

Abstract
The title compound, C11H13N5, is a Schiff base synthesized by the reaction of 4-amino-4H-1,2,4-triazole and 4-(dimethylamino)benzaldehyde. The dihedral angle between the benzene and triazole rings is 43.09 (11)°. The crystal structure displays weak C—H⋯N interactions.
Related literature
For the biological activity of triazole derivatives, see: Modzelewska & Kalabun (1999 ▶); Rollas et al. (1993 ▶); Todoulou et al. (1994 ▶); Demirbas et al. (2002 ▶); Kahveci et al. (2003 ▶). For 4-amino-1,2,4-triazole Schiff bases, see: Desenko & Khim (1995 ▶); Kargin et al. (1988 ▶).
Experimental
Crystal data
C11H13N5
M r = 215.26
Monoclinic,
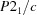
a = 10.3665 (16) Å
b = 11.1585 (19) Å
c = 9.5248 (12) Å
β = 90.257 (1)°
V = 1101.8 (3) Å3
Z = 4
Mo Kα radiation
μ = 0.08 mm−1
T = 298 K
0.52 × 0.15 × 0.11 mm
Data collection
Bruker SMART CCD area-detector diffractometer
Absorption correction: multi-scan (SADABS; Bruker, 2002 ▶) T min = 0.957, T max = 0.991
5465 measured reflections
1940 independent reflections
1184 reflections with I > 2σ(I)
R int = 0.062
Refinement
R[F 2 > 2σ(F 2)] = 0.047
wR(F 2) = 0.119
S = 1.00
1940 reflections
148 parameters
H-atom parameters constrained
Δρmax = 0.17 e Å−3
Δρmin = −0.18 e Å−3
Data collection: SMART (Bruker, 2002 ▶); cell refinement: SAINT (Bruker, 2002 ▶); data reduction: SAINT; program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: SHELXTL (Sheldrick, 2008 ▶); software used to prepare material for publication: SHELXTL.
Supplementary Material
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S1600536812014511/ff2062sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536812014511/ff2062Isup2.hkl
Supplementary material file. DOI: 10.1107/S1600536812014511/ff2062Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| C1—H1⋯N4i | 0.93 | 2.57 | 3.448 (3) | 157 |
| C2—H2⋯N2ii | 0.93 | 2.43 | 3.284 (3) | 152 |
| C11—H11B⋯N1iii | 0.96 | 2.60 | 3.543 (3) | 166 |
Symmetry codes: (i) 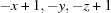 ; (ii)
; (ii) 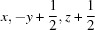 ; (iii)
; (iii) 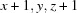 .
.
Acknowledgments
We thank the Instrumental Analysis Center of LiaoCheng University for the data collection on the Bruker SMART CCD facility.
supplementary crystallographic information
Comment
1,2,4-Triazole and their derivatives have been used as starting materials for synthesis of many heterocycles. The aroyl Schiff bases of 4-amino-1,2,4-triazole have received considerable attention over the past few decades (Desenko et al., 1995; Kargin et al., 1988; Modzelewska & Kalabun, 1999). In recent years, various 1,2,4-triazoles and their derivatives have been found to be associated with diverse pharmacological activities such as anticonvulsant, antifungal, anticancer, anti-inflammatory and antibacterial (Rollas et al., 1993; Todoulou et al., 1994). The present X-ray crystal structure analysis was undertaken in order to study the stereochemistry and crystal packing of the title compound (I).
The molecular structure and the atom-numbering scheme of the title compound are shown in Fig. 1. In the molecule, all bond lengths and angles are normal. As shown in Fig 1, the title compound is composed of two planar segments. One segment is a triazole ring, which is contains N3, C1, N2, C2, N1, and another segment is a benzene ring. The dihedral angle between the two planar segments is 43.09 (11)°. In the triazole ring, the N1=C2 and N2=C1 bonds display double-bond character, with bond distances of 1.303 (3) and 1.295 (2) Å, respectively.
Experimental
A mixture of 4-amino-4H-1,2,4-triazole 1 (0.51 g, 6 mmol) and 4-Dimethylaminobenzaldehyde (0.85 g, 6 mmol) was reacted in 40 ml ethanol at 353 K for 0.3 h. Single crystals suitable for X-ray diffraction analysis were obtained by slow evaporation of the ethanol solution.
Refinement
The H atoms were positioned geometrically, with C—H distances of 0.93–0.96 Å for aromatic, methylene and methyl H atoms, respectively, and Uiso(H) = 1.2–1.5Ueq of the parent atom.
Figures
Fig. 1.

The molecular structure of the title compound, showing 30% displacement ellipsoids for the non-hydrogen atoms. Hydrogen atoms are drawn as spheres of arbitrary radius.
