

Abstract
The structure of the title compound, [Mo(C5H5)Cl(CO)3], reveals a pseudo-square-pyramidal piano-stool coordination around the MoII ion, which is surrounded by a cyclopentadienyl ring, three carbonyl groups and a chloride ligand.
Related literature
For related structures, see: Chaiwasie & Fenn (1968 ▶); Churchill & Bueno (1981 ▶); Albright et al. (1978) ▶; Mays & Robb (1968 ▶). For applications of this class of compounds, see: Arzoumanian (1998 ▶); Freund et al. (2006 ▶); Karunadasa et al. (2010 ▶). For the synthesis, see: Amor et al. (2000 ▶); Atwood & Barbour (2003 ▶).
Experimental
Crystal data
[Mo(C5H5)Cl(CO)3]
M r = 280.51
Monoclinic,
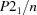
a = 6.4958 (6) Å
b = 11.7671 (10) Å
c = 12.5080 (11) Å
β = 100.064 (2)°
V = 941.36 (14) Å3
Z = 4
Mo Kα radiation
μ = 1.65 mm−1
T = 173 K
0.11 × 0.06 × 0.04 mm
Data collection
Bruker Kappa DUO APEXII diffractometer
Absorption correction: multi-scan (SADABS; Sheldrick, 1997) ▶ T min = 0.840, T max = 0.937
10440 measured reflections
2355 independent reflections
1953 reflections with I > 2σ(I)
R int = 0.035
Refinement
R[F 2 > 2σ(F 2)] = 0.022
wR(F 2) = 0.046
S = 1.01
2355 reflections
118 parameters
H-atom parameters constrained
Δρmax = 0.32 e Å−3
Δρmin = −0.30 e Å−3
Data collection: APEX2 (Bruker, 2006 ▶); cell refinement: SAINT (Bruker, 2006 ▶); data reduction: SAINT; program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008) ▶; molecular graphics: X-SEED (Barbour, 2001 ▶); software used to prepare material for publication: SHELXL97 ▶.
Supplementary Material
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S1600536812008471/kp2389sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536812008471/kp2389Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Selected bond lengths (Å).
| Mo1—C2 | 1.980 (2) |
| Mo1—C3 | 2.008 (2) |
| Mo1—C1 | 2.014 (2) |
| Mo1—Cl1 | 2.5030 (6) |
Acknowledgments
We acknowlege the finacial support from the NRF (THUTHUKA), the University of the Western Cape and Sanate Research.
supplementary crystallographic information
Comment
The oxo-complexes of transition metals, group 6 are very useful in various catalytic applications. Among the numerous transition metal-oxo compounds that have been used as catalysts, molybdenum is probably the element that stands out as the most investigated for oxygen atom transfer reactions (Arzoumanian 1998). Remarkably, most recently, molybdenum derivative has been used to generate hydrogen from water (Karunadasa et al., 2010). While investigating catalytic epoxidation reactions (Freund et al., 2006), we prepared transition metalcarbonyl complexes containing nitrogen bases, chloro- and cyclopentadienyl(Cp)ligands. This compound could easily be oxidized to the dioxo-molybdenum (IV)complexes without losing the attached ligands. Trying to grow crystals of this complex by slow diffusion in a fridge, the titled compound was obtained insted, probably as a decomposition product. In the titled compound, the ligands display a piano stool arrangement. Notably, the carbonyls and the chloride ligands are spaced by the average angle of 77.49°. The Mo-C2 bond trans to the chloride, Cl1, atom [1.980 (2) Å] is noticeably shorter than the others, Mo–C1, 2.014 (2) and M–C3, 2.008 (2) Å, possibly due to the well-known trans effect. The distance between the Mo atom and the C5, C6 and C7 atoms are observed to be shorter than those between Mo and C4 and C8 because of the electronic repulsion between the electronegative Cl atom and the cyclopentadienyl ring electrons. The molecular structure of the title compound (I) is a new polymorph and differ from the structures reported (Chaiwasie et al. 1968; Churchill et al. 1981; Albright et al. 1978; Mays et al.1968). For example, the cell dimensions reported by Chaiwasie et al. (1968) is significantly different from our values (see crystal data).
