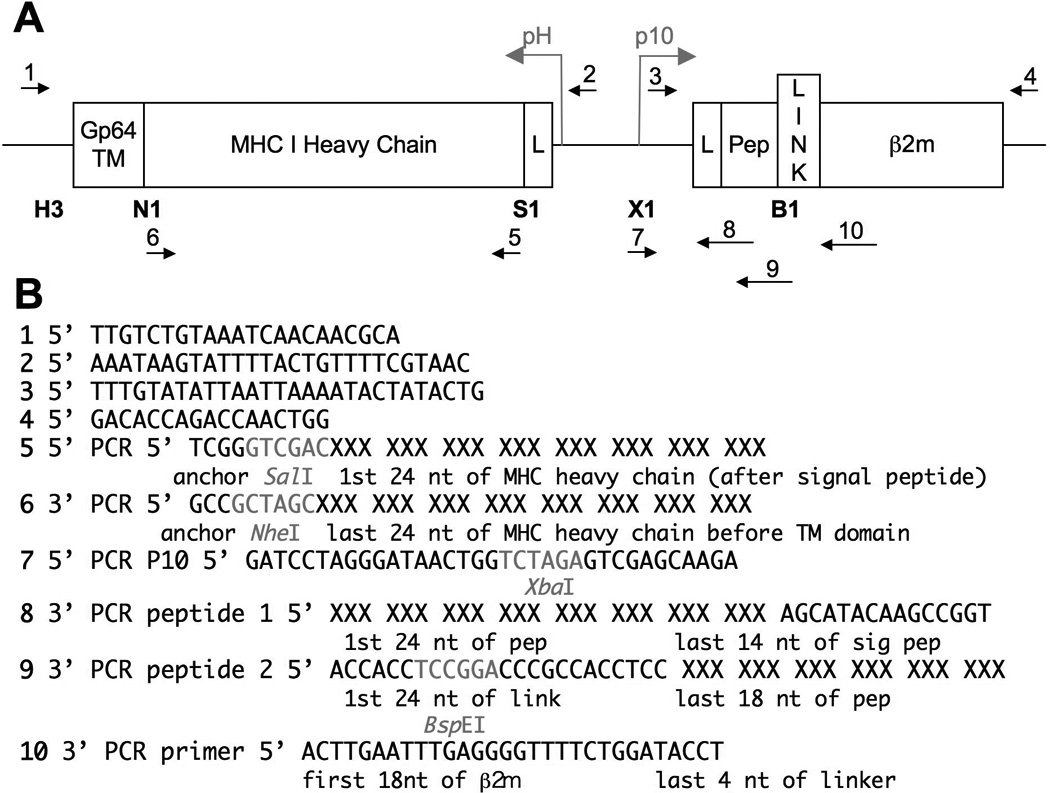
This unit describes the steps required to immunize against recombinant peptide-MHC molecules in baculovirus-infected insect cells (see Figure 2.15.1). However, any recombinant protein can be expressed from the strong baculovirus promoters p10 and pH that are active late in infection (see Basic Protocol 1 and Figure 2.15.2). Although the recombinant baculoviruses described here are designed to stimulate CTL responses, baculovirus-infected insect cell vaccines elicit specific antibody and T cell immune responses. Conveniently, this vaccine strategy eliminates the need to purify recombinant insect cell-produced proteins prior to injection and both soluble and membrane-bound recombinant proteins elicit specific immune responses.
Fig 1.

Fig 2.
The baculoviruses described here are safe to work with because they are attenuated and cannot replicate in mammalian cells, however they are lytic viruses and kill infected insect cells after several days in culture. Therefore, all pipets used for insect cell culture should be disposable and all glassware used to maintain the insect cells in culture should be acid-washed. These viruses are also light-sensitive and should be wrapped with aluminum foil and stored in the dark.
This vaccine strategy may be most convenient for those using baculovirus to produce proteins for other purposes. It is likely that other insect cell lines other than the ones described here (Sf9 and Hi5 cells) will have similar immunological results. A more complete description of the variety of baculoviruses, insect cell culture systems, and plasmid vectors used to produce recombinant proteins in insect cells can be found in the Current Protocols of Molecular Biology, Unit 16.
Basic protocol 1: Construction of baculovirus expression vector encoding recombinant peptide-MHC molecules or antigenic protein
This protocol describes the molecular cloning steps required to construct the baculovirus expression vector that will be homologously recombined with baculovirus DNA (see Basic Protocol 2). More details for success in molecular cloning may be found in UNIT 10 and in the instructions found on product inserts with the molecular reagents purchased.
Materials
cDNA encoding MHC class I molecule of interest
Modified pBacp10pH vector encoding Ldtm-AH1β2m (available from the authors upon request)
Specific oligonucleotides to amplify MHC molecule and peptide (see Figure 2.15.2 for sequence suggestions, Integrated DNA Technologies)
SalI, NheI, XbaI, BspEI restriction enzymes and buffers (New England Biolabs)
5× T4 DNA ligase buffer (Invitrogen)
1 Weiss unit/µl T4 DNA ligase (Invitrogen)
Competent cells, DH5α (homemade or purchased)
LB medium (UNIT 10.3) containing 50 µg/ml ampicillin or carbenicillen
LB plates (UNIT 10.19) containing 50 µg/ml ampicillin or carbenicillen
PCR purification column (S300, GE Healthcare)
Additional reagents and equipment for PCR amplification (UNIT 10.20), agarose gel electrophoresis (UNIT 10.4), restriction enzyme digestion (UNIT 10.8), DNA gel purification and quantitation with ethidium bromide (UNIT 10.5), bacterial transformation by heat shock (UNIT 10.15), isolation of plasmid DNA (miniprep UNIT 10.3), counting cells with trypan blue exclusion (APPENDIX 3B).
Insert restriction enzyme sites into cDNA encoding MHC molecule
-
1.In a 100-µl reaction volume, PCR amplify the cDNA encoding the MHC class I molecule of interest using oligonucleotides that harbor the desired restriction enzyme sites. Use 5′ primers containing a SalI site and 3′ primers containing an NheI site in frame with the end of the α3 domain of the MHC molecule.Specific examples of oligonucleotides used for PCR can be seen in Figure 2.15.2. The reading frame of the restriction sites are shown. A detailed description of PCR reactions can be found in UNIT 10.20, which also discusses variables that are relevant for obtaining optimal yields of DNA in PCR-mediated amplifications.Check that the sequence of the MHC molecule of interest does not harbor Sal1 or Nhe1 sites. If so, other restriction enzyme sites with compatible cohesive ends should be used.
-
2.
Visualize 10 µl of the reaction on an ethidium bromide–stained agarose gel to verify amplification (UNIT 10.4).
-
3.
Purify the amplified MHC class I fragment by column purification.
The details of the instructions for column purification vary by vendor. Typically, columns are first spun to remove the buffer and compact the slurry. Second, the PCR reaction is spun through the slurry to remove unincorporated nucleotides and salts from the buffer.
