

Abstract
The effect of neoplastic transformation on the response to genotoxic stress is of significant clinical interest. In this study, we offer genetic evidence that the apoptotic response of neoplastically transformed cells to DNA damage requires RhoB, a member of the Rho family of actin cytoskeletal regulators. Targeted deletion of the rhoB gene did not affect cell cycle arrest in either normal or transformed cells after exposure to doxorubicin or gamma irradiation, but rendered transformed cells resistant to apoptosis. This effect was specific insofar as rhoB deletion did not affect apoptotic susceptibility to agents that do not damage DNA. However, rhoB deletion also affected apoptotic susceptibility to Taxol, an agent that disrupts microtubule dynamics. We have demonstrated that RhoB alteration mediates the proapoptotic and antineoplastic effects of farnesyltransferase inhibitors, and we show here that RhoB alteration is also crucial for farnesyltransferase inhibitors to sensitize neoplastic cells to DNA damage-induced cell death. We found RhoB to be an important determinant of long-term survival in vitro and tumor response in vivo after gamma irradiation. Our findings identify a pivotal role for RhoB in the apoptotic response of neoplastic cells to DNA damage at a novel regulatory point that may involve the actin cytoskeleton.
Keywords: Rho, Rac, Cdc42, Ras, programmed cell death
RhoB is a member of the Rho family of small GTPases that regulate the actin cytoskeleton and cell adhesion signaling. Although closely related to RhoA, its better-studied relative RhoB differs from RhoA in its localization, regulation, and function. For example, RhoB is localized in early endosome and nuclear membranes and has a specialized role in intracellular receptor trafficking (1–5). In addition, RhoB is short-lived and its levels are elevated by a variety of stimuli, including epidermal growth factor, transforming growth factor β, and UV irradiation (6–9). Lastly, RhoB is unusual in that it is normally either farnesylated or geranylgeranylated (10). Although the significance of these posttranslational modifications is not fully understood, it is clear that different enzymes are responsible for farnesylation or geranylgeranylation in cells and that differential prenylation may have functional consequences in some settings (11).
Recent work has established that alteration in the pattern of RhoB prenylation is a crucial part of the mechanism by which farnesyltransferase inhibitors (FTIs) mediate their selective antineoplastic effects in vitro and in vivo (12). Specifically, FTI treatment elevates and causes mislocalization of the geranylgeranylated isoform, because of the unencumbered action of geranylgeranyltransferase in drug-treated cells, and this gain-of-function event is both necessary and sufficient to mediate apoptotic and antineoplastic effects in vitro and in vivo (13, 14). Thus, although the development and preclinical validation of FTIs was predicated on the farnesylation requirement of Ras proteins, mechanistic investigations have argued against a required role for Ras targeting and, instead, corroborated an alternate model in which RhoB targeting is crucial. Based on promising preclinical results, human clinical trials of FTIs have been initiated (15). One property of FTIs being tested in clinical trials is their ability to sensitize malignant cells to DNA damage-induced cell death (16, 17). In this study, we investigated whether RhoB has a causal role in the response to DNA damage by using cells with different rhoB genotypes. Our findings reveal a link between RhoB function and the genotoxic sensitivity of neoplastic cells.
Materials and Methods
Cell Culture and Gene Transfer.
A detailed characterization of rhoB nullizygous mice will be described elsewhere; cells isolated from such animals have been described and characterized (14). Briefly, mouse embryonic fibroblasts (MEFs) were isolated from embryos of different genotypes at 12–14 days gestation and maintained as described. MEFs were seeded at 5 × 105 cells per dish and transfected with 10 μg each of pT22 (encoding activated H-Ras) and p1A/neo (encoding the adenovirus E1A region) as described by using a calcium phosphate coprecipitation method (14). Single foci were cloned 12–16 days after transfection and expanded into cell lines for analysis. Retroviral complementation of rhoB−/− cells was performed as follows. A retroviral vector encoding a wild-type hemagglutinin epitope-tagged RhoB gene has been described (14). A derivative of this plasmid, termed MSCV-EGFP-RhoB-WT, was made in which the puromycin resistance gene in the parental vector was replaced with a HindIII/ClaI fragment encoding a membrane-bound version of green fluorescent protein (EGFP). The ecotropic packaging cell line Bosc23 was transfected with 20 μg MSCV-EGFP-RhoB-WT or MSCV-EGFP vector essentially as described (18). Viral supernatants were harvested 48 and 56 h after transfection, clarified through 0.45-μm filters (Millipore), supplemented with 8 μg/ml polybrene (Sigma), and frozen at −80°C until use. Cells were infected and harvested for flow cytometric analysis as described (14).
