

Abstract
In the title compound, C17H16O6, the two methyl salicylate moieties are related by crystallographic twofold rotational symmetry with the two benzene rings close to being perpendicular [inter-ring dihedral angle = 86.6 (8)°]. Intramolecular phenolic O—H⋯O hydrogen bonds with carboxyl O-atom acceptors are present, with these groups also involved in centrosymmetric cyclic intermolecular O—H⋯O hydrogen-bonding associations [graph set R 2 2(4)], giving infinite chains extending across (101).
Related literature
For the chemistry and applications of methylene bisphenol derivatives, see: Ogata et al. (1975 ▶); Méric et al. (1993 ▶); Shrestha et al. (2007 ▶); Cameron et al. (2002 ▶). For the preparation, see: Cushman & Kanamathareddy (1990 ▶); Méric et al. (1993 ▶). For the structures of similar compounds, see: Lu et al. (2011 ▶); Zhang et al. (2009 ▶); Liu et al. (2009 ▶). For graph-set analysis, see Etter et al. (1990 ▶). For bond-length data, see: Allen et al. (1987 ▶).
Experimental
Crystal data
C17H16O6
M r = 316.31
Monoclinic,
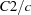
a = 20.4168 (13) Å
b = 4.9300 (3) Å
c = 15.5470 (12) Å
β = 111.290 (3)°
V = 1458.08 (17) Å3
Z = 4
Mo Kα radiation
μ = 0.11 mm−1
T = 150 K
0.38 × 0.30 × 0.24 mm
Data collection
Bruker SMART CCD-detector diffractometer
Absorption correction: multi-scan (SADABS; Bruker, 2008 ▶) T min = 0.953, T max = 0.970
8440 measured reflections
1756 independent reflections
1568 reflections with I > 2σ(I)
R int = 0.025
Refinement
R[F 2 > 2σ(F 2)] = 0.040
wR(F 2) = 0.113
S = 0.96
1756 reflections
109 parameters
H atoms treated by a mixture of independent and constrained refinement
Δρmax = 0.23 e Å−3
Δρmin = −0.33 e Å−3
Data collection: SMART (Bruker, 2008 ▶); cell refinement: SAINT (Bruker, 2008 ▶); data reduction: SAINT; program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: PLATON (Spek, 2009 ▶); software used to prepare material for publication: SHELXL97.
Supplementary Material
Crystal structure: contains datablock(s) global, I. DOI: 10.1107/S1600536812015760/zs2198sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536812015760/zs2198Isup2.hkl
Supplementary material file. DOI: 10.1107/S1600536812015760/zs2198Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| O1—H1⋯O2 | 0.87 (2) | 1.87 (2) | 2.6457 (12) | 147 (2) |
| O1—H1⋯O2i | 0.87 (2) | 2.32 (1) | 3.0067 (11) | 134 (9) |
Symmetry code: (i) 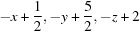 .
.
Acknowledgments
Thanks are due to the University of Aveiro and the Portuguese Fundação para a Ciência e a Tecnologia (FCT) for funding the Organic Chemistry Research Unit (project PEst-C/QUI/UI0062/2011) and the CICECO Associate Laboratory (PEst-C/CTM/LA0011/2011). SG also thanks the FCT for a postdoctoral grant (SFRH/BPD/70702/2010).
supplementary crystallographic information
Comment
The title compound C17H16O6 was first reported as a building block for polymer synthesis (Ogata et al., 1975). It is a useful precursor for organic polymers, metal-organic frameworks, cage compounds (Méric et al., 1993) and biologically active compounds (Shrestha et al., 2007; Cameron et al., 2002). In the title compound (Fig. 1), the two methyl salicylate moieties are related by crystallographic twofold rotational symmetry with the two phenyl rings close to perpendicular [inter-ring dihedral angle = 86.6 (8)°]. Bond lengths and angles are within normal ranges (Allen et al., 1987). Intramolecular phenolic O—H···O hydrogen bonds with carboxyl O-atom acceptors are present, with these groups also involved in centrosymmetric cyclic intermolecular hydrogen-bonding associations [graph set R22(4) (Etter et al., 1990)], making the ester group essentially coplanar with the phenyl ring [torsion angle C1—C6—C7—O3, 178.64 (9)°]. The molecules are involved in centrosymmetric cyclic intermolecular phenolic O—H···Ocarboxyl hydrogen-bonding associations [graph set R22(4) giving infinite chains extending across (101) (Figs. 2, 3).
