

Abstract
In eukaryotic cells, addition of poly(A) tails to transcripts by 3′-end processing/polyadenylation machinery is a critical step in gene expression. The length of the poly(A) tail influences the stability, nuclear export and translation of mRNA transcripts. Control of poly(A) tail length is thus an important mechanism to regulate the abundance and ultimate translation of transcripts. Understanding the global regulation of poly(A) tail length will require dissecting the contributions of enzymes, regulatory factors, and poly(A) binding proteins (Pabs) that all cooperate to regulate polyadenylation. A recent addition to the Pab family is the CCCH-type zinc finger class of Pabs that includes S. cerevisiae Nab2 and its human counterpart, ZC3H14. In S. cerevisiae, Nab2 is an essential nuclear Pab implicated in both poly(A) RNA export from the nucleus and control of poly(A) tail length. Consistent with an important role in regulation of poly(A) tail length, depletion of Nab2 from yeast cells results in hyperadenylation of poly(A) RNA. In this review, we focus on the role of Nab2 in poly(A) tail length control and speculate on potential mechanisms by which Nab2 could regulate poly(A) tail length based on reported physical and genetic interactions. We present models, illustrating how Nab2 could regulate poly(A) tail length by limiting polyadenylation and/or enhancing trimming. Given that mutation of the gene encoding the human Nab2 homologue, ZC3H14, causes a form of autosomal recessive intellectual disability, we also speculate on how mutations in a gene encoding a ubiquitously expressed Pab lead specifically to neurological defects.
Proper gene expression in eukaryotic cells requires the production of properly processed mRNA transcripts in the nucleus and export of those transcripts to the cytoplasm for translation. Following transcription by RNA polymerase II (RNAP II), nascent mRNA transcripts undergo a series of co-transcriptional processing steps to reach maturity. Specifically, mRNA transcripts are bound by processing factors that perform 5′-end capping to add a 7-methylguanosine cap, splicing to remove introns, and 3′-end cleavage and polyadenylation to add a polyadenosine (poly(A)) tail (1,2). Transcripts that are improperly processed and could lead to the production of deleterious proteins are recognized and destroyed by mRNA surveillance factors (3–5). Following nuclear processing, mRNA transcripts are exported through nuclear pore complexes (NPCs) to the cytoplasm where they can be translated (6–8). Throughout their lifespan, mRNA transcripts interface with RNA binding proteins that regulate the processing, export, translation, and degradation of these mRNAs. Importantly, the mechanisms and factors required for mRNA processing and export are evolutionarily conserved from yeast to humans. Thus, experiments in the yeast model system have contributed significantly to our understanding of mRNA processing and export in higher eukaryotes.
Synthesis of poly(A) tails of the right length by the 3′-end processing and polyadenylation machinery is a particularly critical step in eukaryotic gene expression. In yeast and metazoans, poly(A) RNA binding proteins (Pabs) play a key role in regulating poly(A) tail length (9). Most Pabs bind specifically to the polyadenosine RNA via an RNA recognition motif (RRM); however, S. cerevisiae Nab2 (nuclear abundant poly(A) RNA binding protein) binds specifically to polyadenosine RNA via a CCCH-type zinc finger domain (10). Nab2 is therefore the founding member of a novel class of zinc finger Pabs (11). Critically, mutations in genes encoding ubiquitously expressed Pabs cause human disease. Mutations in the gene encoding the nuclear RRM-containing Pab, PABPN1, cause oculopharyngeal muscular dystrophy (OPMD), while mutations in the gene encoding the human Nab2 homologue, ZC3H14, cause a form of autosomal recessive intellectual disability (ARID) (12,13). Proper Pab function and regulation of poly(A) tail length is therefore critical for tissue-specific functions.
As the length of the poly(A) tail impacts the stability, nuclear export, and translation of mRNA transcripts, regulation of poly(A) tail length can control the fate of transcripts. In the yeast Saccharomyces cerevisiae, the average length of poly(A) tails on polyadenylated RNA transcripts is ~60–80 adenosines, whereas in mammalian cells, the average length is approximately 250 adenosines (9). Although shorter than normal poly(A) tails are correlated with decreased RNA stability and translation (14–18), the consequences of longer than normal tails are unknown. A thorough understanding of poly(A) tail length control thus requires identification of the main protein factors that affect poly(A) tail length and elucidation of mechanism by which these factors alter poly(A) tail length.