PCR Genotyping.
An extract from 106 cells was generated by suspending cells in 200 μl of DNA digestion buffer (50 mM KCl/1.5 mM MgCl2/10 mM Tris, pH 8.5/0.01% gelatin/0.045% Nonidet P-40/0.45% Tween-20) and 100 μg/ml proteinase K. After incubation for 3 h at 55°C, the extract was heated 10 min at 100°C and insoluble material was pelleted in a microfuge. One microliter of the clarified extract was used for PCR in a 30-μl reaction containing 3 μl of 10× PCR buffer, 20 μM Neo 5′ primer (5′-GAT CGG CCA TTG AAC AAG ATG), 20 μM Neo 3′ primer (5′-AGA GCA GCC GAT TGT CTG TTG), 20 μM RhoB 5′ primer (5′-ACG GAG CTG GCC CGC ATG AAG), 20 μM RhoB 3′ primer (5′-GGA TCC GTA GCG CTT CTG CAG), 10 mM dNTPs, and 1 unit of Taq polymerase. The reaction was heated to 94°C for 1 min, cycled 35 times at 94°C for 1 min/64°C for 30 sec/72°C for 30 sec, and then heated for 5 min at 72°C. Products were analyzed by electrophoresis on 3.5% agarose gels. The predicted size of the neo product (100 bp) and the RhoB product (150 bp) was confirmed.
Apoptosis Assays.
Cells (5 × 105) were seeded into 60-mm dishes and, 14–16 h later, were irradiated or treated with doxorubicin as indicated. After 16–36 h of incubation, cells were harvested by trypsinization, washed with PBS, and fixed in 70% ethanol. The cells then were stained in PBS containing 0.1% glucose, 10 mg/ml RNase A, and 5 mg/ml propidium iodide. Flow cytometry was performed by using an EPIC/XL cell analyzer (Coulter). The proportion of cells exhibiting sub-G1 phase DNA, indicating DNA degradation, was used as a measurement of the number of apoptotic cells in the population. Apoptotic cell death was confirmed by the production of nucleosomal DNA cleavage, which was monitored in cells as described (19).
Clonogenic Survival Determination.
Radiation survival was determined by clonogenic assay at radiation doses from 1 to 10 Gy on cells from log growth cultures. Clonogenic assays were carried out by plating cells in 60-mm dishes before irradiation with a Mark I cesium irradiator (J. L. Shepherd, San Fernando, CA) at a dose rate of 1.6 Gy per min. Colonies were stained and counted 14–21 days after irradiation. The surviving fraction at a given dose is defined as: colonies formed/(cells plated) × (plating efficiency of untreated cells). Each point on the survival curves represents the mean surviving fraction from at least three dishes.
Xenograft Tumor Irradiation Assay.
Subcutaneous tumors were established by injection of 107 cells suspended in serum-free DMEM into the upper thigh of both legs of severe combined immunodeficient mice. Opposite thighs on the same animal were injected with cells of +/− or −/− genotype. Animals were divided into control and treatment groups 24 h after injection. The injection site of animals in the treatment group was subjected to an irradiation dose of 15 Gy in the same irradiation device noted above with a model 335 Variable Slit Collimator System (J. L. Shepherd, San Fernando, CA). Tumors were measured at times after treatment with a digimatic caliper, and tumor volumes were calculated by the formula volume = width2 × length × 0.52.