Experimental
The title compound was prepared in two steps starting with salicylic acid. 5,5'-Methylenebis(salicylic acid) was prepared according to a known procedure (Cushman et al., 1990), and was then esterified with methanol and a catalytic amount of sulfuric acid (Méric et al., 1993). Slow evaporation of a saturated solution in dichloromethane gave single crystals suitable for X-ray diffraction.
Refinement
The phenolic H-atom (H1) was located in a difference Fourier map and both positional and isotropic displacement parameters were refined. All other H-atoms were placed in geometrically idealized positions and refined using a riding model with C—H = 0.95 Å (aromatic), 0.98 Å (methylene) or 0.97 Å (methyl) and Uiso(H) = 1.2Ueq(C) (aromatic or methylene) or Uiso(H) = 1.5Ueq(C) (methyl).
Figures
Fig. 1.

The molecular structure of the title compound showing atom numbering and displacement ellipsoids drawn at the 30% probability level. The intramolecular hydrogen bonds are shown as dashed lines. Symmetry code: (i) -x + 1, y, -z + 1/2.
Fig. 2.

The one-dimensional hydrogen-bonded chains in the title compound, with hydrogen bonds shown as dashed lines. Displacement ellipsoids are drawn at the 30% probability level.
Fig. 3.

The packing of the title compound in the unit cell viewed down the b axis, with hydrogen bonds and other intermolecular interactions shown as dashed lines.
Crystal data
| C17H16O6 | F(000) = 664 |
| Mr = 316.31 | Dx = 1.441 Mg m−3 |
| Monoclinic, C2/c | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -C 2yc | Cell parameters from 8440 reflections |
| a = 20.4168 (13) Å | θ = 2.8–27.9° |
| b = 4.9300 (3) Å | µ = 0.11 mm−1 |
| c = 15.5470 (12) Å | T = 150 K |
| β = 111.290 (3)° | Block, colourless |
| V = 1458.08 (17) Å3 | 0.38 × 0.30 × 0.24 mm |
| Z = 4 |
Data collection
| Bruker SMART CCD-detector diffractometer | 1756 independent reflections |
| Radiation source: fine-focus sealed tube | 1568 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.025 |
| ω and φ scans | θmax = 27.9°, θmin = 4.1° |
| Absorption correction: multi-scan (SADABS; Bruker, 2008) | h = −26→21 |
| Tmin = 0.953, Tmax = 0.970 | k = −6→6 |
| 8440 measured reflections | l = −19→20 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.040 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.113 | H atoms treated by a mixture of independent and constrained refinement |
| S = 0.96 | w = 1/[σ2(Fo2) + (0.0724P)2 + 0.9516P] where P = (Fo2 + 2Fc2)/3 |
| 1756 reflections | (Δ/σ)max = 0.049 |
| 109 parameters | Δρmax = 0.23 e Å−3 |
| 0 restraints | Δρmin = −0.33 e Å−3 |
Special details
| Geometry. All s.u.'s (except the s.u. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell s.u.'s are taken into account individually in the estimation of s.u.'s in distances, angles and torsion angles; correlations between s.u.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell s.u.'s is used for estimating s.u.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > 2σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | Occ. (<1) | |