Insert the cDNA into Ldtm-AH1β2m vector
-
4.
Digest the PCR-generated MHC class I gene with SalI and NheI (UNIT 10.8), resolve on an agarose gel, and isolate the desired fragment from the gel by electroelution (UNIT 10.5) or using a commercially available gel extraction kit such as the Qiagen Gel Extraction kit.
Other commercially available kits such as the Bio101 gel extraction kit also work. However, kits that use glassmilk or bead isolation are more likely to shear large DNA.
-
5.Digest Ldtm-AH1β2m plasmid with SalI and NheI and purify the 9-kb vector fragment as in step 4.Ldtm-AH1β2m digested with MluI and XhoI will yield two fragments: a 9-kb fragment encoding the vector, and a 1-kb fragment encoding the H-2Ld sequence insert.
-
6.
Run digested purified PCR fragment and digested purified Ldtm-AH1β2m vector on an agarose gel to quantitate DNA (UNIT 10.5).
-
7.
Set up the following ligation reaction with the purified fragments in a total volume of 15 µl and incubate overnight at 14°C.
50 ng digested pBacp10pH vector
50 ng isolated MHC fragment
3 µl 5× T4 DNA ligation buffer containing 5 mM ATP
1 µl 1 U/µl T4 DNA ligase.
Adjust the volume to 15 µl with water
The insert-to-vector molar ratio should be ~10 to 1.
-
8.
Transform competent E. coli by heat shock using 1 to 2 µl ligation mixture as described in the manufacturer’s instructions.
-
9.
Spread transformed bacteria onto LB plates containing 50 µg/ml ampicillin or carbenicillen. Incubate overnight at 37°C.
-
10.
Pick colonies, transfer each to 5 ml LB medium containing 50 µg/ml ampicillin or carbenicillen, and grow overnight at 37°C with shaking.
-
11.
Isolate plasmid DNA by alkaline lysis miniprep (UNIT 10.3).
Commerically available miniprep kits also work. Purification by CsCl gradient is not required.
-
12.Screen for the presence of desired insert by restriction enzyme mapping and confirm by sequencing (see sequencing primers 1 and 2 in Figure 2.15.2).Unless previously established in house, DNA sequencing can be performed in a core facility or commercially.
Sub-clone antigenic peptide into MHCtm-AH1β2m vector
-
13.
Similar to steps 1–3, in a 100-µl reaction volume PCR amplify the Ldtm-AH1β2m vector using the #7 5’ PCR p10 primer and the #8 3’ PCR peptide 1 primer (see Figure 2.15.2).
-
14.
Gel purify the 378 base-pair fragment (if the peptide encodes a nonomer) as in step 4.
-
15.In a 100-µl reaction volume PCR amplify the first fragment from step 14 using the #7 5’ p10 primer and #9 3’ peptide primer 2 (see Figure 2.15.2).Use 10 ng of fragment from step 14 as the template in the PCR reaction.
-
16.
Column purify the 400 base-pair fragment as in step 4.
-
17.
Digest the PCR-generated fragment with XbaI and BspEI (UNIT 10.8), resolve on an agarose gel, and isolate the desired fragment as in step 4.
-
18.
Digest MHCtm-AH1β2m plasmid from step 12 with XbaI and BspEI and purify the 10-kb vector fragment as in steps 4 and 5.
-
19.
Run digested purified PCR fragment and digested purified MHCtm-AH1β2m vector on an agarose gel to quantitate DNA (UNIT 10.5) as in step 6 above.
-
20.
Ligate, transform, pick colonies, screen for inserts, and sequence as in steps 7–12 above.
-
21.Prepare the DNA construction using the Qiagen Midiprep kit (or similar product) according to the manufacturer’s protocol. 3 µg of a 0.5–1 µg/µl stock solution is required in the next protocol.The alkaline lysis miniprep protocol does not yield clean enough DNA to produce baculovirus in the following protocol, although cesium chloride purified DNA (Unit 10.3) does.
Basic protocol 2: Transfection of the peptide-MHC (MHCtm-pepβ2m) construction into Sf9 insect cells and recombination of plasmid DNA with baculovirus DNA
This protocol details cotransfection of the plasmid DNA construction from Basic Protocol 1 with purchased linearized DNA. Homologous recombination between the plasmid and the viral DNA restores the virus and the circularized DNA results in productive infectious baculovirus particles that encodes the antigenic protein of interest. An alternative to this procotol is to use a commercially available homologous recombination kit.
Materials
Plasmid encoding MHCtm-pepβ2m from Basic Protocol 1
Sf9 insect cells between 0.5–2×106 cells/ml (see Supporting Protocol 2)
Supplemented Insect Cell Medium (see Supporting Protocol 2 and Reagents and Solutions)
Unsupplemented Grace’s Insect Cell Medium (Invitrogen)
Linearized Sapphire™ Baculovirus DNA (Orbigen)
Transfection Buffers A and B (see Reagents and Solutions)
6-well tissue culture dishes
0.2 µm syringe filter
FACS buffer (see Reagents and Solutions)
Monoclonal antibodies specific for MHC or β2m
Co-transfect insect cells with the MHCtm-pepβ2m plasmid DNA and linearized baculovirus DNA
-
1.Transfer 2×106 cells Sf9 insect cells into 3 wells each of a 6-well dish. Adjust the volume to 4 ml with Supplemented Insect Cell Medium. Allow the cells to adhere to the plate for 30 min to 1 h at room temperature.Well 1 is for the homologous recombination reaction. This well will yield the recombinant baculovirus of interest.Well 2 is a transfection control, containing the Transfection Buffers and linearized baculovirus DNA. Well 3 is also a transfection control, containing only the Transfection Buffers. Wells 2 and 3 will not yield recombinant baculovirus and are used to help distinguish infected from uninfected wells in step 11.
-
2.
Remove the medium using a pipet, being careful not to dislodge the cells.
-
3.
Rinse the cells 1× with Unsupplemented Grace’s Insect Cell Medium by tilting the plate and carefully applying 2 ml along the edge of the well. Gently swirl the plate.
-
4.In 1.5 ml microcentrifuge tubes corresponding to wells 1–3 from step 1, combine 500 µl of Transfection Buffer B, 0.5 µg of linearized Baculovirus DNA, and 3 µg of plasmid DNA (see Basic Protocol 1). In a second tube, combine 500 µl of Transfection Buffer B with 0.5 µg of linearized Baculovirus DNA. In the third tube, add 500 µl of Transfection Buffer B.The linearized baculovirus DNA is very large and prone to shearing. Handle the DNA with care and do not pipet up and down or vortex to mix.
-
5.
Remove the Grace’s Unsupplemented Medium from the insect cells in the 6-well dish using a pipet.
-
6.
Add 500 µl of Transfection Buffer A dropwise to each well while gently swirling the plate.