Crystal data
| C11H13N5 | F(000) = 456 |
| Mr = 215.26 | Dx = 1.298 Mg m−3 |
| Monoclinic, P21/c | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -P 2ybc | Cell parameters from 1214 reflections |
| a = 10.3665 (16) Å | θ = 2.7–23.0° |
| b = 11.1585 (19) Å | µ = 0.08 mm−1 |
| c = 9.5248 (12) Å | T = 298 K |
| β = 90.257 (1)° | Cuboid, colourless |
| V = 1101.8 (3) Å3 | 0.52 × 0.15 × 0.11 mm |
| Z = 4 |
Data collection
| Bruker SMART CCD area-detector diffractometer | 1940 independent reflections |
| Radiation source: fine-focus sealed tube | 1184 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.062 |
| phi and ω scans | θmax = 25.0°, θmin = 2.0° |
| Absorption correction: multi-scan (SADABS; Bruker, 2002) | h = −12→12 |
| Tmin = 0.957, Tmax = 0.991 | k = −13→9 |
| 5465 measured reflections | l = −11→11 |
Refinement
| Refinement on F2 | Secondary atom site location: difference Fourier map |
| Least-squares matrix: full | Hydrogen site location: inferred from neighbouring sites |
| R[F2 > 2σ(F2)] = 0.047 | H-atom parameters constrained |
| wR(F2) = 0.119 | w = 1/[σ2(Fo2) + (0.0462P)2] where P = (Fo2 + 2Fc2)/3 |
| S = 1.00 | (Δ/σ)max < 0.001 |
| 1940 reflections | Δρmax = 0.17 e Å−3 |
| 148 parameters | Δρmin = −0.18 e Å−3 |
| 0 restraints | Extinction correction: SHELXL97 (Sheldrick, 2008), Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 |
| Primary atom site location: structure-invariant direct methods | Extinction coefficient: 0.086 (5) |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| N1 | 0.20167 (17) | 0.23656 (19) | 0.4358 (2) | 0.0665 (6) | |
| N2 | 0.24339 (17) | 0.13578 (17) | 0.36475 (19) | 0.0599 (5) | |
| N3 | 0.34969 (14) | 0.14765 (14) | 0.56204 (17) | 0.0461 (4) | |
| N4 | 0.43764 (14) | 0.11434 (15) | 0.66817 (17) | 0.0498 (5) | |
| N5 | 0.87803 (15) | 0.16192 (15) | 1.14369 (18) | 0.0590 (5) | |
| C1 | 0.33047 (19) | 0.08539 (19) | 0.4428 (2) | 0.0540 (6) | |
| H1 | 0.3740 | 0.0154 | 0.4195 | 0.065* | |
| C2 | 0.26690 (19) | 0.2406 (2) | 0.5530 (2) | 0.0588 (6) | |
| H2 | 0.2576 | 0.2996 | 0.6212 | 0.071* | |
| C3 | 0.49452 (17) | 0.20274 (18) | 0.7270 (2) | 0.0469 (5) | |
| H3 | 0.4751 | 0.2797 | 0.6959 | 0.056* | |
| C4 | 0.58726 (17) | 0.18934 (17) | 0.8392 (2) | 0.0437 (5) | |
| C5 | 0.61841 (19) | 0.07978 (19) | 0.8997 (2) | 0.0539 (6) | |
| H5 | 0.5744 | 0.0114 | 0.8707 | 0.065* | |
| C6 | 0.7122 (2) | 0.06956 (19) | 1.0008 (2) | 0.0581 (6) | |
| H6 | 0.7301 | −0.0051 | 1.0396 | 0.070* | |
| C7 | 0.78200 (17) | 0.17091 (18) | 1.0468 (2) | 0.0458 (5) | |
| C8 | 0.74731 (17) | 0.28123 (18) | 0.9884 (2) | 0.0491 (6) | |
| H8 | 0.7890 | 0.3504 | 1.0186 | 0.059* | |
| C9 | 0.65320 (18) | 0.28964 (18) | 0.8876 (2) | 0.0477 (6) | |
| H9 | 0.6329 | 0.3644 | 0.8505 | 0.057* | |
| C10 | 0.9193 (3) | 0.0473 (2) | 1.1988 (3) | 0.0853 (9) | |
| H10A | 0.8525 | 0.0144 | 1.2566 | 0.128* | |