Experimental
A solution of cyclopentadienyl molybdenum (II) tricarbonyl dimer, [Cp (CO)3Mo]2, (0.506 g, 1.03 mmol) in THF (10 mL) was added to Na/Hg amalgam in a Schlenck tube with a tap at the bottom. The mixture was stirred until the brick red solution of, [Cp(CO)3Mo]2 turned pale-green to confirm the formation of [Cp(CO)3Mo]- anions. The reduced dimer solution was filtered under nitrogen to another Schlenk tube. An excess CCl4 was added and vigorously stired for 30 min. The solvent was removed under vaccum to give a light yellow solid. Yield:0.55 g (61%). The solution of the product in a minimum volume of dichloromethane was allowed to undergo a slow diffusion in an excess of hexane at 277 K for a few days. Block red single crystals suitable for X-ray analysis were obtained.
Refinement
All non-hydrogen atoms were refined anisotropically. All the hydrogen peaks could be found in the difference electron density maps but were finally placed in idealized positions and refined in riding models with Uiso assigned 1.2 times those of their parent atoms and the constraint distances of C—H equal to 0.95 Å. The structure was refined to R factor of 0.0221.
Figures
Fig. 1.

A view of the molecular structure with numbering scheme. Displacement ellipsoids are drawn at the 40% probability level for non-H atoms.
Crystal data
| [Mo(C5H5)Cl(CO)3] | F(000) = 544 |
| Mr = 280.51 | F(000) = 544 |
| Monoclinic, P21/n | Dx = 1.979 Mg m−3 |
| Hall symbol: -P 2yn | Mo Kα radiation, λ = 0.71073 Å |
| a = 6.4958 (6) Å | Cell parameters from 10440 reflections |
| b = 11.7671 (10) Å | θ = 3.3–28.4° |
| c = 12.5080 (11) Å | µ = 1.65 mm−1 |
| β = 100.064 (2)° | T = 173 K |
| V = 941.36 (14) Å3 | Block, red |
| Z = 4 | 0.11 × 0.06 × 0.04 mm |
Data collection
| Bruker Kappa DUO APEXII diffractometer | 2355 independent reflections |
| Radiation source: fine-focus sealed tube | 1953 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.035 |
| 0.5° φ 0.5° φ scans and ω scans | θmax = 28.4°, θmin = 3.3° |
| Absorption correction: multi-scan (SADABS; Sheldrick, 1997) | h = −8→8 |
| Tmin = 0.840, Tmax = 0.937 | k = −15→15 |
| 10440 measured reflections | l = −16→16 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.022 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.046 | H-atom parameters constrained |