The topic of poly(A) tail length control by RRM-containing Pabs has recently been reviewed (9,19). Here, we focus primarily on the role of the nuclear zinc finger Pab, Nab2, in the control of poly(A) tail length in S. cerevisiae. Importantly, key aspects of our current understanding of poly(A) tail length control have come from in vitro cleavage and polyadenylation assays using yeast lysate coupled with functional studies of S. cerevisiae mutants. We discuss the function of Nab2 and speculate on the potential mechanisms by which Nab2 could modulate 3′-end processing and the polyadenylation machinery to control poly(A) tail length based on the physical and genetic interactions of Nab2. We also discuss the function of the Drosophila Nab2 counterpart, dNab2, and the human Nab2 counterpart, ZC3H14. Given that mutations in ZC3H14 cause a form of intellectual disability (12), we comment on how defects in the function of a ubiquitously expressed Pab, ZC3H14, could lead to neurological defects in humans.
Nab2 Structure and Function
The Nab2 protein contains four key domains: an N-terminal domain (residues 1-97), a Q-rich domain (residues 104-169), an RGG domain (residues 201-261), and a C-terminal tandem zinc finger domain (residues 262-473) (Fig. 1) (20,21). The N-terminal domain of Nab2, which facilitates poly(A) RNA export, forms a five alpha-helix bundle with a proline-tryptophan-isoleucine (PWI)-like fold (20,22). The RGG domain, which is important for nuclear import, mediates interaction with the import receptor, Kap104 (20,23–25). The Q-rich domain, which is not essential, has not been ascribed a function (20). The C-terminal zinc finger domain, which contains seven tandem CCCH-type zinc fingers (ZnF), mediates high affinity binding to polyadenosine RNA (10,20,21,26). The functionally important N- and C-terminal domains of Nab2 are the best characterized domains and play roles in mRNA export and poly(A) tail length control, respectively.
Fig. 1. Functional domains of Nab2.

Nab2 is a 525 residue protein that possesses four key domains: an N-terminal domain (residues 1-97), a Q-rich domain (residues 104-169), an RGG domain (residues 201-261), and C-terminal tandem zinc finger domain (residues 262-473). The N-terminal domain of Nab2 (Nab2-N) facilitates nuclear export of poly(A) RNA and interacts with nuclear pore-associated Mlp1 protein in the nucleus and the nuclear rim-associated Gfd1 protein in the cytoplasm (20,22,28,30). The crystal structure of the N-terminal domain of Nab2 reveals that Nab2-N forms a five alpha-helix bundle with a proline-tryptophan-isoleucine (PWI)-like fold (20,22). The key residue Phe73 (red) that is important for interaction with Mlp1 and Tyr34 (magenta) that is critical for interaction with Gfd1 are highlighted (22,30,31). The RGG domain mediates interaction with the Nab2 import receptor, Kap104 (20,23–25). The Q-rich domain is not essential and currently has no characterized function (20). The C-terminal zinc finger domain contains seven tandem CCCH-type zinc fingers (ZnF) and mediates specific high affinity binding to polyadenosine RNA (10,20,21,26).
The N-terminal domain of Nab2 (Nab2-N) is functionally important and critical for poly(A) RNA export in yeast cells (20,27). Specifically, nab2-ΔN/nab2-1 mutant cells, which express a nab2 mutant that lacks the N-terminal domain (residues 4-97) as the sole copy of Nab2, exhibit severely impaired growth and significant accumulation of bulk poly(A) RNA in the nucleus (20,27). In addition, nab2-ΔN cells also show extended poly(A) tails on bulk RNA, suggesting that Nab2-N may play a role in restriction of poly(A) tail length (26). Alternatively, the longer poly(A) RNA tails in nab2-ΔN cells may reflect an indirect effect of the block to mRNA export which could give rise to increased nuclear residence time and thus hyperadenylation of transcripts. The N-terminal domain of Nab2 (Nab2-N) physically interacts with two key nuclear pore-associated proteins, Mlp1 and Gfd1 (22,28–31). One face of Nab2-N directly interacts with the large myosin-like platform protein, Mlp1, which localizes to the nuclear side of the nuclear pore complex (NPC) and functions in pre-mRNA retention (22,32–34) (Fig. 1 & 2). A second face of Nab2-N directly interacts with the small coiled-coil protein, Gfd1, which localizes to the cytoplasmic face of the nuclear pore and helps to facilitate mRNP disassembly (29,31,35) (Fig. 1 & 2). Nab2 may therefore bind to Mlp1 to concentrate properly processed mRNAs at the nuclear side of the pore for nuclear export and bind to Gfd1 to tether the mRNP at the cytoplasmic face of the pore for disassembly (28–31).