-
7.Add 500 µl of the solutions containing Transfection Buffer B from step 4 dropwise to the respective wells while gently swirling the plate.The buffer may turn white after adding the Transfection Buffer B, but should not be opaque (see Critical Parameters and Troubleshooting).
-
8.
Place the plate in a plastic baggie and incubate the insect cells for 4 hours at 27°C.
-
9.
Remove the transfection buffers with a pipet while tilting the plate. Carefully rinse the cells as in step 3 with Unsupplemented Grace’s Insect Cell Medium.
-
10.
Add 5 ml of Supplemented Insect Cell Medium and place the plate in a plastic baggie to prevent evaporation. Incubate the cells at 27°C for 7–10 days.
Harvest the co-transfection and determine if the baculovirus and plasmid DNA recombined
-
11.7–10 days after the transfection, observe the insect cells using a light microscope with a 4× or 10× objective.The second and third wells, which contain the Transfection Buffers and linearized Baculovirus DNA, should be healthy and adherent. The cells in the first well, which were co-transfected with the plasmid DNA and linearized Baculovirus DNA, should be non-adherent and more granular in appearance, indicating viral infection.And alternative to visualizing dead insect cells using a microscope is to count the insect cells using Trypan Blue exclusion (APPENDIX 3B). The two control wells should have fewer Trypan Blue positive cells than the well that was incubated with plasmid DNA.
-
12.Harvest the supernatant from each well and transfer to 15 ml conical tube. Spin the tube for 10 min at 500 × g to remove the insect cells.If the transfection was successful the supernatant should contain infectious baculovirus particles.
-
13.Remove the cellular debris using a 0.2 µm syringe filter and transfer to a new 15 ml conical tube. Store the virus in the dark at 4°C.This supernatant will be referred to as the Uncloned Viral Stock in the remaining protocols.
Confirm the baculovirus encodes the peptide-MHC
The following steps are to confirm that the baculoviruses encode the proteins of interest by flow cytometric analysis. For the further details on how to achieve successful flow cytometry, see UNIT 5. The samples suggested here are stained with 1) the primary MHC antibody and secondary antibody, 2) the secondary antibody only, 3) or no antibody as an unstained control.
-
14.Plate 2×106 Sf9 insect cells in 4 ml in a 6-well plate as in step 1. Add 10 µl of the Uncloned Viral Stock from step 13 to the new plate of insect cells. Incubate the cells for 3 days at 27°C.14b: Plate the Sf9 insect cells and allow them to adhere for 30 min to 1 h. Rinse the cells as in steps 2–3 with Unsupplemented Grace’s Insect Medium. Add 2 ml of Grace’s Insect Medium and 10 µl of the Uncloned Viral Stock and incubate for 1 h at room temperature. Remove the supernatant and wash the insect cells as in steps 2–3. Add 4 ml of Supplemented Insect Cell Medium and incubate for 3 days at 27°C.
-
15.
Harvest the infected insect cells by pipeting up and down with a 1 ml pipet to remove the adherent cells. Transfer the cells and medium to a 15 ml conical tube and spin at 500 × g for 5 min.
-
16.
Remove the supernatant and wash the infected insect cells with 10 ml of Unsupplemented Insect Cell Medium. Spin at 500 × g for 5 min.
-
17.Remove the supernatant and resuspend cells in 2 ml of FACS buffer. Transfer 100 µl into 3 wells of a 96-well plate and spin at 500 × g for 5 min.The first well will be incubated with both the primary MHC antibody and secondary antibody, the second well will be incubated with only the secondary antibody, and the third well will be unstained.
-
18.Resuspend the cell pellet in the first well with 50 µl of FACS buffer containing 1 µg of a monoclonal antibody specific for the MHC or β2m. Resuspend the second and third wells in 50 µl of FACS buffer. Incubate for 1 h at 4°C.1 µg may be excess for some antibodies. The concentration of antibody required for optimal staining intensities can be determined by titrating both the primary and secondary antibodies (see UNIT 5).
-
19.
Wash the cells with 150 µl of FACS buffer and spin at 500 × g and repeat.
-
20.
Resuspend the cell pellets in the first and second wells with 50 µl FACS buffer containing 1 µg of a fluorescently-labeled secondary antibody specific for the Ig-class of the primary MHC antibody or β2m antibody. Resuspend the third well in 50 µl of FACS buffer. Incubate for 30 min at 4°C.
-
21.
Wash the cells 2× as in step 19.
-
22.Resuspend the cells in 150 µl FACS buffer and visualize the cells by flow cytometry.If the transfection was successful, the peptide-MHC complexes should be easily detected by flow cytometry as described above. If peptide-MHC complexes are not detected, it may be informative to stain the infected insect cells with an antibody specific for a baculovirus envelope protein, gp64 (clone AcV1, eBioscience). If the cells are positive for gp64, but negative for the peptide-MHC, there may be a problem with the plasmid construction.
Supporting Protocol 1: Conformation of the antigenic peptide sequence encoded by the baculovirus
The following protocol describes how to confirm that the baculoviruses encode the predicted sequences. The baculoviruses are propagated, DNA is isolated, and the DNA fragment harboring the sequence of interest is amplified by PCR and sequenced. Particular attention should be paid to the sequence of the antigenic peptide, especially when multiple similar peptides are being compared.
Materials
24-well culture dishes
Sf9 insect cells
Supplemented Insect Cell Medium (see Supporting Protocol 2 and Reagents and Solutions)
Uncloned Viral Stock (from step 13 of Basic Protocol 2)
Extraction Buffer (see Reagents and Solutions)
-
10 mg/ml Proteinase K (Promega, prepared according to the manufacturer’s instructions)
56°C water bath
PCR tubes
Taq DNA polymerase and reaction buffer (Invitrogen)
dNTPs (Invitrogen)
Oligonucleotides #3 and 4 (see Figure 2.15.2)
PCR purification column (S300, GE Healthcare)
Microcentrifuge
Infect insect with the transfection medium
-
1.
Transfer 3×105 Sf9 insect cells into a 24-well plate. Adjust the volume to 1 ml with Supplemented Insect Cell Medium.
-
2.
Add 20 µl of filtered Uncloned Viral Stock from step 13 of Basic Protocol 2.
-
3.Place the plate in a plastic baggie and incubate for 2 days at 27°C.At 2 days post-infection DNA production is favored relative to DNA degredation and protein production.
Harvest the infected insect cells and extract DNA
-
4.
Transfer the infected insect cells from step 3 into a 1.5 ml microcentrifuge tube and spin at 500 × g in a microcentrifuge (under 4,000 rpm) for 5 min.
-
5.
Resuspend the cell pellet in 50 µl of extraction buffer and vortex on high for 30 s.
-
6.
Add 1 µl of Proteinase K and mix.
-
7.Incubate the mixture for 2 h at 56°C and then for 20 m at 95°C to inactivate the Proteinase K.Samples can be frozen at this point for future analysis.
PCR amplify the peptide DNA for sequencing
-
8.