Results
RhoB Is Crucial for the Apoptotic Response of Neoplastically Transformed Cells to DNA Damage and Blunts the Response to Taxol.
MEFs were isolated from mice that were heterozygous or homozygous null for rhoB, the characterization of which will be described elsewhere (A.X.L., N. Rane, J.-P. Liu, and G.C.P., unpublished results). Previous investigations have indicated that +/+ and +/− MEFs are identical in phenotype and that genotype does not affect the susceptibility of MEFs to neoplastic transformation by adenovirus E1A plus mutated H-Ras (14). This study presents differences between +/− and −/− MEFs, the rhoB expression of which has been documented previously; genotypes were determined by PCR and confirmed in all primary MEFs and E1A + Ras-transformed MEF cell lines used (ref. 14; Fig. 1A). Primary MEFs are resistant to cell death caused by DNA damage, but MEFs that express E1A or are transformed by E1A + Ras undergo apoptosis (20–22).
Figure 1.

RhoB is dispensable for arrest response of primary cells after DNA damage. (A) PCR genotyping. Genotypes were determined by multiplex PCR for rhoB or neo transgene sequences as described (14). Products were fractionated on agarose gels, stained with ethidium bromide, and photographed. (B) Flow cytometry. Primary MEFs (passage 3) were subjected to treatment with 1,000 ng/ml doxorubicin (Dox) or a 20-Gy dose of gamma irradiation and processed for flow cytometry 24 h or 36 h later, respectively. MEFs prepared from different embryos responded similarly.
We used two clinical modalities that are effective in causing DNA damage and tumor regressions in this study: doxorubicin and gamma irradiation. Doxorubicin causes cell death by irreversibly inhibiting DNA topoisomerase II, leading to the production of double-strand breaks, whereas gamma irradiation directly causes double-stranded DNA breaks as well as indirectly damages DNA through production of free radicals. +/− or −/− primary MEFs exposed to either modality accumulated in the G2/M-phase cell cycle, consistent with the anticipated G2/M arrest (Fig. 1B). Apoptosis was not apparent in primary cells at the doses that are sufficient to completely kill malignant cell populations (see below). The quantitative response of −/− MEFs with regard to cell cycle profile was statistically similar to +/− MEFs. Consistent with previous observations (22), neoplastic transformation by E1A + Ras sensitized +/− cells to doxorubicin-induced apoptosis, a phenomenon that was readily discernible by substratum detachment, cell shrinkage, and plasma membrane blebbing (Fig. 2A). In contrast, neoplastic transformation by E1A + Ras did not sensitize −/− cells to doxorubicin-induced apoptosis. Flow cytometric analysis indicated that transformed +/− cells underwent apoptosis after passage through a transient arrest in the G2/M phase of the cell cycle (Fig. 2B). At lower drug concentrations, there was an increase in the number of cells exhibiting G2/M-phase arrest. At higher drug concentrations, the number of these cells was reduced but accompanied by an increased number of cells displaying sub-G1 DNA, indicative of DNA degradation and apoptotic cell death. In transformed −/− MEFs, no signs of cells exhibiting sub-G1 DNA were apparent. Instead, increased concentrations of doxorubicin led to the appearance of increasing numbers of cells exhibiting G2/M-phase arrest, in the absence of apoptotic cell death. The apparent resistance of transformed −/− cells was not due to clonal variation, because all of the E1A + Ras-transformed clones and pooled cell focus populations tested reacted similarly (data not shown).
Figure 2.

RhoB is crucial for the apoptotic response of neoplastically transformed cells after DNA damage. (A) Morphology after doxorubicin (Dox) treatment. E1A + Ras-transformed cells were treated for 18 h with 200 ng/ml doxorubicin before being photographed. (B) Resistance to doxorubicin. Cells were treated for 18 h with doxorubicin at the concentrations indicated and processed for flow cytometry. (C) Resistance to gamma irradiation. Cells were gamma-irradiated to the dose indicated and processed for flow cytometry. The proportion of the population exhibiting sub-G1-phase DNA is presented on the y axis. (D) Long-term survival after irradiation. Cells (4 × 103) were seeded into 100-mm dishes and gamma-irradiated at the dose indicated. Two weeks later, cell colonies were fixed in methanol and stained with crystal violet. (E) Irradiation survival curve. The mean and SE of the number of colonies remaining is shown, relative to control, after gamma irradiation at the dose indicated. One +/− and two −/− transformed cell clones were tested.