Fig. 2. Function of Nab2 in mRNA biogenesis.

During RNA polymerase II (RNAPII) transcription, the THO transcription elongation complex (Tho2, Hpr1, Mft1, and Thp2), the ATPase, Sub2, the mRNA export cofactor, Yra1, the mRNA binding SR protein, Npl3, the poly(A) binding protein, Nab2, and other mRNA processing factors are all recruited to the nascent transcript (6–8,38). Following capping and splicing, the polyadenylation signal (PAS) in the 3′-UTR of the transcript is recognized by the cleavage machinery, cleavage factor IA ((CFIA); Rna14, Rna15, Clp1 and Pcf11), cleavage factor IB ((CFIB); Hrp1/Nab4), and the cleavage and polyadenylation factor ((CPF); a complex including riboendonuclease, Ysh1/Brr5 (CPSF73 in mammals), the poly(A) polymerase, Pap1, and the Pap1 regulation factor, Fip1) (39). For cleavage at the poly(A) site (pA) in the 3′-UTR of the transcript, CFIA protein, Rna15, which contains a single RNA recognition motif (RRM), together with other CFIA subunits recognizes the A-rich positioning element (e.g. AAUAAN) in the PAS to position CPF to cleave the poly(A) site (39–41). Hrp1, which contains two RRMs, binds to the AU-rich efficiency element (e.g. UAUAUAU) in the PAS and influences the efficiency of the cleavage reaction (39,42,43). Following cleavage, CPF-stimulated Pap1 processively synthesizes the poly(A) tail. During polyadenylation, Nab2 likely binds to the nascent poly(A) tail. Transcripts that are improperly processed (e.g. contain short poly(A) tails) due to defective 3′-end processing and polyadenylation are recognized, retained in the nucleus and degraded by the nuclear exosome riboexonuclease complex (Exo), containing the 3′-5′ riboexonuclease subunit, Rrp6 (3,5,51,52). Recognition of RNA targets by the exosome is facilitated by exosome cofactors, such as TRAMP (3,5,51,52). Together with the mRNA export cofactor, Yra1, Nab2 helps to recruit the heterodimeric mRNA export receptor, Mex67-Mtr2, to facilitate the nuclear export of the transcript (53). Nab2 also interacts with the nuclear pore complex-associated protein, Mlp1, to facilitate targeting of the transcript to the pore (22,28,30). Following Mex67-mediated export to the cytoplasm, Gfd1 helps tether Nab2 to the cytoplasmic face of the pore, while the RNA helicase, Dbp5, facilitates mRNP dissociation of Nab2, Npl3, Mex67, and other RNA-binding proteins from the transcript (29,31,37,54). During mRNP remodeling, the cytoplasmic Pab, Pab1, may bind to the poly(A) tail of the transcript to replace Nab2 in an exchange step. As Pab1 shuttles between the nucleus and cytoplasm and can bind the Rna15 CFIA cleavage factor, Pab1 likely initially loads onto the transcript in the nucleus (15,16,55,56). The cytoplasmic Pab1-bound transcript can then be translated or turned over. For cytoplasmic mRNA decay, the transcript is first deadenylated by the Ccr4-Not complex deadenylases, Ccr4 and Caf1. The transcript body is then decapped by Dcp2 and degraded from the 5′-end by the 5′-3′ riboexonuclease, Xrn1, or degraded from the 3′-end by the cytoplasmic exosome (4,5,58,59). Pab1 can also recruit the Pan2-Pan3 deadenylase complex (Pan2 is the deadenylase) to trim the poly(A) tails of specific transcripts (60–63).
The essential C-terminal CCCH-type zinc finger (ZnF) domain of Nab2 is a novel polyadenosine RNA binding domain that specifically recognizes polyadenosine RNA and plays an important role in the regulation of poly(A) tail length (10,20,21,26,36). Biochemical and genetic analyses of the seven zinc fingers of the Nab2 ZnF domain have revealed that ZnF5-7 are necessary and sufficient for high affinity binding to polyadenosine RNA (20,26). Critically, cells expressing nab2 ZnF domain mutants, nab2-21 (ΔZnF6-7), nab2-C437S (C437S in ZnF6), or nab2-C5-7→A (C415A, C437A, & C458A in ZnF5-7) as the sole copy of Nab2 exhibit cold-sensitive growth and poly(A) RNA with extended poly(A) tails (26,36). Notably, nab2-C5-7→A cells do not show nuclear accumulation of poly(A) RNA at 16°C, but nab2-21 cells do exhibit nucleolar accumulation of poly(A) RNA at 14°C (26,36), indicating that further study is required to decipher how Nab2 interconnects with polyadenylation and poly(A) RNA export. As the nab2 ZnF mutant, nab2-C437S, shows reduced binding to polyadenosine RNA in vitro (10), impairment of Nab2 interaction with poly(A) RNA is correlated with the longer poly(A) tail phenotype observed in these nab2 ZnF mutant cells (10,26,37). The combined data on Nab2 structure and function thus indicate that Nab2 binds to the poly(A) tails of transcripts during their polyadenylation via the C-terminal ZnF domain and then facilitates targeting of these transcripts to the nuclear pore for nuclear export via the N-terminal PWI-like domain (Fig. 1).