| H10B | 0.9963 | 0.0579 | 1.2539 | 0.128* | |
| H10C | 0.9366 | −0.0064 | 1.1224 | 0.128* | |
| C11 | 0.9429 (2) | 0.2687 (2) | 1.1945 (2) | 0.0691 (7) | |
| H11A | 0.9861 | 0.3074 | 1.1180 | 0.104* | |
| H11B | 1.0049 | 0.2468 | 1.2652 | 0.104* | |
| H11C | 0.8806 | 0.3225 | 1.2340 | 0.104* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| N1 | 0.0658 (12) | 0.0829 (14) | 0.0507 (13) | 0.0148 (10) | −0.0210 (10) | −0.0039 (11) |
| N2 | 0.0641 (11) | 0.0706 (13) | 0.0448 (12) | −0.0003 (10) | −0.0169 (9) | 0.0003 (10) |
| N3 | 0.0464 (9) | 0.0559 (11) | 0.0359 (10) | −0.0001 (8) | −0.0119 (8) | 0.0019 (9) |
| N4 | 0.0521 (10) | 0.0569 (11) | 0.0403 (11) | 0.0002 (8) | −0.0165 (8) | 0.0024 (9) |
| N5 | 0.0578 (10) | 0.0678 (13) | 0.0512 (12) | 0.0076 (10) | −0.0251 (9) | −0.0019 (10) |
| C1 | 0.0651 (13) | 0.0536 (13) | 0.0432 (14) | −0.0038 (11) | −0.0132 (11) | −0.0005 (11) |
| C2 | 0.0547 (13) | 0.0712 (16) | 0.0503 (15) | 0.0103 (12) | −0.0140 (11) | −0.0079 (12) |
| C3 | 0.0441 (11) | 0.0530 (13) | 0.0436 (14) | 0.0054 (10) | −0.0059 (10) | −0.0006 (11) |
| C4 | 0.0454 (11) | 0.0481 (12) | 0.0374 (13) | 0.0048 (9) | −0.0075 (9) | −0.0018 (10) |
| C5 | 0.0642 (13) | 0.0516 (13) | 0.0456 (14) | −0.0066 (10) | −0.0164 (11) | −0.0038 (11) |
| C6 | 0.0744 (14) | 0.0508 (13) | 0.0488 (14) | 0.0075 (11) | −0.0204 (12) | 0.0025 (11) |
| C7 | 0.0472 (11) | 0.0536 (13) | 0.0364 (13) | 0.0077 (10) | −0.0068 (9) | −0.0039 (10) |
| C8 | 0.0462 (11) | 0.0535 (13) | 0.0475 (14) | −0.0016 (10) | −0.0104 (10) | −0.0063 (11) |
| C9 | 0.0489 (11) | 0.0493 (13) | 0.0448 (14) | 0.0051 (9) | −0.0099 (10) | −0.0002 (10) |
| C10 | 0.0898 (17) | 0.089 (2) | 0.0766 (19) | 0.0287 (15) | −0.0392 (15) | 0.0001 (16) |
| C11 | 0.0548 (13) | 0.0922 (18) | 0.0602 (17) | −0.0066 (12) | −0.0212 (12) | −0.0041 (14) |
Geometric parameters (Å, º)
| N1—C2 | 1.303 (3) | C4—C9 | 1.389 (3) |
| N1—N2 | 1.383 (2) | C5—C6 | 1.370 (3) |
| N2—C1 | 1.295 (2) | C5—H5 | 0.9300 |
| N3—C1 | 1.346 (2) | C6—C7 | 1.411 (3) |
| N3—C2 | 1.349 (2) | C6—H6 | 0.9300 |
| N3—N4 | 1.408 (2) | C7—C8 | 1.397 (3) |
| N4—C3 | 1.277 (2) | C8—C9 | 1.369 (3) |
| N5—C7 | 1.358 (2) | C8—H8 | 0.9300 |
| N5—C10 | 1.446 (3) | C9—H9 | 0.9300 |
| N5—C11 | 1.450 (3) | C10—H10A | 0.9600 |
| C1—H1 | 0.9300 | C10—H10B | 0.9600 |
| C2—H2 | 0.9300 | C10—H10C | 0.9600 |
| C3—C4 | 1.442 (3) | C11—H11A | 0.9600 |
| C3—H3 | 0.9300 | C11—H11B | 0.9600 |
| C4—C5 | 1.389 (3) | C11—H11C | 0.9600 |
| C2—N1—N2 | 106.54 (17) | C5—C6—C7 | 120.83 (19) |
| C1—N2—N1 | 106.92 (17) | C5—C6—H6 | 119.6 |
| C1—N3—C2 | 104.55 (17) | C7—C6—H6 | 119.6 |
| C1—N3—N4 | 124.23 (17) | N5—C7—C8 | 121.48 (18) |
| C2—N3—N4 | 131.21 (17) | N5—C7—C6 | 121.64 (18) |
| C3—N4—N3 | 114.03 (17) | C8—C7—C6 | 116.88 (18) |
| C7—N5—C10 | 121.78 (18) | C9—C8—C7 | 121.39 (19) |
| C7—N5—C11 | 120.24 (17) | C9—C8—H8 | 119.3 |
| C10—N5—C11 | 117.98 (18) | C7—C8—H8 | 119.3 |