Irradiation experiments established that the resistance of transformed −/− cells was related to DNA damage and not other effects of doxorubicin. Neoplastic transformation also sensitized +/− but not −/− cells to apoptosis after gamma irradiation, as measured by the proportion of cells exhibiting sub-G1 DNA (Fig. 2C). To determine whether the short-term resistance of transformed −/− cells resulted in differences in long-term proliferative potential, we compared colony-formation efficiencies after exposure to increasing doses of gamma irradiation (Fig. 2D). As anticipated, transformed +/− cells were highly sensitive to colony inhibition after irradiation at doses of 2.5–10 Gy and were killed essentially completely at the highest dose of 10 Gy. In contrast, the cloning efficiency of transformed −/− cells was affected only ≈50% at the highest dose tested. Quantitative measurements determined by survival curves confirmed that the effect of RhoB loss was significant (Fig. 2E).
To confirm that cell death caused by doxorubicin or gamma irradiation was by apoptosis, we examined cells for evidence of nucleosomal DNA cleavage, a hallmark of this process. Transformed +/− cells but not −/− cells exhibited a characteristic DNA ladder on agarose gels after doxorubicin treatment or gamma irradiation (Fig. 3). We concluded that loss of RhoB abolished the sensitization to DNA damage-induced apoptosis that is caused by neoplastic transformation in this system.
Figure 3.
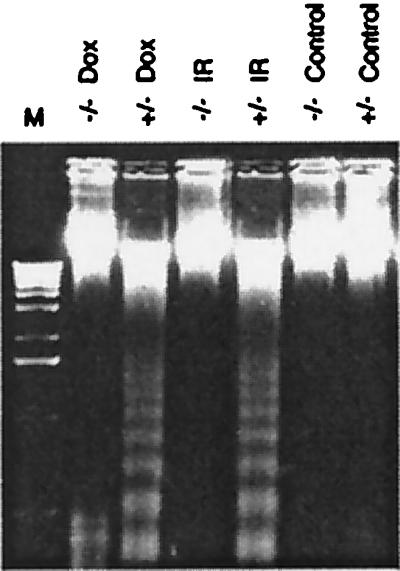
DNA-laddering assay. Genomic DNA was prepared from E1A + Ras-transformed cells that were treated for 18 h with 200 ng/ml doxorubicin (Dox) or 5 Gy gamma irradiation (IR). DNAs were fractionated on agarose gels, stained with ethidium bromide, and photographed. M, DNA molecular weight marker.
To establish the specificity of these effects, we compared the susceptibility of transformed +/− and −/− cells with cell death-inducing agents that do not cause DNA damage. Cell treatment with the tyrosine kinase inhibitor staurosporine, which induces apoptosis, or the electron transport poison sodium azide, which induces necrosis, elicited cell deaths with similar efficiency regardless of rhoB genotype (Fig. 4). These results supported the conclusion that the resistance to DNA damage-induced cell death caused by RhoB loss was specifically associated with mechanisms related to DNA damage, rather than intrinsic cell death mechanisms. The staurosporine result also ruled out the possibility that resistance was due to mutation of central apoptotic regulators (e.g., p53, Bax, etc.), because such events would render cells more resistant to staurosporine as well as DNA-damaging agents. In support of this interpretation, Western blot analysis revealed no defects in transformed −/− cells in p53 response or in the p53-regulated genes Bax and p21WAF1 after various insults (data not shown).
Figure 4.

Specificity of RhoB in cell death. E1A + Ras-transformed cells were treated for 24 h with 10 ng/ml staurosporine (STSP) or 250 μg/ml sodium azide (SA) and processed for flow cytometry. The mean and SE of the proportion of cells exhibiting sub-G1-phase DNA is presented on the y axis. Three trials using three different transformed cell lines were performed.
