

Abstract
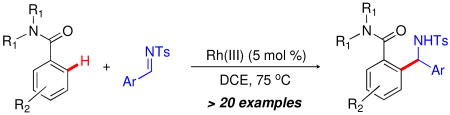
Rhodium-catalyzed addition of benzamide C-H bonds to a range of aromatic N-sulfonyl aldimines has been developed and proceeds with high functional group compatibility. The synthetic utility of the resulting branched amine products has also been demonstrated by the preparation of isoindoline and isoindolinone frameworks.
Transition-metal catalyzed methods for the direct functionalization of C−H bonds have emerged as powerful alternatives to more traditional reactions that rely heavily on stoichiometric substrate pre-activation. While significant progress has been documented for the addition of sp2 C−H bonds across alkenes and alkynes,1 the identification of analogous methods for the arylation of C−N multiple bonds, such as imines,2 isocyanates,3 nitriles,4 and isocyanides,5 have seen considerably less development. The ability to selectively install nitrogen-based functional groups into molecules through the direct addition of C−H bonds to C−N π-bonds represents a powerful method for rapid and convergent amine synthesis.
Recently, the synthesis of α-branched amines by the Rh(III)-catalyzed addition of 2-arylpyridine C−H bonds to N-Boc and N-sulfonyl imines has been reported.2a,b While these studies serve as excellent proof-of-principle models for C−H additions to C−N multiple bonds, the pyridyl directing group is of limited utility for the synthesis of biologically interesting drugs and natural products. With this limitation in mind, we have focused our efforts on expanding the repertoire of directing groups that are effective for the Rh(III)-catalyzed addition of C−H bonds to imines. In particular, we have become interested in the use of benzamide derivatives as directing groups6 - an important motif that provides rapid access to organic frameworks that are well-represented in natural products and drugs. Herein, we report the preparation of α-branched amines using the Rh(III)-catalyzed, amide-directed arylation of aromatic N-sulfonyl imines. In addition, we have demonstrated the utility of these amine products for the preparation of isoindoline and isoindolinone frameworks.7
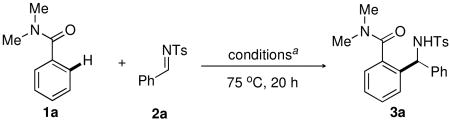
Our initial investigations focused on the identification of a suitable catalyst and reaction conditions for the regioselective coupling of N,N-dimethyl benzamide (1a) with N-tosyl imine 2a to afford the branched amine product 3a (Table 1). Although the use of 2.5 mol % of [Cp*RhCl2]2 proved unsuccessful in catalyzing the reaction (entry 1), employing a mixture of [Cp*RhCl2]2 (2.5 mol %) and AgSbF6 (5 mol %) in CH2Cl2 at 75 °C provided the desired adduct 3a in a 30% yield (entry 2). In comparison, the use of the pre-formed cationic Rh(III) precursor [Cp*Rh(MeCN)3](SbF6)2 showed negligible catalytic activity (entry 3), which we postulate is due to competitive coordination of the acetonitrile ligands with the N-tosyl imine to the Cp*Rh catalyst.8 Although the use of alternative solvents, such as THF and t-BuOH, provided inferior yields, employing DCE as the reaction solvent led to reactivity similar to that obsevred with CH2Cl2 and so DCE was used for all subsequent optimization studies due to its higher boiling point (entries 4 to 6). Further optimization studies showed that improved yields of 3a could be achieved under higher concentration conditions and that the stoichiometry of reactants had no effect on the reaction outcome (entries 7 and 8). In search of further yield improvements, a series of alternative halide abstracting reagents were screened in combination with [Cp*RhCl2]2 for the preparation of 3a (entries 9 to 12). From this survey it was observed that the use of the more non-coordinating tetrakis(pentafluorophenyl)borate9 anion, in place of SbF6, provided a modest increase in 3a with a 67% 1H NMR yield (entry 12).