Set up a PCR reaction containing the following:
1.5 µl of template DNA from step 7 (approximately 20 ng)
1× Taq DNA polymerase buffer
1.25 units Taq DNA polymerase
0.2 mM each dNTP
1 µM 5’ primer (#7)
1 µM 3’ primer (#10)
See Figure 2.15.2 for primer sequences.
-
9.
Amplify the reactions in a PCR thermocycler using the following conditions:
95°C for 1 minute
52°C for 1 minute
72°C for 1 minute
Repeat for 35–40 cycles.
-
10.
Remove the nucleotides and salts from the PCR reaction using a PCR purification column according to the manufacturer’s instructions.
-
11.
Verify PCR amplification on an agarose gel as in step 12 of Basic Protocol 1. Submit the sample for sequencing using the appropriate sequencing primer (#3, see Figure 2.15.1).
Supporting protocol 2: Growing insect cells
The following protocols describe how to expand and maintain insect cells. As with all cell culture, it is important to work in a clean environment and to ensure that all culture media and supplies are sterile. As in the previous protocols in this unit, the optimal temperature for maintenance of insect cells is 27°C in the absence of additional CO2.
The Supplemented Insect Cell Medium is described first. Choosing the Fetal Bovine Serum for the cells is the most important step in making the medium. An alternative is Expression System’s ESF921 Insect Cell Culture Medium, Protein-free 96–001 (www.expressionsystems.com), which avoids using fetal bovine serum. Thawing and freezing insect cells is a similar process as with other standard cells lines, although maintaining consistent growth of insect cells differs. Once insect cells are consistently dividing, they are transferred from a flat bottom flask to a spinner flask in which large quantities can be harvested for infection and vaccination.
Materials
Grace’s Supplemented Insect Cell Medium (Invitrogen)
Fetal Bovine Serum (FBS)
Antibiotic/Antimycotic (100×, Invitrogen)
Pluronic F-68, Invitrogen (10%, Invitrogen)
0.2 µM sterile filter devices
Sf9 or Hi5 insect cells (Invitrogen)
Acid-washed 1 L spinner bottle (Bellco, see Supporting Protocol 3 for acid washing procedure)
Freezing medium (see Reagents and Solutions)
75 mm2 tissue culture flask
175 mm2 tissue culture flask
15 ml conical tube
Table-top centrifuge
Magnetic stirrer (Bellco, 4-position, 115v-500 ml)
Prepare 500 ml of Supplemented Insect Cell Medium
-
1.In a biosafety cabinet, combine 500 ml of Grace’s Supplemented insect cell medium with 50 ml of FBS, 5 ml of Pluronic F-68, and 5 ml of Antibiotic/Antimycotic.The brand, type, and lot of serum is very important. Even variations between lots of the same type of serum can result in low confluency, low infectivity, and low productivity. Test several types and lots of serum by growing the cells in small flasks and counting overtime. Choose the serum they grow the fastest in, and perform several test infections to ensure the insect cells produce protein in the presence of that serum.Store the Antibiotic/Antimycotic at −20°C in aliquots.
-
2.
Sterile filter the medium and store at 4°C.
Starting insect cells from a frozen vial
-
3.
Thaw cells rapidly by shaking in a 37°C waterbath and immediately dilute the cells in a 15 ml conical tube with 10 ml of Supplemented Insect Cell Medium. Spin the cells at 500 × g for 5 min.
-
4.
Resuspend the cell pellet in 10 ml of Supplemented Insect Cell Medium and repeat.
-
5.Resuspend the cell pellet in 15 ml of Supplemented Insect Cell Medium and transfer to a 75mm2 flask.This volume of medium and size of flask is appropriate for 1×107 cells, adjust for different cell numbers.
-
6.Incubate the cells at 27°C for 2–3 days, or until the cells are confluent on the bottom of the flask. The cells are semi-adherent and will attach to the bottom of the flask.Depending on the condition of the insect cells when they were frozen, they may take up to two weeks to become fully confluent. Change the medium in the flask weekly until they are confluent.
-
7.
Remove the cells from the bottom of the flask by banging it several times with the palm of your hand. Transfer all 15 ml to a larger 175 mm2 flask and add an additional 30 ml of Supplemented Insect Cell Medium.
-
8.
Incubate at 27°C until the cells are confluent on the bottom of the flask.
Maintaining insect cells in a 1 L spinner flask
-
9.
Remove the cells from the 175 mm2 flask as in step 5 and transfer to an acid-washed 1 L Bellco spinner flask (See Supporting Protocol 3 for acid washing).
-
10.Add approximately 300 ml of Supplemented Insect Cell medium. Add enough medium to cover the spinner bar.An alternative to a Bellco Spinner flask is to use an acid-washed 1 L bottle with a medium-sized magnetic spinner bar. These bottles only support a 200 ml culture volume.
-
11.Incubate the flask at 27°C using a medium spinner speed.For the 4-position Bellco magnetic stirrer, a setting of 3–4 is appropriate.
-
12.Split the cells every 3–4 days, maintaining the cells between 2×105 and 2×106 cells/ml.Do not use the cells for experiments or infections until they are growing consistently. It can take several weeks to two months for them to grow consistently from a frozen vial. It’s best to start fresh cultures from frozen aliquots approximately every 6 months or less. The cells become less efficient at protein production and grow inconsistently over time.
Freezing insect cells
-
13.
Transfer 1×107 insect cells to a 15 ml conical tube and spin at 500 × g for 5 min.
-
14.
Remove the medium and resuspend the cell pellet in 1 ml freezing medium.
-
15.Transfer the cells to a 1.5 ml cryovial and incubate at −80°C for 12 to 24 h in an isopropanol chamber.The temperature of the isopropanol chamber or Mr. Frosty decreases slowly, about one degree per min, to maintain the integrity of the cell membranes.
-
16.
Transfer the cryovial to a liquid nitrogen tank for long-term storage.
Supporting protocol 3: Acid-washing glassware
Any glassware that contacts insect cells, insect cell media, or baculovirus should be acid-washed. Acid-washing the glassware used to grow the insect cells is important since baculoviruses are lytic and kill the insect cell cultures. Acid washing also ensures that the baculoviruses will not cross-contaminate cultures. Wear protective rubber gloves and a lab coat or apron throughout this procedure to protect yourself and the glassware.
Materials
Large aluminum tub or sink
diH2O
Bleach (6 % sodium hypochlorite)
7× cleaning solution (Fisher Scientific)
Anhydrous HCl
-
1.
Rinse the glassware with water to remove residual cells, protein, viruses, and medium. Fill with diluted bleach (1:10).
-
2.After 10 min, rinse the dishes once with diH2O and disassemble the glassware.The stir bar shafts are very fragile, so be careful when remove the stir bars from the spinner flasks.
-
3.
Fill the sink and dishes with diH2O and dilute 7× soap approximately 1:500 (approximately 100 ml per 10 1L Bellco spinner flasks).
-
4.After 2 h (or overnight), drain the sink and rinse the dishes 6× with diH2ORemember to rinse the lids and stir bars.