To probe further the specificity of the effect of rhoB deletion, we examined its effects on apoptosis induced by Taxol, a chemotherapeutic agent that inhibits microtubule dynamics. Taxol elicited cell death with high efficiency in transformed +/− cells. Interestingly, transformed −/− cells displayed a significantly reduced sensitivity to Taxol-induced cell death in this system (Fig. 5). This result suggested the possibility of a broad role for RhoB in cancer cell deaths induced by important therapeutic modalities.
Figure 5.

RhoB has an important role in the apoptotic response to Taxol, an agent that disrupts microtubule dynamics. E1A + Ras-transformed cells were treated with the indicated concentrations of Taxol and processed for flow cytometry 24 h later. The mean and SE of the proportion of cells exhibiting sub-G1-phase DNA is presented on the y axis.
To establish that resistance to DNA damage-induced apoptosis was caused by the absence of rhoB, rather than a distal effect of rhoB deletion (e.g., one that emerged during mouse development or in vitro cell culture), we attempted to complement the apoptotic defect by retroviral gene transfer. −/− cells were infected with retroviral vectors encoding no insert or wild-type RhoB. Each vector also included a EGFP to readily allow the identification of virally infected cells. Forty-eight hours after infection, cells were irradiated at a dose of 10 Gy, and the relative number of green cells in the infected cell population was scored by viable immunofluorescence microscopy at 24, 48, and 72 h (Fig. 6). Cells infected with empty vector exhibited no relative decrease in green cell number after irradiation. In contrast, cells infected with RhoB vector exhibited a marked relative decrease in green cell number after irradiation by 72 h. At this time, a >90% reduction in the relative number of green cells had occurred. The results of this experiment indicated that rhoB deletion was directly responsible for the lack of sensitization to DNA damage, because ectopic RhoB could reverse the resistant phenotype of transformed null cells. These results provided further evidence against the possibility that nonspecific alterations of central apoptotic regulators were responsible for the phenotype. We concluded that RhoB had a specific role in mediating the sensitization to DNA damage caused by neoplastic transformation.
Figure 6.

Complementation of the null cell defect. E1A + Ras-transformed −/− cells were infected with GFP-tagged retroviruses that expressed wild-type RhoB or no insert. Forty-eight hours after infection, cells were irradiated to a dose of 5 Gy and the number of live green cells was monitored at various times afterward. Open symbols, vector; filled symbols, RhoB.
Farnesyltransferase Inhibitors Must Alter RhoB to Sensitize Neoplastically Transformed Cells to DNA Damage-Induced Apoptosis.
Previous work has established that RhoB alteration is crucial for the proapoptotic effects of FTIs, a novel class of cancer chemotherapeutic agents in clinical trials that selectively target the growth and survival of malignant cells (reviewed in ref. 23). Specifically, FTI treatment elevates the level of the geranylgeranylated isoform of RhoB, leading to its mislocalization in cells, and this event is necessary and sufficient for apoptosis and antineoplastic effects in vitro and in vivo (13, 14, 24). One of the interesting properties of FTIs is that they sensitize neoplastically transformed cells to DNA damage-induced cell deaths (16, 17). We reasoned that if RhoB alteration was crucial for this effect, then −/− cells would be resistant to FTI-induced sensitization, because in the absence of the rhoB gene it would be impossible for FTIs to elevate geranylgeranylated RhoB in cells. As anticipated, FTI treatment increased the sensitivity of transformed +/− cells to apoptosis by doxorubicin (Fig. 7). At the relatively low concentrations of 12.5 ng/ml or 25 ng/ml doxorubicin, which induced cell cycle arrest but not cell death (see Fig. 2), the coaddition of FTI increased the number of apoptotic cells significantly. In contrast, FTI treatment did not sensitize transformed −/− cells to doxorubicin-induced apoptosis (Fig. 7). We concluded that RhoB alteration was crucial for the ability of FTIs to sensitize cells to DNA damage-induced apoptosis. This conclusion confirmed a primary role for RhoB in the antineoplastic mechanism engaged by FTI and offered a second line of support for the conclusion that RhoB had an important function in the apoptotic response of transformed cells to DNA damage.