| S = 1.01 | w = 1/[σ2(Fo2) + (0.017P)2 + 0.2175P] where P = (Fo2 + 2Fc2)/3 |
| 2355 reflections | (Δ/σ)max = 0.002 |
| 118 parameters | Δρmax = 0.32 e Å−3 |
| 0 restraints | Δρmin = −0.30 e Å−3 |
Special details
| Experimental. crystal mounted on a cryoloop |
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Special detailsGeometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R– factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| Mo1 | 0.09443 (3) | 0.787273 (14) | 0.244289 (15) | 0.02342 (6) | |
| Cl1 | 0.41891 (9) | 0.66982 (5) | 0.25627 (6) | 0.04278 (15) | |
| O1 | 0.0004 (3) | 0.58701 (16) | 0.39597 (16) | 0.0539 (5) | |
| O2 | −0.1151 (3) | 0.91712 (18) | 0.41495 (16) | 0.0580 (5) | |
| O3 | 0.4393 (3) | 0.95672 (15) | 0.35712 (18) | 0.0560 (5) | |
| C1 | 0.0387 (4) | 0.6605 (2) | 0.34363 (19) | 0.0355 (5) | |
| C2 | −0.0421 (4) | 0.8694 (2) | 0.35115 (19) | 0.0366 (5) | |
| C3 | 0.3158 (4) | 0.89401 (19) | 0.3179 (2) | 0.0344 (5) | |
| C4 | −0.1349 (4) | 0.7141 (2) | 0.09365 (19) | 0.0388 (6) | |
| H4 | −0.1983 | 0.6414 | 0.0946 | 0.047* | |
| C5 | −0.2134 (4) | 0.8169 (2) | 0.1275 (2) | 0.0363 (5) | |
| H5 | −0.3391 | 0.8258 | 0.1557 | 0.044* | |
| C6 | −0.0732 (4) | 0.90483 (19) | 0.1122 (2) | 0.0385 (6) | |
| H6 | −0.0886 | 0.9832 | 0.1273 | 0.046* | |
| C7 | 0.0940 (4) | 0.8552 (2) | 0.07028 (19) | 0.0419 (6) | |
| H7 | 0.2126 | 0.8937 | 0.0531 | 0.050* | |
| C8 | 0.0520 (4) | 0.7379 (2) | 0.05865 (19) | 0.0446 (6) | |
| H8 | 0.1380 | 0.6837 | 0.0312 | 0.053* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| Mo1 | 0.02314 (10) | 0.02310 (9) | 0.02324 (10) | −0.00117 (7) | 0.00194 (7) | 0.00350 (8) |
| Cl1 | 0.0329 (3) | 0.0364 (3) | 0.0588 (4) | 0.0086 (2) | 0.0075 (3) | 0.0075 (3) |
| O1 | 0.0611 (13) | 0.0507 (11) | 0.0479 (11) | −0.0172 (9) | 0.0045 (10) | 0.0232 (9) |
| O2 | 0.0513 (12) | 0.0742 (14) | 0.0516 (12) | 0.0026 (10) | 0.0174 (10) | −0.0223 (11) |
| O3 | 0.0424 (11) | 0.0369 (10) | 0.0785 (15) | −0.0112 (8) | −0.0177 (10) | 0.0079 (10) |
| C1 | 0.0326 (13) | 0.0396 (13) | 0.0320 (13) | −0.0052 (10) | −0.0005 (11) | 0.0033 (10) |
| C2 | 0.0300 (13) | 0.0454 (14) | 0.0330 (13) | −0.0002 (11) | 0.0018 (11) | −0.0007 (12) |
| C3 | 0.0275 (12) | 0.0299 (12) | 0.0426 (14) | 0.0008 (9) | −0.0028 (11) | 0.0081 (11) |