-
5.
Fill the sink and dishes with diH2O and dilute HCl approximately 1:150 (approximately 300 ml of HCl per 10 1L Bellco spinner flasks).
-
6.After 2 h (or overnight), drain the sink and rinse the dishes 6× with diH2O.Be sure to rinse the outsides, edges, and spouts of the dishes, stir bars, and lids. Any residual detergent or acid will kill the insect cells.
-
7.
Dry on clean paper towels overnight.
-
8.
Reassemble the glassware and cover the lids with aluminum foil. Autoclave the glassware, being sure to vent caps.
Basic protocol 3: Cloning baculoviruses that express peptide-MHC by limiting dilution
Cloning the baculoviruses by limiting dilution eliminates viruses that incorporated an incomplete plasmid construction during homologous recombination. These infectious particles may not express the protein of interest and may decrease the protein yield.
Materials
Filtered Uncloned Viral Stock (from step 13 of Basic Protocol 2)
500 ml acid-washed bottle (see Supporting Protocol 3 for acid washing procedure)
96-well flat-bottom tissue culture plates
15 ml conical tubes
Hi5 insect cells (Invitrogen)
Supplemented Insect Cell Medium (see Supporting Protocol 2 and Reagents and Solutions)
Serially dilute the baculovirus in the Uncloned Viral Stock
-
1.Transfer 4×106 Hi5 insect cells into a 50 ml conical tube. Adjust the cell concentration to 1.1×106 cells/ml with Supplemented Insect Cell Medium and mix.Typically, Hi5 insect cells are used for large-scale protein production and Sf9 insect cells are used for viral production. Using Hi5 insect cells is suggested here because it is much easier to visually distinguish infected and uninfected cells.
-
2.
Transfer 12 ml of Supplemented Insect Cell Medium to a 15 ml conical tube (label 1×103) and 10.8 ml to 6 additional 15 ml conical tubes (label 1×104 – 1×109).
-
3.Add 12 µl of the Uncloned Viral Stock from step 13 of Basic Protocol 2 to the 1×103 tube containing 12 ml of Supplemented Insect Cell Medium and mix well.This is a 1:1000 dilution of the Uncloned Viral Stock.
-
4.
Remove 1.2 ml of medium from the 1×103 tube and transfer to the 1×104 tube and mix well.
-
5.
Continue serial diluting as in step 4, until the 1×109 conical tube.
-
6.Transfer 200 µl of diluted Hi5 insect cells from step 1 to each virus dilution tube.Each 15 ml conical tube containing the baculovirus dilutions should have 2.2×105 Hi5 insect cells at a final concentration of 2×104 cells/ml.
-
7.Using a multichannel pipet, transfer 100 µl of each dilution into ½ of a 96-well plate.6 columns of a plate will be used for each dilution of the baculovirus.
-
8.Transfer 2×105 Hi5 insect cells to a 15 ml conical tube and adjust the volume to 10 ml with Supplemented Insect Cell Medium. As a control for uninfected Hi5 insect cells, transfer 100 µl of the diluted cells from this tube into . of a 96-well plate.The final concentration of Hi5 insect cells in this control tube is 2×104 cells/ml. This is a control containing no baculovirus to help distinguish infected from uninfected cells in the next section of the protocol.
-
9.
To reduce evaporation, place the plates inside a plastic baggie. Incubate the cells at 27°C for 7–10 days.
Identify infected wells and choose a baculoviral clone
-
10.Visualize the Hi5 insect cells under a light microscope with a 4× or 10× objective. Begin by looking at the cells in the control wells containing no baculovirus.These cells should be healthy, adherent, and in contact with each other. Most of the cells will also have a dendrite-like appearance. None of these cells should be infected.
-
11.Visualize the infected cells from the 1×103 dilution of baculovirus as in step 10.These cells should be smaller, round, non-adherent, not in contact with each other, and much fewer in number. All of these cells should be infected.
-
12.Identify the infected wells on the plates from the 1×109, 1×108, and 1×107 dilutions of baculovirus.Usually all of the wells in the 1×103 – 1×106 dilutions are infected, so it may be unnecessary to count the infected wells on these plates.A clone should come from a dilution in which less than 30% of the wells are infected. If greater than 30% of the wells are infected on the 1×109 plate, the virus will need to be diluted further (this is rare). Repeat steps 1–9, but dilute the virus 3 additional times, making the last plate a 1×1012 dilution.
-
13.
Choose 6–8 baculovirus clones for further testing. Its best to choose clones from the lowest dilution in which there are infected wells.
-
14.Transfer 2×106 Sf9 insect cells to a 6-well dish and adjust the volume to 4 ml with Supplemented Insect Cell Medium.Sf9 insect cells are used here for the generation of a viral stock in the next steps.
-
15.
Add 10 µl of supernatant from the clone wells on the 96-well plate that were chosen for further analysis in step 13 and incubate for 7 days at 27°C.
-
16.
Seal the 96-well plates with plastic wrap or parafilm and store in the dark at 4°C.
-
17.Harvest the baculoviral supernatant from step 15 by transferring the medium to a 15 ml conical tube and spinning at 500 × g for 5 min to remove cells and debris. Filter the supernatant using a 0.2 µm syringe filter device.This is a small stock of the cloned baculoviral supernatant. This will yield enough virus to test each clone for protein production and DNA sequence in the subsequent steps.
-
18.
Test the baculovirus clones for protein production by following steps 14–23 of Basic Protocol 2. Test the baculovirus clones for the appropriate DNA sequence by following the steps in Supporting Protocol 1.
Growing up a viral stock of the cloned baculovirus for experimentation
-
19.
Transfer 3×107 Sf9 insect cells into a 175 cm2 culture flask and adjust the volume to 50 ml with Supplemented Insect Cell Medium.
-
20.
Choose the viral clone that produces the appropriate peptide-MHC protein and encodes the correct DNA sequence (from step 18). Add 150 µl of the baculovirus supernatant from step 17 and mix gently.
-
21.
Incubate at 27°C for 7 to 10 days, or until most of the cells are Trypan Blue positive.
-
22.
Harvest the baculoviral supernatant as in step 17.
-
23.
Store the cloned viral stocks at 4°C in the dark.
Titer the baculovirus stock
-
24.
Serially dilute the cloned baculoviral stock as in steps 1–6.
-
25.
Using a multichannel pipet, transfer 100 µl of the 1×106 – 1×109 dilutions into a 96-well plate. Use a full plate for each dilution of the baculovirus.
-
26.As a control, transfer uninfected Hi5 insect cells to a 96-well plate as in step 8.This uninfected control is to help distinguish infected from uninfected cells in the next section of the protocol.
-
27.
Incubate the cells at 27°C for 7–10 days. To avoid evaporation, place the plates inside a plastic baggie.
-
28.
Visualize and count the infected wells as in steps 10–12.