Table 1. Catalyst and reaction optimization.
 | ||||
|---|---|---|---|---|
| entry | catalyst | solvent | concn (M) | yield (%)b |
| 1 | [Cp*RhCl2]2 | CH2Cl2 | 0.2 | <5 |
| 2 | [Cp*RhCl2]2, AgSbF6 | CH2Cl2 | 0.2 | 30 |
| 3 | [Cp*Rh(MeCN)3] (SbF6)2 | CH2Cl2 | 0.2 | <5 |
| 4 | [Cp*RhCl2]2, AgSbF6 | THF | 0.2 | 17 |
| 5 | [Cp*RhCl2]2, AgSbF6 | t-BuOH | 0.2 | <5 |
| 6 | [Cp*RhCl2]2, AgSbF6 | DCE | 0.2 | 32 |
| 7 | [Cp*RhCl2]2, AgSbF6 | DCE | 0.75 | 46 |
| 8c | [Cp*RhCl2]2, AgSbF6 | DCE | 0.75 | 45 |
| 9c | [Cp*RhCl2]2, AgOTf | DCE | 0.75 | 33 |
| 10c | [Cp*RhCl2]2, AgBF4 | DCE | 0.75 | 42 |
| 11c | [Cp*RhCl2]2, AgNTf2 | DCE | 0.75 | 53 |
| 12c | [Cp*RhCl2]2, AgB(C6F5)4 | DCE | 0.75 | 67 |
Conditions: 1a:2a = 2:1, 0.15 mmol scale, 5 mol % of Rh; Rh:Ag = 1:2; 75 °C for 20 h.
Determined by 1H NMR relative to 2,6- dimethoxytoluene as an internal standard.
1a:2a = 1:1.5.
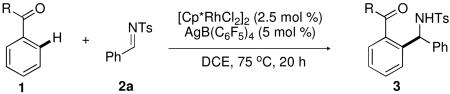
The influence of the electronic and steric properties of the amide directing group on the progress of the C−H arylation of imine 2a was next assessed (Table 2). Replacement of the N,N-dimethyl substituents in 1a with more bulky diethyl (1b) or dibenzyl (1c) groups resulted in a marked decrease in yield (entries 2 and 3, 40% and <5%, respectively). In addition, the use of a secondary amide directing group (1d), in place of the tertiary amide, provided only trace amine product (entry 4).
Table 2. Substrate scope for benzamide directing groupa.
 | ||||
|---|---|---|---|---|
| entry | R | yield (%)b | ||
| 1 | NMe2 (1a) | 58 | ||
| 2 | NEt2 (1b) | 40c | ||
| 3 | NBn2 (1c) | <5c | ||
| 4 | NHMe (1d) | <5c | ||
| 5 | morpholinyl (1e) | 19c | ||
| 6 | piperidinyl (1f) | 34 | ||
| 7 | pyrrolidinyl (1g) | 80 | ||
| 8 | 2-methyl pyrrolidinyl (1h) | 73d | ||
Conditions: 1:2a = 1:1.5, 0.15 mmol (0.75 M DCE) scale, 5 mol % of Rh, Rh:Ag = 1:2, 75 °C for 20 h.
Isolated yield.
Determined by 1H NMR relative to 2,6-dimethoxytoluene as an internal standard.
Diastereomeric ratio = 1:1.
Given the observed sensitivity of product formation to the steric profile of the directing group, a series of cyclic tertiary amides were surveyed (entries 5 to 8). The use of both morpholino (1e) and piperidinyl (1f) amides provided inferior yields of amine product (19% and 34%, respectively) relative to the N,N-dimethyl amide 1a. In contrast to these results, the pyrrolidine amide afforded significant gains in product yield, providing the α-branched amine product in an 80% isolated yield (entry 7). In a study by Tanaka and co-workers,6g a similar result was observed for the amide-directed, Rh(I)-catalyzed alkenylation of aromatic and α, β-unsaturated C−H bonds, where substitution of an N,N-dimethyl amide for a pyrrolidine amide provided dramatic reaction rate enhancement. In entry 8, substitution of the pyrrolidine amide with rac-2-methyl pyrrolidine (1h) afforded the desired product with activity similar to that observed with amide 1g; however, negligible diastereoselectivity was achieved for this transformation.
Having defined a highly effective catalyst system and reaction conditions for the addition of pyrrolidine benzamide 1g to N-tosyl imine 2a, we sought to further explore the reaction scope for pyrrolidine benzamides and a broad range of aromatic N-sulfonyl imines (Scheme 1). In addition to the use of N-tosyl imines, the more electronegative N-nosyl10 protecting group also served as an effective imine substituent under the standard reaction conditions providing 3i in a 85% isolated yield. While the use of electron-deficient aromatic imines possessing nitro (3j), carbomethoxy (3k), trifluoromethyl (3l), and chloro (3n) para substituents delivered branched amine products in good to excellent yields, imines with electron-donating substitutents provided only moderate yields (3o−3p). In addition, aromatic imines with meta- and ortho-fluoro groups were effective coupling partners under optimized conditons (3q−3r). The 2-thienyl-subtituted branched amine product 3s was isolated in a 58% yield from the corresponding heteroaromatic imine substrate.