| C4 | 0.0473 (15) | 0.0336 (12) | 0.0294 (12) | −0.0057 (11) | −0.0105 (11) | 0.0013 (11) |
| C5 | 0.0310 (13) | 0.0451 (14) | 0.0286 (12) | 0.0008 (10) | −0.0066 (11) | 0.0023 (11) |
| C6 | 0.0492 (15) | 0.0291 (12) | 0.0312 (13) | 0.0024 (10) | −0.0096 (12) | 0.0081 (10) |
| C7 | 0.0450 (15) | 0.0535 (15) | 0.0261 (12) | −0.0093 (12) | 0.0031 (11) | 0.0133 (12) |
| C8 | 0.0574 (18) | 0.0514 (16) | 0.0226 (12) | 0.0123 (13) | 0.0008 (12) | −0.0036 (11) |
Geometric parameters (Å, º)
| Mo1—C2 | 1.980 (2) | O3—C3 | 1.136 (3) |
| Mo1—C3 | 2.008 (2) | C4—C8 | 1.390 (4) |
| Mo1—C1 | 2.014 (2) | C4—C5 | 1.407 (3) |
| Mo1—C6 | 2.280 (2) | C4—H4 | 0.9500 |
| Mo1—C5 | 2.288 (2) | C5—C6 | 1.413 (3) |
| Mo1—C7 | 2.318 (2) | C5—H5 | 0.9500 |
| Mo1—C4 | 2.352 (2) | C6—C7 | 1.412 (4) |
| Mo1—C8 | 2.363 (2) | C6—H6 | 0.9500 |
| Mo1—Cl1 | 2.5030 (6) | C7—C8 | 1.410 (4) |
| O1—C1 | 1.138 (3) | C7—H7 | 0.9500 |
| O2—C2 | 1.145 (3) | C8—H8 | 0.9500 |
| C2—Mo1—C3 | 75.80 (10) | C8—Mo1—Cl1 | 82.84 (7) |
| C2—Mo1—C1 | 78.15 (10) | O1—C1—Mo1 | 176.8 (2) |
| C3—Mo1—C1 | 111.84 (10) | O2—C2—Mo1 | 177.9 (2) |
| C2—Mo1—C6 | 88.85 (10) | O3—C3—Mo1 | 177.9 (2) |
| C3—Mo1—C6 | 99.54 (9) | C8—C4—C5 | 107.7 (2) |
| C1—Mo1—C6 | 141.41 (9) | C8—C4—Mo1 | 73.28 (14) |
| C2—Mo1—C5 | 84.97 (10) | C5—C4—Mo1 | 69.89 (13) |
| C3—Mo1—C5 | 132.35 (9) | C8—C4—H4 | 126.2 |
| C1—Mo1—C5 | 106.00 (9) | C5—C4—H4 | 126.2 |
| C6—Mo1—C5 | 36.04 (8) | Mo1—C4—H4 | 122.4 |
| C2—Mo1—C7 | 122.57 (10) | C4—C5—C6 | 108.2 (2) |
| C3—Mo1—C7 | 95.63 (9) | C4—C5—Mo1 | 74.84 (14) |
| C1—Mo1—C7 | 149.74 (10) | C6—C5—Mo1 | 71.69 (14) |
| C6—Mo1—C7 | 35.74 (9) | C4—C5—H5 | 125.9 |
| C5—Mo1—C7 | 59.35 (9) | C6—C5—H5 | 125.9 |
| C2—Mo1—C4 | 115.08 (9) | Mo1—C5—H5 | 119.4 |
| C3—Mo1—C4 | 154.06 (9) | C7—C6—C5 | 107.7 (2) |
| C1—Mo1—C4 | 93.79 (9) | C7—C6—Mo1 | 73.59 (14) |
| C6—Mo1—C4 | 59.08 (9) | C5—C6—Mo1 | 72.27 (13) |
| C5—Mo1—C4 | 35.26 (9) | C7—C6—H6 | 126.2 |
| C7—Mo1—C4 | 58.49 (9) | C5—C6—H6 | 126.2 |
| C2—Mo1—C8 | 142.61 (10) | Mo1—C6—H6 | 119.8 |
| C3—Mo1—C8 | 123.76 (10) | C8—C7—C6 | 107.2 (2) |
| C1—Mo1—C8 | 114.93 (10) | C8—C7—Mo1 | 74.20 (14) |
| C6—Mo1—C8 | 58.54 (9) | C6—C7—Mo1 | 70.67 (13) |
| C5—Mo1—C8 | 58.07 (9) | C8—C7—H7 | 126.4 |
| C7—Mo1—C8 | 35.04 (9) | C6—C7—H7 | 126.4 |
| C4—Mo1—C8 | 34.29 (9) | Mo1—C7—H7 | 120.6 |