-
29.Using a dilution of virus in which less than 30% of the wells are infected, calculate the approximate viral titer:(# of wells infected) × (dilution of the plate) × 0.1 ml/well = Units/ml.Most viral stocks grown up in 50 ml with 3×107 cells are around 2–6×108 Units/ml.
Basic protocol 4: Vaccination of mice with baculovirus-infected insect cells
In the following steps, mice are vaccinated using Sf9 cells infected with the baculovirus produced in the previous Basic Protocols. Institutional Animal Care and Use Committee protocols should be prepared according to institutional regulations. Using the injection regiment of two injections one week apart, the antigen-specific T cell response peaks approximately on day 14. At this time the responding T cells and other parts of the adaptive immune response can be analyzed. See UNIT 1 for details on handling and injecting experimental mice.
Materials
3×107 Sf9 insect cells per one vaccination of 5 mice
Supplemented Insect Cell Medium (see Supporting Protocol 2 and Reagents and Solutions)
Cloned Baculovirus Stock (from step 23 of Basic Protocol 3)
175 cm2 tissue culture flasks
1× Hank’s Balanced Salt Solution (HBSS, Cellgro)
1 ml syringes
27 gauge needles (or similar)
Inbred experimental mice
Infecting the Sf9s for vaccination
-
1.Transfer 3×107 cells to a 175 cm2 culture flask. Adjust the volume to 50 ml with Supplemented Insect Cell Medium.This is enough cells to vaccinate 5 mice, with a little extra for the void volume of the syringe. Infect multiple flasks to obtain enough cells to vaccinate more mice. For fewer mice, reduce the number of cells, the size of flask, and the total volume proportionally.
-
2.Add 2 units of baculovirus stock per cell (see steps 24–29 of Basic Protocol 3 to titer the virus stock).Usually 2 units/cell of baculovirus stock is between 100–200 µl for one flask of Sf9s. Using more or less virus will affect the antigen dose in the vaccine.
-
3.
Incubate the cells for 3 days at 27°C.
Prepare the infected insect cell vaccine
-
4.
Dislodge the infected cells by banging the side of the flask several times with the palm of your hand. Transfer the cells and medium to a 50 ml conical tube.
-
5.
Spin the cells at 500 × g for 5 min and remove the supernatant. Resuspend the cell pellet in 30 ml of sterile HBSS.
-
6.
Repeat step 5 two more times.
-
7.Resuspend the pellet in 1.2 ml of sterile HBSS (2.5×107 cells/ml) and transfer to a 1.5 ml microcentrifuge tube.There is very little cell loss during the preparation of the vaccine. If you count the cells, you’ll notice that some of the cells are dead due to infection. The vaccine includes the live and dead cells, the baculovirus still attached to the cells, and any cell debris.
-
8.
Mix the microcentrifuge tube well and transfer to a 1 ml syringe.
-
9.Carefully inject the infected insect cells intraperitoneally (see UNIT 1.6).Intraperitoneal injections are the easiest injection route and give the most consistent T cell responses. Intravenous injections result in similar responses. Subcutaneous injections result in poorer T cell responses.
-
10.
Mix the microcentrifuge tube again before reloading the syringe.
-
11.
Prepare Sf9s 4 days after the initial injection as in steps 1–3.
-
12.
Harvest the infected insect cells and prepare the vaccine 7 days after the initial injection as in steps 4–7.
-
13.Inject the second vaccine 7 days after the initial injection as in steps 8–10.Tetramer-positive CD8 T cells are not detectable in most mice prior to the second injection. They are detectable in the blood on day 10 (3 days after the second injection), but peak at day 14 (7 days after the second injection). They begin to contract by day 17 (10 days after the second injection).
Support protocol 4: Quantitation of peptide-MHC expression in baculovirus infected insect cells
This support protocol is useful for several applications: (1) to determine the concentration of peptide-MHC molecules generated in the Basic Protocols 2 and 3 to aid in selecting the best producing clones, and (2) to determine the amount of antigen in each vaccine dose. This method is important to ensure that a consistent amount of antigen is being delivered by different preparations of infected insect cells, and that there is little or no variation in the concentration of different peptides. This method involves infecting the insect cells in the same manner as for the vaccine (see Basic Protocol 4), extracting the membrane-associated peptide-MHC compexes in a lysis buffer containing detergent, and performing an ELISA reaction using MHC-specific antibodies. For the sandwich ELISA to be performed, two different MHC-specific antibodies are needed. Alternatives to ELISA quantification include Western Blot analysis (see UNIT 8.10) or Immunoprecipitation (see UNIT 8.3), which require only one MHC-specific antibody. However, these assays are less quantitative.
Materials
Sf9 insect cells
-
175 cm2 tissue culture flask
1× Hank’s Balanced Salt Solution (HBSS, Cellgro)
Insect Cell Lysis Buffer (see Reagents and Solutions)
Anti-MHC class I monoclonal antibody (30.7.5S for H-2Ld, ATCC) or anti-β2m monoclonal antibody
Secondary anti–MHC class I monoclonal antibody conjugated to biotin (e.g., 28.18.4S for H-2Ld protein; ATCC)
Streptavidin HRP conjugate (Pierce)
Blocking buffer: 1% (v/v) FBS in carbonate buffer
Wash buffer: PBS (APPENDIX 2A) containing 1% (v/v) FBS and 0.5% (v/v) Tween 20
Diluent buffer: PBS (APPENDIX 2A) containing 1% (v/v) FBS
Standard: previously generated peptide-MHC
Final wash buffer: PBS (APPENDIX 2A) containing 0.5% (v/v) Tween 20
TMB One-Step Substrate System (Promega)
0.18 M Sulfuric Acid
96-well ELISA plates: high-protein-binding, 1/2-area plates for enzyme immunoassay/radioimmunoassay (EIA/RIA; Costar)
96-well plate reader
Prepare insect cells for quantification
-
1.
Transfer 3×107 Sf9 insect cells into a 175 cm2 tissue culture flask and infect as described in steps 1–3 of Basic Protocol 4.
-
2.
After 3 days, dislodge the infected insect cells by banging the flask with the palm of your hand. Transfer to a 50 ml conical tube and spin at 500 × g for 5 min.
-
3.
Wash the cells 2× with HBSS as described in steps 5–6 of Basic Protocol 4.
-
4.
Resuspend the cell pellet at 1×107 cells/ml of Insect Cell Lysis Buffer.
-
5.Vortex the tube for 1 min and incubate while rotating at 4°C for 4 h.Incubating the cells overnight at 4°C does not affect the amount of protein extracted or the final results of the ELISA.
Prepare coated plate
-
6.
Coat wells of a 96-well ELISA plate with 50 µl 10 µg/ml Anti-MHC class I monoclonal antibody in PBS. Allow two wells per plate as blanks. Incubate for 1 hr at room temperature.
-
7.Remove the coating antibody and add 50 µl blocking buffer to each well and incubate for 1 hr at room temperature.The coating antibody can be reused 6–7 more times. Store at 4°C.