| N2—C1—N3 | 111.1 (2) | C8—C9—C4 | 121.70 (19) |
| N2—C1—H1 | 124.4 | C8—C9—H9 | 119.1 |
| N3—C1—H1 | 124.4 | C4—C9—H9 | 119.1 |
| N1—C2—N3 | 110.9 (2) | N5—C10—H10A | 109.5 |
| N1—C2—H2 | 124.6 | N5—C10—H10B | 109.5 |
| N3—C2—H2 | 124.6 | H10A—C10—H10B | 109.5 |
| N4—C3—C4 | 123.37 (19) | N5—C10—H10C | 109.5 |
| N4—C3—H3 | 118.3 | H10A—C10—H10C | 109.5 |
| C4—C3—H3 | 118.3 | H10B—C10—H10C | 109.5 |
| C5—C4—C9 | 117.30 (18) | N5—C11—H11A | 109.5 |
| C5—C4—C3 | 123.47 (18) | N5—C11—H11B | 109.5 |
| C9—C4—C3 | 119.19 (18) | H11A—C11—H11B | 109.5 |
| C6—C5—C4 | 121.84 (19) | N5—C11—H11C | 109.5 |
| C6—C5—H5 | 119.1 | H11A—C11—H11C | 109.5 |
| C4—C5—H5 | 119.1 | H11B—C11—H11C | 109.5 |
| C2—N1—N2—C1 | 0.1 (2) | C3—C4—C5—C6 | 176.31 (19) |
| C1—N3—N4—C3 | 143.42 (19) | C4—C5—C6—C7 | −0.5 (3) |
| C2—N3—N4—C3 | −38.0 (3) | C10—N5—C7—C8 | −176.5 (2) |
| N1—N2—C1—N3 | −0.3 (2) | C11—N5—C7—C8 | 3.2 (3) |
| C2—N3—C1—N2 | 0.5 (2) | C10—N5—C7—C6 | 3.8 (3) |
| N4—N3—C1—N2 | 179.38 (16) | C11—N5—C7—C6 | −176.49 (19) |
| N2—N1—C2—N3 | 0.2 (2) | C5—C6—C7—N5 | −177.92 (19) |
| C1—N3—C2—N1 | −0.4 (2) | C5—C6—C7—C8 | 2.4 (3) |
| N4—N3—C2—N1 | −179.24 (17) | N5—C7—C8—C9 | 177.99 (18) |
| N3—N4—C3—C4 | 179.31 (16) | C6—C7—C8—C9 | −2.3 (3) |
| N4—C3—C4—C5 | −3.8 (3) | C7—C8—C9—C4 | 0.4 (3) |
| N4—C3—C4—C9 | 173.89 (18) | C5—C4—C9—C8 | 1.5 (3) |
| C9—C4—C5—C6 | −1.4 (3) | C3—C4—C9—C8 | −176.33 (18) |
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| C1—H1···N4i | 0.93 | 2.57 | 3.448 (3) | 157 |
| C2—H2···N2ii | 0.93 | 2.43 | 3.284 (3) | 152 |
| C11—H11B···N1iii | 0.96 | 2.60 | 3.543 (3) | 166 |
Symmetry codes: (i) −x+1, −y, −z+1; (ii) x, −y+1/2, z+1/2; (iii) x+1, y, z+1.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: FF2062).
References
- Bruker (2002). SADABS, SAINT and SMART Bruker AXS Inc., Madison, Wisconsin, USA.
- Demirbas, N., Ugurluoglu, R. & Demirbas, A. (2002). Bioorg. Med. Chem. 10, 3717–3723. [DOI] [PubMed]
- Desenko, S. M. & Khim, G. S. (1995). Chem. Heterocycl. Comput pp. 2–24.
- Kahveci, B., Bekircan, O., Serdar, M. & Ikizler, A. A. (2003). Indian J. Chem. Sect. B, 42, 1527–1530.
- Kargin, Y. M., Kitaeva, M. Y., Latypova, V. Z., Vafina, A. A., Zaripova, R. M. & Il’yasov, A. V. (1988). Izv. Akad. Nauk SSSR Ser. Khim. 3, 607–611.
- Modzelewska, B. & Kalabun, J. (1999). Pharmazie, 54, 503–505. [PubMed]
- Rollas, S., Kalyoncuoglu, N., Sur-Altiner, D. & Yegenoglu, Y. (1993). Pharmazie, 48, 308–309. [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Todoulou, O. G., Papadaki-Valiraki, A. E., Ikeda, S. & Clercq, E. D. (1994). Eur. J. Med. Chem. 29, 611–620.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S1600536812014511/ff2062sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536812014511/ff2062Isup2.hkl
Supplementary material file. DOI: 10.1107/S1600536812014511/ff2062Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report