| C2—Mo1—Cl1 | 134.49 (7) | C4—C8—C7 | 109.2 (2) |
| C3—Mo1—Cl1 | 77.86 (7) | C4—C8—Mo1 | 72.43 (14) |
| C1—Mo1—Cl1 | 78.15 (7) | C7—C8—Mo1 | 70.76 (13) |
| C6—Mo1—Cl1 | 131.96 (7) | C4—C8—H8 | 125.4 |
| C5—Mo1—Cl1 | 139.05 (7) | C7—C8—H8 | 125.4 |
| C7—Mo1—Cl1 | 96.29 (7) | Mo1—C8—H8 | 123.0 |
| C4—Mo1—Cl1 | 104.76 (6) | ||
| C2—Mo1—C1—O1 | 111 (4) | C4—C5—C6—C7 | −0.9 (3) |
| C3—Mo1—C1—O1 | −180 (100) | Mo1—C5—C6—C7 | 65.60 (17) |
| C6—Mo1—C1—O1 | 38 (4) | C4—C5—C6—Mo1 | −66.49 (17) |
| C5—Mo1—C1—O1 | 30 (4) | C2—Mo1—C6—C7 | 161.76 (16) |
| C7—Mo1—C1—O1 | −26 (4) | C3—Mo1—C6—C7 | 86.37 (16) |
| C4—Mo1—C1—O1 | −4 (4) | C1—Mo1—C6—C7 | −129.00 (18) |
| C8—Mo1—C1—O1 | −32 (4) | C5—Mo1—C6—C7 | −115.2 (2) |
| Cl1—Mo1—C1—O1 | −108 (4) | C4—Mo1—C6—C7 | −77.83 (16) |
| C3—Mo1—C2—O2 | −31 (6) | C8—Mo1—C6—C7 | −37.53 (15) |
| C1—Mo1—C2—O2 | 86 (6) | Cl1—Mo1—C6—C7 | 4.16 (18) |
| C6—Mo1—C2—O2 | −131 (6) | C2—Mo1—C6—C5 | −83.02 (16) |
| C5—Mo1—C2—O2 | −167 (6) | C3—Mo1—C6—C5 | −158.41 (15) |
| C7—Mo1—C2—O2 | −119 (6) | C1—Mo1—C6—C5 | −13.8 (2) |
| C4—Mo1—C2—O2 | 174 (6) | C7—Mo1—C6—C5 | 115.2 (2) |
| C8—Mo1—C2—O2 | −159 (6) | C4—Mo1—C6—C5 | 37.39 (14) |
| Cl1—Mo1—C2—O2 | 26 (6) | C8—Mo1—C6—C5 | 77.69 (16) |
| C2—Mo1—C3—O3 | −94 (6) | Cl1—Mo1—C6—C5 | 119.38 (14) |
| C1—Mo1—C3—O3 | −165 (6) | C5—C6—C7—C8 | 1.0 (3) |
| C6—Mo1—C3—O3 | −7 (6) | Mo1—C6—C7—C8 | 65.74 (17) |
| C5—Mo1—C3—O3 | −24 (6) | C5—C6—C7—Mo1 | −64.72 (17) |
| C7—Mo1—C3—O3 | 28 (6) | C2—Mo1—C7—C8 | −136.98 (16) |
| C4—Mo1—C3—O3 | 25 (7) | C3—Mo1—C7—C8 | 146.30 (16) |
| C8—Mo1—C3—O3 | 51 (6) | C1—Mo1—C7—C8 | −9.3 (3) |
| Cl1—Mo1—C3—O3 | 124 (6) | C6—Mo1—C7—C8 | −115.2 (2) |
| C2—Mo1—C4—C8 | 150.55 (15) | C5—Mo1—C7—C8 | −76.95 (17) |
| C3—Mo1—C4—C8 | 40.5 (3) | C4—Mo1—C7—C8 | −35.54 (15) |
| C1—Mo1—C4—C8 | −130.76 (16) | Cl1—Mo1—C7—C8 | 67.94 (15) |
| C6—Mo1—C4—C8 | 78.38 (16) | C2—Mo1—C7—C6 | −21.80 (19) |
| C5—Mo1—C4—C8 | 116.6 (2) | C3—Mo1—C7—C6 | −98.53 (16) |
| C7—Mo1—C4—C8 | 36.33 (15) | C1—Mo1—C7—C6 | 105.9 (2) |
| Cl1—Mo1—C4—C8 | −52.03 (15) | C5—Mo1—C7—C6 | 38.23 (14) |
| C2—Mo1—C4—C5 | 33.94 (18) | C4—Mo1—C7—C6 | 79.64 (16) |
| C3—Mo1—C4—C5 | −76.1 (3) | C8—Mo1—C7—C6 | 115.2 (2) |
| C1—Mo1—C4—C5 | 112.63 (15) | Cl1—Mo1—C7—C6 | −176.89 (14) |
| C6—Mo1—C4—C5 | −38.23 (14) | C5—C4—C8—C7 | 0.2 (3) |