Figure 7.

FTIs must alter RhoB to sensitize cells to DNA damage. E1A + Ras-transformed cells were treated for 24 h with 10 μM FTI L-744,832 (30) and then treated for an additional 18 h where indicated with doxorubicin at the concentrations noted. The concentrations of doxorubicin used in this experiment are insufficient to cause cell death (see Fig. 3B). Control cells were untreated. The mean and SE of the proportion of cells exhibiting sub-G1-phase DNA, indicating apoptosis, derived from multiple trials by using different MEF preparations is shown.
RhoB Has a Significant Role in the Radio Response of Neoplastic Cells in Vivo.
To assess the in vivo significance of the radio resistance elicited by rhoB deletion, we compared the response of murine tumor xenografts after gamma irradiation. Transformed +/− or −/− cells were injected s.c. into opposite flanks of individual severe combined immunodeficient mice. Tumors that formed after several days were left untreated (control group) or irradiated at the injection site with a single dose of 15 Gy (treatment group). Tumor size was monitored at various times after treatment. Control group tumors achieved statistically similar sizes within 20 days. In contrast, the tumors in the treatment group differed significantly in both size and growth rate throughout this period (Fig. 8). At 20 days after treatment, tumors generated by +/− cells were ≈5-fold smaller than control tumors, whereas tumors generated by −/− cells were only ≈50% smaller. The more limited reduction in the latter case was consistent with the retention of a growth arrest response mechanism after DNA damage in the transformed −/− cells. We concluded that RhoB had a significant role in the radio response of neoplastically transformed cells in vivo.
Figure 8.

RhoB is critical for the radio response of tumor xenografts. Severe combined immunodeficient mouse tumor xenografts were generated by s.c. injection of 107 transformed +/− or −/− cells into opposite thighs of the same mouse. The next day, the animals were divided into control or treatment groups and the latter group was irradiated at the injection site with a dose of 15 Gy. Tumor growth was monitored at various times afterward. Each point represents the mean and SE of the tumor volume as calculated by the formula volume = width2 × length × 0.52.
Discussion
Rho proteins are key regulators of cytoskeletal actin that participate in many cellular processes, including neoplastic transformation (25, 26). This report establishes a causal link between Rho activity and the DNA damage response. Our findings are a logical extension of reports that RhoB is regulated by and contributes to genotoxic stress (7, 27) and that RhoB plays a crucial role in mediating the proapoptotic effects of FTIs in a manner that is coordinated with effects on the actin cytoskeleton (14, 28). Here, we have extended the role of RhoB to provide an explanation for how FTIs sensitize transformed cells to DNA damage-induced cell death (16). In summary, our findings offer insights into how neoplastic transformation alters the cellular response to DNA damage.
Our findings uncover an additional player in cancer pathophysiology and therapeutic response, the alteration or suppression of which may be important in the development of drug resistance. One intriguing aspect of the link to RhoB identified here is its potential relationship to the histopathology of cancer cells and their drug-resistant variants, which long has been recognized but not understood. Through the effects on the actin cytoskeleton, RhoB and perhaps other Rho proteins may not only regulate cell shape but also aspects of the malignant phenotype that dictate clinical status, progression, and resistance to therapeutics. This study focused primarily on therapeutics believed to act by damaging DNA, but also offered evidence of a role for RhoB in the response to antimicrotubule agents. Together, these two classes of cancer therapeutics constitute a major part of the arsenal of weapons presently available to the clinical oncologist. One prediction of our study is that RhoB or the signaling pathways in which it functions might be suppressed or mutated in cancer cells that are resistant to such therapeutics.