| H1 | 0.2434 (10) | 1.009 (5) | 0.9251 (15) | 0.070 (6)* | |
| O2 | 0.18996 (4) | 1.12860 (16) | 0.99304 (5) | 0.0260 (2) | |
| O1 | 0.24736 (4) | 0.89366 (17) | 0.88455 (6) | 0.0265 (2) | |
| O3 | 0.08604 (4) | 0.98089 (17) | 0.99095 (6) | 0.0284 (2) | |
| C5 | 0.07384 (5) | 0.6081 (2) | 0.85532 (7) | 0.0212 (2) | |
| H5 | 0.0372 | 0.6288 | 0.8789 | 0.025* | |
| C6 | 0.13366 (5) | 0.7717 (2) | 0.89054 (7) | 0.0189 (2) | |
| C2 | 0.18118 (6) | 0.5483 (2) | 0.78810 (8) | 0.0269 (3) | |
| H2 | 0.2178 | 0.5240 | 0.7648 | 0.032* | |
| C7 | 0.14042 (5) | 0.9762 (2) | 0.96221 (7) | 0.0196 (2) | |
| C3 | 0.12105 (6) | 0.3904 (2) | 0.75449 (8) | 0.0277 (3) | |
| H3 | 0.1170 | 0.2602 | 0.7078 | 0.033* | |
| C1 | 0.18813 (5) | 0.7425 (2) | 0.85599 (7) | 0.0212 (2) | |
| C4 | 0.06631 (5) | 0.4174 (2) | 0.78730 (7) | 0.0238 (2) | |
| C9 | 0.0000 | 0.2479 (3) | 0.7500 | 0.0296 (4) | |
| H9A | −0.0029 | 0.1295 | 0.7999 | 0.036* | 0.50 |
| H9B | 0.0029 | 0.1295 | 0.7001 | 0.036* | 0.50 |
| C8 | 0.09263 (7) | 1.1785 (3) | 1.06230 (9) | 0.0350 (3) | |
| H8A | 0.0510 | 1.1700 | 1.0796 | 0.053* | |
| H8B | 0.0966 | 1.3604 | 1.0393 | 0.053* | |
| H8C | 0.1347 | 1.1389 | 1.1165 | 0.053* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| O2 | 0.0226 (4) | 0.0255 (4) | 0.0290 (4) | −0.0081 (3) | 0.0084 (3) | −0.0051 (3) |
| O1 | 0.0221 (4) | 0.0265 (4) | 0.0335 (5) | −0.0035 (3) | 0.0132 (3) | −0.0013 (3) |
| O3 | 0.0247 (4) | 0.0324 (5) | 0.0315 (4) | −0.0085 (3) | 0.0142 (3) | −0.0101 (3) |
| C5 | 0.0180 (4) | 0.0180 (5) | 0.0235 (5) | 0.0006 (4) | 0.0027 (4) | 0.0024 (4) |
| C6 | 0.0178 (4) | 0.0171 (5) | 0.0188 (5) | 0.0007 (3) | 0.0031 (4) | 0.0023 (4) |
| C2 | 0.0289 (5) | 0.0259 (5) | 0.0266 (5) | 0.0049 (4) | 0.0110 (4) | 0.0009 (4) |
| C7 | 0.0175 (4) | 0.0200 (5) | 0.0191 (5) | −0.0013 (3) | 0.0043 (4) | 0.0025 (4) |
| C3 | 0.0335 (6) | 0.0207 (5) | 0.0235 (5) | 0.0050 (4) | 0.0039 (4) | −0.0024 (4) |
| C1 | 0.0200 (5) | 0.0199 (5) | 0.0218 (5) | 0.0014 (4) | 0.0052 (4) | 0.0045 (4) |
| C4 | 0.0227 (5) | 0.0157 (5) | 0.0242 (5) | 0.0020 (4) | −0.0021 (4) | 0.0026 (4) |
| C9 | 0.0247 (7) | 0.0162 (7) | 0.0350 (8) | 0.000 | −0.0045 (6) | 0.000 |
| C8 | 0.0396 (7) | 0.0382 (7) | 0.0338 (6) | −0.0085 (5) | 0.0211 (5) | −0.0124 (5) |
Geometric parameters (Å, º)
| O2—C7 | 1.2104 (13) | C2—C1 | 1.3938 (15) |
| O1—C1 | 1.3508 (13) | C2—H2 | 0.9500 |
| O1—H1 | 0.87 (2) | C3—C4 | 1.3932 (17) |
| O3—C7 | 1.3390 (12) | C3—H3 | 0.9500 |
| O3—C8 | 1.4455 (14) | C4—C9 | 1.5157 (13) |
| C5—C4 | 1.3811 (15) | C9—C4i | 1.5157 (13) |
| C5—C6 | 1.3989 (14) | C9—H9A | 0.9900 |
| C5—H5 | 0.9500 | C9—H9B | 0.9900 |
| C6—C1 | 1.4071 (14) | C8—H8A | 0.9800 |
| C6—C7 | 1.4715 (14) | C8—H8B | 0.9800 |
| C2—C3 | 1.3857 (16) | C8—H8C | 0.9800 |
| C1—O1—H1 | 106.9 (13) | O1—C1—C6 | 123.74 (10) |
| C7—O3—C8 | 114.23 (8) | C2—C1—C6 | 118.82 (10) |
| C4—C5—C6 | 122.07 (10) | C5—C4—C3 | 117.65 (10) |
| C4—C5—H5 | 119.0 | C5—C4—C9 | 120.26 (10) |
| C6—C5—H5 | 119.0 | C3—C4—C9 | 122.08 (9) |