| C7—Mo1—C4—C5 | −80.28 (16) | Mo1—C4—C8—C7 | −61.57 (17) |
| C8—Mo1—C4—C5 | −116.6 (2) | C5—C4—C8—Mo1 | 61.80 (16) |
| Cl1—Mo1—C4—C5 | −168.64 (13) | C6—C7—C8—C4 | −0.8 (3) |
| C8—C4—C5—C6 | 0.4 (3) | Mo1—C7—C8—C4 | 62.62 (18) |
| Mo1—C4—C5—C6 | 64.41 (17) | C6—C7—C8—Mo1 | −63.39 (17) |
| C8—C4—C5—Mo1 | −64.00 (17) | C2—Mo1—C8—C4 | −47.2 (2) |
| C2—Mo1—C5—C4 | −149.49 (16) | C3—Mo1—C8—C4 | −160.02 (14) |
| C3—Mo1—C5—C4 | 144.94 (15) | C1—Mo1—C8—C4 | 56.46 (17) |
| C1—Mo1—C5—C4 | −73.36 (16) | C6—Mo1—C8—C4 | −80.10 (16) |
| C6—Mo1—C5—C4 | 115.5 (2) | C5—Mo1—C8—C4 | −37.46 (14) |
| C7—Mo1—C5—C4 | 77.63 (16) | C7—Mo1—C8—C4 | −118.4 (2) |
| C8—Mo1—C5—C4 | 36.40 (14) | Cl1—Mo1—C8—C4 | 129.80 (14) |
| Cl1—Mo1—C5—C4 | 16.90 (19) | C2—Mo1—C8—C7 | 71.2 (2) |
| C2—Mo1—C5—C6 | 94.97 (16) | C3—Mo1—C8—C7 | −41.62 (19) |
| C3—Mo1—C5—C6 | 29.4 (2) | C1—Mo1—C8—C7 | 174.86 (15) |
| C1—Mo1—C5—C6 | 171.11 (15) | C6—Mo1—C8—C7 | 38.30 (15) |
| C7—Mo1—C5—C6 | −37.90 (15) | C5—Mo1—C8—C7 | 80.93 (17) |
| C4—Mo1—C5—C6 | −115.5 (2) | C4—Mo1—C8—C7 | 118.4 (2) |
| C8—Mo1—C5—C6 | −79.13 (16) | Cl1—Mo1—C8—C7 | −111.81 (16) |
| Cl1—Mo1—C5—C6 | −98.64 (15) |
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: KP2389).
References
- Albright, M. J., Glick, M. D. & Oliver, J. P. (1978). J. Organomet. Chem. 161, 221–231.
- Amor, F., Royo, P., Spaniol, T. P. & Okuda, J. (2000). J. Organomet. Chem. 604, 126–131.
- Arzoumanian, H. (1998). Coord. Chem. Rev. 178–180, 191–202.
- Atwood, J. L. & Barbour, L. J. (2003). Cryst. Growth Des. 3, 3–8.
- Barbour, L. J. (2001). J. Supramol. Chem. 1, 189–191.
- Bruker (2006). APEX2 and SAINT Bruker AXS Inc., Madison, Wisconsin, USA.
- Chaiwasie, S. & Fenn, R. H. (1968). Acta Cryst. B24, 525–529.
- Churchill, M. R. & Bueno, C. (1981). Inorg. Chem. 20, 2197–2202.
- Freund, C., Abrantes, M. & Kühn, F. E. (2006). J. Organomet. Chem. 691, 3718–3729.
- Karunadasa, H. I., Chang, C. J. & Long, J. R. (2010). Nature (London), 464, 1329–1323. [DOI] [PubMed]
- Mays, M. J. & Robb, J. D. (1968). J. Chem. Soc. A, pp. 329–332.
- Sheldrick, G. M. (1997). SADABS University of Göttingen, Germany.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S1600536812008471/kp2389sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536812008471/kp2389Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report