The mechanism of resistance to genotoxic stress caused by RhoB loss differs significantly from other mechanisms that have been described. One can rule out differential drug exposure as its basis because nullizygous cells similarly were susceptible to cell cycle arrest. For the same reason, one also can argue against a defect in the intrinsic DNA-damage-signaling machinery. Molecular support was offered by evidence of an intact p53 response in −/− cells after DNA damage or other insults, consistent with the ability of the cells to undergo cell cycle arrest (unpublished results). The resistance mechanism described here was peculiarly associated with neoplastic status and was not associated with a general loss of susceptibility to apoptosis. These features differ from losses of the intrinsic elements of the apoptotic machinery or the DNA-damage-signaling apparatus in cells. We therefore favor the interpretation that RhoB participates at some level downstream of the DNA-damage-signaling machinery, as a death effector or death signal modifier, rather than upstream in the DNA-damage-signaling process itself. An additional finding was that RhoB appeared to be involved in the apoptotic response to Taxol, which disrupts microtubule dynamics. Microtubules can indirectly affect adhesion-dependent processes that involve actin stress fibers (29), so this observation suggests the possibility that at least some of the apoptotic effects of Taxol might be mediated via RhoB-dependent effects on the actin cytoskeleton.
How RhoB acts is not yet clear. Given evidence that RhoB has a specialized function in intracellular receptor trafficking (5), it is tempting to speculate that the resistance phenotype reported here reflects an inability to appropriately traffic elements of a death-effector pathway(s) that is activated by DNA damage. In any case, our findings suggest that alterations in the actin cytoskeleton may have a causative role in manifesting the sensitization to DNA damage that is associated with neoplastic transformation of cells. Further investigations of the role of Rho proteins in mediating proapoptotic effects of DNA damage may lead to new insights into cancer cell death mechanisms that are pathophysiologically specific and therefore of interest for therapeutic exploitation.
Acknowledgments
We thank Allen Oliff for providing FTI L-744,832 and Neena Rane, Robert Grafstrom, Alexander Muller, and George Farmer for critically reading the manuscript. This work was supported by National Institutes of Health Grant CA82222 (to G.C.P.).
Abbreviations
- FTI
farnesyltransferase inhibitor
- MEF
mouse embryonic fibroblast
- EGFP
membrane-bound version of green fluorescent protein
References
- 1.Adamson P, Paterson H F, Hall A. J Cell Biol. 1992;119:617–627. doi: 10.1083/jcb.119.3.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lebowitz P F, Davide J P, Prendergast G C. Mol Cell Biol. 1995;15:6613–6622. doi: 10.1128/mcb.15.12.6613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zalcman G, Closson V, Linares-Cruz G, Leregours F, Honore N, Tavitian A, Olofsson B. Oncogene. 1995;10:1935–1945. [PubMed] [Google Scholar]
- 4.Lebowitz P, Prendergast G C. Cell Adhes Commun. 1998;4:1–11. doi: 10.3109/15419069809010787. [DOI] [PubMed] [Google Scholar]
- 5.Gampel A, Parker P J, Mellor H. Curr Biol. 1999;9:955–958. doi: 10.1016/s0960-9822(99)80422-9. [DOI] [PubMed] [Google Scholar]