| C5—C6—C1 | 119.36 (9) | C4—C9—C4i | 113.07 (12) |
| C5—C6—C7 | 121.34 (9) | C4—C9—H9A | 109.0 |
| C1—C6—C7 | 119.30 (9) | C4i—C9—H9A | 109.0 |
| C3—C2—C1 | 120.27 (10) | C4—C9—H9B | 109.0 |
| C3—C2—H2 | 119.9 | C4i—C9—H9B | 109.0 |
| C1—C2—H2 | 119.9 | H9A—C9—H9B | 107.8 |
| O2—C7—O3 | 122.15 (9) | O3—C8—H8A | 109.5 |
| O2—C7—C6 | 124.07 (9) | O3—C8—H8B | 109.5 |
| O3—C7—C6 | 113.78 (8) | H8A—C8—H8B | 109.5 |
| C2—C3—C4 | 121.81 (10) | O3—C8—H8C | 109.5 |
| C2—C3—H3 | 119.1 | H8A—C8—H8C | 109.5 |
| C4—C3—H3 | 119.1 | H8B—C8—H8C | 109.5 |
| O1—C1—C2 | 117.44 (9) | ||
| C8—O3—C7—O2 | −1.07 (15) | C3—C4—C5—C6 | 0.43 (15) |
| C8—O3—C7—C6 | 179.17 (9) | C9—C4—C5—C6 | −178.93 (9) |
| O1—C1—C2—C3 | −178.69 (10) | C3—C4—C9—C4i | −123.32 (10) |
| C6—C1—C2—C3 | 1.01 (16) | C5—C4—C9—C4i | 56.01 (11) |
| O1—C1—C6—C5 | 178.95 (10) | C4—C5—C6—C1 | 0.00 (16) |
| O1—C1—C6—C7 | −0.29 (15) | C4—C5—C6—C7 | 179.24 (10) |
| C2—C1—C6—C5 | −0.73 (15) | C1—C6—C7—O2 | 1.60 (16) |
| C2—C1—C6—C7 | −179.97 (10) | C1—C6—C7—O3 | −178.64 (9) |
| C1—C2—C3—C4 | −0.58 (17) | C5—C6—C7—O2 | −177.63 (10) |
| C2—C3—C4—C5 | −0.15 (16) | C5—C6—C7—O3 | 2.13 (14) |
| C2—C3—C4—C9 | 179.20 (10) |
Symmetry code: (i) −x, y, −z+3/2.
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| O1—H1···O2 | 0.87 (2) | 1.87 (2) | 2.6457 (12) | 147 (2) |
| O1—H1···O2ii | 0.87 (2) | 2.32 (1) | 3.0067 (11) | 134 (9) |
Symmetry code: (ii) −x+1/2, −y+5/2, −z+2.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: ZS2198).
References
- Allen, F. H., Kennard, O., Watson, D. G., Brammer, L., Orpen, A. G. & Taylor, R. (1987). J. Chem. Soc. Perkin Trans. 2, pp. 1–19.
- Bruker (2008). SMART, SAINT and SADABS Bruker AXS Inc., Madison, Wisconsin, USA.
- Cameron, K. S., Fielding, L., Mason, R., Muir, A. W., Rees, D. C., Thorn, S. & Zhang, M.-Q. (2002). Bioorg. Med. Chem. Lett. 12, 753–755. [DOI] [PubMed]
- Cushman, M. & Kanamathareddy, S. (1990). Tetrahedron, 46, 1491–1498.
- Etter, M. C., MacDonald, J. C. & Bernstein, J. (1990). Acta Cryst. B46, 256–262. [DOI] [PubMed]
- Liu, Y.-Z., Li, Y.-X., Zhang, L. & Li, X. (2009). Acta Cryst. E65, o1716. [DOI] [PMC free article] [PubMed]
- Lu, J., Han, L.-W., Lin, J.-X. & Cao, R. (2011). Cryst. Growth Des. 11, 3551–3557.
- Méric, R., Vigneron, J.-P. & Lehn, J.-M. (1993). J. Chem. Soc. Chem. Commun. pp. 129–131.
- Ogata, N., Sanui, K., Kanasugi, K. & Ohira, N. (1975). Polym. J. 7, 544–549.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Shrestha, S., Bhattarai, B. R., Chang, K. J., Lee, K.-H. & Cho, H. (2007). Bioorg. Med. Chem. Lett. 17, 2760–2764. [DOI] [PubMed]
- Spek, A. L. (2009). Acta Cryst. D65, 148–155. [DOI] [PMC free article] [PubMed]
- Zhang, Z.-H., Tan, X. & Chen, S.-C. (2009). Acta Cryst. C65, o457–o459. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) global, I. DOI: 10.1107/S1600536812015760/zs2198sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536812015760/zs2198Isup2.hkl
Supplementary material file. DOI: 10.1107/S1600536812015760/zs2198Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report