-
8.
Wash plate three times with 200 µl/well wash buffer. After the last wash, remove all residual liquid and add 50 µl Diluent buffer to each well.
Bind MHC-peptide to plates
-
9.Prepare standard and sample from step 5 in Diluent buffer. Dilute the MHC standard to 100 ng/ml and samples to fit in the linear range of the standard curve.Most insect cell lysates are diluted 100-fold. This dilution will vary depending on the antibodies used in the ELISA and the amount of protein produced by the insect cells. An initial standardizing ELISA should be performed to determine the optimal dilution of insect cell lysates.
-
10.
Perform two-fold serial dilutions in ELISA plates by adding 50 µl of standard or insect cell lysates to the first row. Transfer 50 µl from the first row to the second, from the second row to the third, and so on. Discard 50 µl from the last row. Leave two wells/plate as blanks (containing 50 µl Diluent buffer from step 8).
-
11.
Incubate 1 hr at room temperature.
-
12.
Wash plate three times with 200 µl/well Wash buffer.
Add the detecting antibody
-
13.
Add 50 µl/well of biotinylated detecting antibody at 1 µg/ml (for example, 28.14.8S-biotin) in diluent buffer. Incubate 30 to 45 min at room temperature.
-
14.
Wash plate three times with 200 µl/well wash buffer.
Develop and analyze plates
-
15.
Add 50 µl of streptavidin-HRP conjugate to each well and incubate for 30 min at room temperature.
-
16.
Wash the plate three times with 200 µl/well with Final wash buffer.
-
17.
Add 50 µl of TMB substrate solution to each well and wait 2 to 15 min until the standards turn blue.
-
18.Add 50 µl of 0.18 M Sulfuric Acid per well to stop color reaction.The wells will turn from blue to yellow upon the addition of sulfuric acid.
-
19.
Clean bottom of plate and eliminate bubbles from wells if necessary. Read at 450 nm in a 96-well plate reader within 10 min of stopping the reaction. Determine the concentration of the MHC molecules by comparing to the standard curve.
REAGENTS AND SOLUTIONS
Extraction Buffer
10 mM Tris Buffer pH 8.5
0.01% gelatin
0.45% TritonX-100
0.45% Tween-20
50 mM KCl
Insect Cell Lysis Buffer
10mM NaH2PO4
10% glycerol
0.6% CHAPs
0.5 mM DTT
PMSF
Leupeptin
Pepsinase A
FACS Buffer
1× Hank’s Balanced Salt Solution (HBSS, Cellgro)
2% Fetal Bovine Serum (FBS)
10 mM HEPES solution (Cellgro)
0.02% Sodium Azide
Freezing Medium
Grace’s Supplemented Insect Cell Medium (Invitrogen)
10% Fetal Bovine Serum
10% Dimethyl sulfoxide (DMSO)
Supplemented Insect Cell Medium
500 ml Grace’s Supplemented Insect Cell Medium (Invitrogen)
5 ml Pluronic, F-68 (10%, Invitrogen)
5 ml Antibiotic/Antimycotic (100×, Invitrogen)
10% Fetal Bovine Serum (FBS)
Transfection Buffer A
500 ml Grace’s Unsupplemented Insect Cell Medium (Invitrogen)
5% FBS (this varies between lots of serum, see Critical Parameters and Troubleshooting)
Transfection Buffer B
25 mM Hepes
125 mM CaCl2
140 mM NaCl, pH 7.1
Commentary
Although this vaccine strategy has been used to generate both antibody and T cell responses, it has been more thoroughly characterized for the responding peptide-specific cytotoxic T cell responses, as summarized below. Non-specific responses to the vaccine vehicle are controlled for by vaccinating with insect cells infected with baculoviruses encoding irrelevant antigens or no antigen. The baculovirus-infected insect cells alone are an effective immune adjuvant to elicit antigen-specific T cells.
The method of antigen presentation has been analyzed (1). Although baculovirus constructions require MHC molecules on the cell surface to characterize peptides in vitro, they are not required to elicit specific T cells in vivo. Vaccination with insect cells infected with either baculoviruses encoding peptide-MHC complexes or only peptide-β2m complexes elicit a similar frequency of specific T cells. In addition, CD11c+ cells from mice vaccinated with insect cells expressing peptide-MHC molecules stimulate transgenic T cells to proliferate in an antigen-specific manner ex vivo. Therefore, like other effective CD8+ T cell responses (2, 3), most responses generated by vaccination with baculovirus-infected insect cells are initiated by cross-priming.
It is likely that toll-like receptors (TLRs) play a role in the adjuvant effects of baculovirus-infected insect cells. Most TLR, besides TLR3 and sometimes TLR4, signal through the adapter protein, MyD88 (4). Antigen-specific T cells are elicited by this vaccine in MyD88-knock out mice to the same level as in wild type mice (unpublished). An unknown ligand from the infected insect cells may signal through TLR4 in a MyD88-independent signaling cascade or dendritic cells may be activated through interactions between mannose receptors and the insect cell proteins known to expose mannose. Since CD8 T cell memory is generated within 45 days after vaccination, CD4 T cells or Type I interferons may be positively contributing to the response (unpublished) (5–9).
This vaccination strategy compares with other more conventional vaccination strategies such as peptide-loaded dendritic cells (10) and the LANAC adjuvant with peptide (11, 12). All of the vaccination strategies elicit functional specific T cell responses. However, the baculovirus-infected insect cell vaccine has a number of advantages. 1) The peptides are genetically generated; they do not have to be synthesized prior to testing, which eliminates complications with solubility and oxidation associated with many residues [Trp, Cys, Met] and makes this method financially reasonable for testing large numbers of mimotopes. 2) No additional manipulations to the peptides are necessary after they are identified from baculoviruses-encoded peptide libraries, making the evaluation of these peptides straightforward (13, 14). 3) Pools of enriched library peptides that bind to particular T cells can be evaluated for immune responses prior to individual cloning and before sequence information is obtained. 4) Large specific T cell populations are generated using this vaccination strategy, so in vitro stimulation is not required to analyze antigen-specific T cell responses.
Common problems with insect cells and troubleshooting
The following section describes the common problems encountered during the cell culture and infection of insect cells and discusses potential solutions. If these solutions do not adequately address the issues, its best to contact the manufacturer of the insect cells, plasmid vectors, or baculoviruses in use.
Insect cells are particular about the type of serum they are grown in. Many different types and lots of serum may need to be tested in order to achieve optimal growth, infection, and protein production. To test the serum, grow small batches of insect cells in 75 cm2 flasks for one to two weeks and record the growth and death rates. Once a lot of serum has been shown to support the growth of the insect cells, it must also be tested for infection and protein production (see Basic Protocol 2). Finally, the serum needs to tested in a transfection reaction (see Basic Protocol 2). Make Transfection Buffer A containing a titration of serum starting from 1% to 10%, and following the transfection protocol in Basic Protocol 2. The buffer should turn cloudy white after the addition of Transfection Buffer B, but should not be opaque and there should not be any visible white precipitates. Continue following Basic Protocol 2 through step 10. Check the viability of the cells after 7 days in culture. Since no baculovirus or plasmid DNA was used in this titration, the cells should be healthy and not Trypan Blue positive. Choose the concentration of serum for Transfection Buffer A in which the buffer turned white, but the cells were still viable.