Scheme 1. Substrate scopea.

aConditions: 1:2 = 1:1.5, 0.15 mmol (0.75 M DCE) scale, 5 mol % of Rh, Rh:Ag = 1:2, 75 °C for 20 h; yields represent isolated material. bDetermined by 1H NMR relative to 2,6-dimethoxytoluene as an internal standard.
Electron-rich or -poor pyrrolidine amides featuring meta- or para-substitution gave the desired branched amine products with good to excellent yields (3t−3x, 49-85%). Moreover, when empoying meta-substituted amide substrates exclusive reaction at the less-hindered C-H site occurred (3w−3x).
The aggregate collection of benzamide and imine substrates investigated also established a high level of functional group compatibility. Only the nitrile group (3m) resulted in poor yields, presumably due to competitive coordination to the metal. Otherwise, nitro (3i), ester (3k), keto (3v), chloro (3n), fluoro (3q), bromo (3w), thienyl (3s) methoxy (3t), and trifluoromethyl (3u) funtional groups were all compatible with the reaction conditions.
The product regiochemistry observed for the Rh(III)-catalyzed addition of benzamide substrates to imines is most consistent with a Rh-mediated C−H cleavage step directed by the Lewis basic amide group rather than a more traditional electrophilic aromatic substitution (EAS) mechanism. Specifically, exclusive ortho-functionalization is observed in all cases, as opposed to the expected meta-substitution for EAS of a deactivated amide substrate.
To demonstrate the synthetic versatility of the C−H addition products, several transformations of 3g were performed (Scheme 2). Using a previously developed protocol for the reduction of tertiary amides to the corresponding aldehydes,11 treatment of 3g with Cp2Zr(H)Cl (Schwartz's reagent) promoted reduction of the pyrrolidine amide to a cyclic aminal intermediate,12 which could be further reduced with NaBH4 in trifluoroacetic acid to the isoindoline 4 in reasonable overall yield over the two steps. The isoindolinone 5 was also obtained in excellent yield by the p-TsOH mediated transamidation/cyclization of 3g. Cleavage of the N-Ts protecting group in 5 was readily achieved by exposure to Na/naphthalene. Furthermore, reduction of in situ prepared 5 with LiAlH4 provided access to the corresponding alcohol 7 in a 83% yield, which is poised for further synthetic elaboration.
Scheme 2. Synthetic Transformations of Branched Amine 3g.

In summary, mixtures of [Cp*RhCl2]2 and AgB(C6F5)4 have been shown to catalyze the addition of N,N-dialkyl benzamide C−H bonds to a variety of aromatic N-sulfonyl imines to provide branched amine products. The obtained products were also shown to be easily transformed into isoindoline and isoindolinone frameworks. Further investigations to extend the reaction scope and illustrate applications of this process in organic synthesis are underway.
Supplementary Material
Acknowledgments
This work was supported by NIH Grant GM069559 (to J.A.E.). R.G.B. acknowledges funding from The Director, Office of Energy Research, Office of Basic Energy Sciences, Chemical Sciences Division, U.S. Department of Energy, under Contract DE-AC02-05CH11231. K.D.H. is grateful to the National Sciences and Engineering Research Council of Canada (NSERC) for a post-doctoral fellowship.
Footnotes
Supporting Information Available. Full experimental details and characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
Contributor Information
Robert G. Bergman, Email: rbergman@berkeley.edu.
Jonathan A. Ellman, Email: jonathan.ellman@yale.edu.
References
- 1.For recent reviews on C-H alkenylation/alkynylation, see: Wencel-Delord J, Dröge T, Liu F, Glorius F. Chem Soc Rev. 2011;40:4740. doi: 10.1039/c1cs15083a.Satoh T, Miura M. Chem –Eur J. 2010;16:11212. doi: 10.1002/chem.201001363.Colby DA, Bergman RG, Ellman JA. Chem Rev. 2010;110:624. doi: 10.1021/cr900005n.Karimi B, Behzadnia H, Elhamifar D, Akhavan PF, Zamani A. Synthesis. 2010:1399.Chen X, Engle KM, Wang DH, Yu JQ. Angew Chem Int, Ed. 2009;48:5094. doi: 10.1002/anie.200806273.