- 6.Jahner D, Hunter T. Mol Cell Biol. 1991;11:3682–3690. doi: 10.1128/mcb.11.7.3682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fritz G, Kaina B, Aktories K. J Biol Chem. 1995;270:25172–25177. doi: 10.1074/jbc.270.42.25172. [DOI] [PubMed] [Google Scholar]
- 8.Fritz G, Kaina B. J Biol Chem. 1997;272:30637–30644. doi: 10.1074/jbc.272.49.30637. [DOI] [PubMed] [Google Scholar]
- 9.Engel M E, Datta P K, Moses H L. J Biol Chem. 1998;273:9921–9926. doi: 10.1074/jbc.273.16.9921. [DOI] [PubMed] [Google Scholar]
- 10.Adamson P, Marshall C J, Hall A, Tilbrook P A. J Biol Chem. 1992;267:20033–20038. [PubMed] [Google Scholar]
- 11.Lebowitz P, Casey P J, Prendergast G C, Thissen J. J Biol Chem. 1997;272:15591–15594. doi: 10.1074/jbc.272.25.15591. [DOI] [PubMed] [Google Scholar]
- 12.Prendergast G C. Curr Opin Cell Biol. 2000;12:166–173. doi: 10.1016/s0955-0674(99)00072-1. [DOI] [PubMed] [Google Scholar]
- 13.Du W, Prendergast G C. Cancer Res. 1999;59:5924–5928. [PubMed] [Google Scholar]
- 14.Liu A-X, Du W, Liu J-P, Jessell T M, Prendergast G C. Mol Cell Biol. 2000;20:6105–6113. doi: 10.1128/mcb.20.16.6105-6113.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rowinsky E K, Windle J J, Von Hoff D D. J Clin Oncol. 1999;17:3631–3652. doi: 10.1200/JCO.1999.17.11.3631. [DOI] [PubMed] [Google Scholar]
- 16.Bernhard E J, Kao G, Cox A D, Sebti S M, Hamilton A D, Muschel R J, McKenna W G. Cancer Res. 1996;56:1727–1730. [PubMed] [Google Scholar]
- 17.Bernhard E J, McKenna W G, Hamilton A D, Sebti S M, Qian Y, Wu J M, Muschel R J. Cancer Res. 1998;58:1754–1761. [PubMed] [Google Scholar]
- 18.Pear W, Nolan G, Scott M, Baltimore D. Proc Natl Acad Sci USA. 1993;90:8392–8396. doi: 10.1073/pnas.90.18.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sakamuro D, Eviner V, Elliott K, Showe L, White E, Prendergast G C. Oncogene. 1995;11:2411–2418. [PubMed] [Google Scholar]
- 20.Rao L, Debbas M, Sabbatini P, Hockenbery D, Korsmeyer S, White E. Proc Natl Acad Sci USA. 1992;89:7742–7746. doi: 10.1073/pnas.89.16.7742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lowe S W, Ruley H E. Genes Dev. 1993;7:535–545. doi: 10.1101/gad.7.4.535. [DOI] [PubMed] [Google Scholar]
- 22.Lowe S W, Ruley H E, Jacks T, Housman D E. Cell. 1993;74:957–967. doi: 10.1016/0092-8674(93)90719-7. [DOI] [PubMed] [Google Scholar]
- 23.Prendergast G C, Oliff A. Semin Cancer Biol. 2000;10:443–452. doi: 10.1006/scbi.2000.0335. [DOI] [PubMed] [Google Scholar]
- 24.Du W, Lebowitz P, Prendergast G C. Mol Cell Biol. 1999;19:1831–1840. doi: 10.1128/mcb.19.3.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Prendergast G C, Khosravi-Far R, Solski P, Kurzawa H, Lebowitz P, Der C J. Oncogene. 1995;10:2289–2296. [PubMed] [Google Scholar]
- 26.Qiu R G, Chen J, McCormick F, Symons M. Proc Natl Acad Sci USA. 1995;92:11781–11785. doi: 10.1073/pnas.92.25.11781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fritz G, Kaina B. Biochem Biophys Res Commun. 2000;268:784–789. doi: 10.1006/bbrc.2000.2211. [DOI] [PubMed] [Google Scholar]
- 28.Lebowitz P F, Sakamuro D, Prendergast G C. Cancer Res. 1997;57:708–713. [PubMed] [Google Scholar]
- 29.Bershadsky A, Chausovsky A, Becker E, Lyubimova A, Geiger B. Curr Biol. 1996;6:1279–1289. doi: 10.1016/s0960-9822(02)70714-8. [DOI] [PubMed] [Google Scholar]
- 30.Kohl N E, Omer C A, Conner M W, Anthony N J, Davide J P, deSolms S J, Giuliani E A, Gomez R P, Graham S L, Hamilton K. Nat Med. 1995;1:792–797. doi: 10.1038/nm0895-792. [DOI] [PubMed] [Google Scholar]