Since baculovirus is a lytic virus and kills infected cells, all glassware used to grow insect cells should be acid-washed and autoclaved (see Supporting Protocol 3). In addition, we recommend using plastic disposable pipets and filter pipet tips for transferring insect cells, media, or baculovirus. If the insect cells die while being maintained in culture, it is likely they are contaminated with a virus or the glassware they maintained in is contaminated with detergents. Overgrowth can also be an issue, and it is difficult for the cells to be recovered once they have grown above 3×106 cells/ml. Starting a fresh culture from a frozen stock and maintaining the cells between 2×105 and 2×106 cells/ml is recommended (see Supporting Protocol 2). If baculoviruses is found to have a low titer (less than 1×108 Units/ml), a new stock of baculovirus should be made (see Basic Protocol 3). For highest titers, infected Sf9s should be grown until most or all of the cells stain with Trypan Blue.
Careful titration of the baculoviruses is recommended before beginning large-scale infections for vaccines (see Basic Protocol 3). Adding too much virus to the cells can be detrimental, as the cells may die before achieving the peak of protein production. Adding too little virus may result in a poor infection and low protein yield. If the insect cells do not produce the protein of interest, there is likely a problem with the original plasmid construction. We recommend confirming the sequence of the plasmid construction prior to making baculovirus (see Basic Protocol 1) and confirming the sequencing of the final baculovirus clone prior to beginning large-scale infections (see Supporting Protocol 1). There are commercially available positive controls for baculovirus production if further trouble-shooting is necessary.
Time Considerations
If the investigators are already producing recombinant proteins using baculovirus expression systems, then setting up the vaccine technology will not be as cumbersome. The time requirements of the first step, molecular cloning of the antigens to be analyzed in the immune response (Basic Protocol 1), are the least predictable. If the MHC and peptide sequences are inserted into the baculovirus expression vector without any obstacles, the process will take about 2 weeks. The following steps, producing baculoviruses that reliably express the proteins of interest (Basic Protocols 2–3), take about 2 months to complete. The vaccination protocol requires approximately two and a half weeks before T cells can be analyzed (Basic Protocol 4).
Literature Cited
- 1.Jordan KR, McMahan RH, Oh JZ, Pipeling MR, Pardoll DM, Kedl RM, Kappler JW, Slansky JE. Baculovirus-infected insect cells expressing peptide-MHC complexes elicit protective antitumor immunity. J. Immunol. 2008;180:188–197. doi: 10.4049/jimmunol.180.1.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huang AYC, Bruce AT, Pardoll DM, Levitsky HI. In vivo cross-priming of MHC class I-restricted antigens requires the TAP transporter. Immunity. 1996;4:349–355. doi: 10.1016/s1074-7613(00)80248-4. [DOI] [PubMed] [Google Scholar]
- 3.Valmori D, Souleimanian NE, Tosello V, Bhardwaj N, Adams S, O'Neill D, Pavlick A, Escalon JB, Cruz CM, Angiulli A, Angiulli F, Mears G, Vogel SM, Pan L, Jungbluth AA, Hoffmann EW, Venhaus R, Ritter G, Old LJ, Ayyoub M. Vaccination with NY-ESO-1 protein and CpG in Montanide induces integrated antibody/Th1 responses and CD8 T cells through cross-priming. Proc. Natl. Acad. Sci. USA. 2007;104:8947–8952. doi: 10.1073/pnas.0703395104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 5.Janssen EM, Lemmens EE, Wolfe T, Christen U, von Herrath MG, Schoenberger SP. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature. 2003;421:852–856. doi: 10.1038/nature01441. [DOI] [PubMed] [Google Scholar]
- 6.Sun JC, Bevan MJ. Defective CD8 T cell memory following acute infection without CD4 T cell help. Science. 2003;300:339–342. doi: 10.1126/science.1083317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shedlock DJ, Shen H. Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science. 2003;300:337–339. doi: 10.1126/science.1082305. [DOI] [PubMed] [Google Scholar]
- 8.Curtsinger JM, Valenzuela JO, Agarwal P, Lins D, Mescher MF. Type I IFNs provide a third signal to CD8 T cells to stimulate clonal expansion and differentiation. J. Immunol. 2005;174:4465–4469. doi: 10.4049/jimmunol.174.8.4465. [DOI] [PubMed] [Google Scholar]
- 9.Aichele P, Unsoeld H, Koschella M, Schweier O, Kalinke U, Vucikuja S. CD8 T cells specific for lymphocytic choriomeningitis virus require type I IFN receptor for clonal expansion. J. Immunol. 2006;176:4525–4529. doi: 10.4049/jimmunol.176.8.4525. [DOI] [PubMed] [Google Scholar]
- 10.Slansky JE, Rattis FM, Boyd LF, Fahmy T, Jaffee EM, Schneck JP, Margulies DH, Pardoll DM. Enhanced antigen-specific antitumor immunity with altered peptide ligands that stabilize the MHC-peptide-TCR complex. Immunity. 2000;13:529–538. doi: 10.1016/s1074-7613(00)00052-2. [DOI] [PubMed] [Google Scholar]
- 11.Zaks K, Jordan M, Guth A, Sellins K, Kedl R, Izzo A, Bosio C, Dow S. Efficient Immunization and Cross-Priming by Vaccine Adjuvants Containing TLR3 or TLR9 Agonists Complexed to Cationic Liposomes. J. Immunol. 2006;176:7335–7345. doi: 10.4049/jimmunol.176.12.7335. [DOI] [PubMed] [Google Scholar]
- 12.McMahan RH, McWilliams JA, Jordan KR, Dow SW, Wilson DB, Slansky JE. Relating MHC-Peptide-TCR affinity to immunogenicity for the rational design of tumor vaccines. J. Clin. Invest. 2006;116:2543–2551. doi: 10.1172/JCI26936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crawford F, Jordan KM, Stadinsky B, Wang Y, Huseby E, Marrack P, Slansky JE, Kappler JW. Use of baculovirus MHC/peptide display libraries to characterize T-cell receptor ligands. Immunol. Rev. 2006;210:156–170. doi: 10.1111/j.0105-2896.2006.00365.x. [DOI] [PubMed] [Google Scholar]
- 14.Wang Y, Rubtsov A, Heiser R, White J, Crawford F, Marrack P, Kappler JW. Using a baculovirus display library to identify MHC class I mimotopes. Proc. Natl. Acad. Sci. USA. 2005;102:2476–2481. doi: 10.1073/pnas.0409798102. [DOI] [PMC free article] [PubMed] [Google Scholar]