- 2.(a) Tsai AS, Tauchert ME, Bergman RG, Ellman JA. J Am Chem Soc. 2011;133:1248. doi: 10.1021/ja109562x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Li Y, Li BJ, Wang WH, Huang WP, Zhang XS, Chen K, Shi ZJ. Angew Chem, Int Ed. 2011;50:2115. doi: 10.1002/anie.201007464. [DOI] [PubMed] [Google Scholar]; (c) Qian B, Guo S, Shao J, Zhu Q, Yang L, Xia C, Huang H. J Am Chem Soc. 2010;132:3650. doi: 10.1021/ja910104n. [DOI] [PubMed] [Google Scholar]; (d) Gao K, Yoshikai N. Commun. 2012;48:4305. doi: 10.1039/c2cc31114c. [DOI] [PubMed] [Google Scholar]
- 3.(a) Hesp KD, Bergman RG, Ellman JA. J Am Chem Soc. 2011;133:11430. doi: 10.1021/ja203495c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kuninobu Y, Tokunaga Y, Kawata A, Takai K. J Am Chem Soc. 2006;128:202. doi: 10.1021/ja054216i. [DOI] [PubMed] [Google Scholar]; (c) Hong P, Yamazaki H, Sonogashira K, Hagihara N. Chem Lett. 1978:535. [Google Scholar]
- 4.Zhou C, Larock RC. J Am Chem Soc. 2004;126:2302. doi: 10.1021/ja038687l. [DOI] [PubMed] [Google Scholar]
- 5.Zhu C, Xie W, Falck JR. Chem –Eur J. 2011;17:12591. doi: 10.1002/chem.201102475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Selected recent examples of the use of benzamides as directing groups in C-H addition chemistry: Sharma S, Park E, Park J, Kim IS. Org Lett. 2012;14:906. doi: 10.1021/ol2034228.Patureau FW, Besset T, Glorius F. Angew Chem, Int Ed. 2011;50:1064. doi: 10.1002/anie.201006222.Du Y, Hyster TK, Rovis T. Chem Commun. 2011;47:12074. doi: 10.1039/c1cc15843k.Park J, Park E, Kim A, Lee Y, Chi KW, Kwak JH, Jung YH, Kim IS. Org Lett. 2011;13:4390. doi: 10.1021/ol201729w.Yoo EJ, Ma S, Mei TS, Chan KSL, Yu JQ. J Am Chem Soc. 2011;133:7652. doi: 10.1021/ja202563w.Wasa M, Worrell BT, Yu JQ. Angew Chem, Int Ed. 2010;49:1275. doi: 10.1002/anie.200906104.Shibata Y, Otake Y, Hirano M, Tanaka K. Org Lett. 2009;11:689. doi: 10.1021/ol802767s.
- 7.For selected examples of isoindoline/isoindolinone sturctural motifs in drugs and drug candidates, see: Li E, Jiang L, Guo L, Zhang H, Che Y. Bioorg Med Chem. 2008;16:7894. doi: 10.1016/j.bmc.2008.07.075.Stuk TL, Assink BK, Bates RC, Erdman DT, Fedij V, Jennings SM, Lassig JA, Smith RJ, Smith TL. Org Process Res Dev. 2003;7:851.Kukkola PJ, Bilci NA, Ikler T, Savage P, Shetty SS, DelGrande D, Jeng AY. Bioord Med Chem Lett. 2001;11:1737. doi: 10.1016/s0960-894x(01)00273-6.Zhuang ZP, Kung MP, Mu M, Kung HF. J Med Chem. 1998;41:157. doi: 10.1021/jm970296s.
- 8.For a study on the mechanism of the Rh(III)-catalyzed arylation of 2-phenylpyridine with N-carbamate or N-Ts imines, see: Tauchert ME, Incarvito CD, Rheingold AL, Bergman RG, Ellman JA. J Am Chem Soc. 2012;134:1482. doi: 10.1021/ja211110h.Li Y, Zhang XS, Li H, Wang WH, Chen K, Li BJ, Shi ZJ. Chem Sci. 2012;3:1634.
- 9.Kuprat M, Lehmann M, Schulz A, Villinger A. Organometallics. 2010;29:1421. [Google Scholar]
- 10.Kan T, Fukuyama T. Chem Commun. 2004:353. doi: 10.1039/b311203a. [DOI] [PubMed] [Google Scholar]
- 11.(a) Leighty MW, Spletstoser JT, Georg GI, Umihara H, Fukuyama T. Org Synth. 2011;88:427. [Google Scholar]; (b) Spletstoser JT, White JM, Tunoori AR, Georg GI. J Am Chem Soc. 2007;129:3408. doi: 10.1021/ja066362+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.See Supporting Information for further details.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.