Nab2, mRNA Processing/Export, and Poly(A) Tail Length Control
Throughout mRNA biogenesis, RNA processing steps are intimately coupled to one another through RNA binding proteins. Nab2 is one of several factors that contribute to this coupling of RNA processing steps. Nab2 facilitates targeting of mRNA transcripts to the nuclear pore for export via the nuclear pore-associated protein, Mlp1 (28),(30). Additional RNA binding proteins, including the key mRNA export receptor protein, Mex67, also help to concentrate transcripts at the pore (6,8). Mex67 then translocates the mRNP through the pore to the cytoplasm, where the mRNP is disassembled at the cytoplasmic face of the pore (6,8). Nuclear mRNA export is thus a relatively well characterized process; however, the steps leading up to export and the connections between polyadenylation and export factors are less well understood. To understand how Nab2 may link mRNA processing to export in S. cerevisiae, it is critical to visualize Nab2 in the context of these highly-coupled mRNA processing and export steps. A comprehensive model for mRNA processing and export is shown in Figure 2.
RNA binding proteins are loaded onto a nascent transcript during transcription to ensure the proper processing and maturation of the transcript (Fig. 2). These RNA binding proteins are temporally and spatially connected to facilitate the coupling of mRNA processing steps. During RNA polymerase II transcription, the RNA binding proteins that associate with the nascent transcript include the THO transcription complex, splicing factors, 3′-end processing proteins, and export factors (6–8,38). Nab2 is co-transcriptionally recruited to the nascent transcript (38), primed to associate with the message at an appropriate time together with other RNA binding proteins.
Following capping and splicing of the transcript, the polyadenylation signal (PAS), containing the AU-rich efficiency element (EE), A-rich positioning element (PE), and U-rich element (UE), is recognized by the 3′-end cleavage machinery, which comprises cleavage factor IA (CFIA), a complex containing Rna14, Rna15, Clp1 and Pcf11, cleavage factor IB (CFIB), Hrp1/Nab4, and the cleavage and polyadenylation factor (CPF), a complex including the riboendonuclease, Ysh1/Brr5 (CPSF73 in mammals), the poly(A) polymerase, Pap1, and the Pap1 regulation factor, Fip1 (39). Distantly resembling CPSF recognition of the AAUAAA polyadenylation signal and cleavage of the poly(A) site as it occurs in mammals, the single RRM-containing protein, Rna15, associates with the scaffold protein, Rna14, which binds to the other CFIA subunits, Pcf11/Clp1, and recognizes the A-rich PE (e.g. AAUAAN) to position CPF to cleave the poly(A) site (39–41). The dual RRM-containing protein, Hrp1/Nab4, however, binds to the AU-rich EE (e.g. UAUAUAU) and contacts Rna14 and Rna15 to influence the efficiency of the cleavage reaction (39,42,43). After the cleavage reaction, poly(A) polymerase, Pap1, synthesizes a poly(A) tail of ~60–80 adenosines in a template-independent manner (9). As Pap1 is an inefficient distributive enzyme alone, Pap1 processivity requires stimulation/regulation by factors that bind Pap1 and tether it to the RNA (9). The mammalian RRM-containing Pab, PABPN1, enhances the processivity of PAP in this manner (44–46), and another mammalian component, nucleophosmin, influences polyadenylation (47). In addition, the S. cerevisiae CPF component, Fip1, and its human CPSF counterpart, hFip, directly bind Pap and tether it to CPF/CPSF to stimulate/regulate Pap activity (48–50). A current challenge is to understand how the zinc finger-containing Pab, Nab2/ZC3H14, interfaces with these polyadenylation factors to regulate poly(A) tail length.
Any errors in mRNA processing are quickly resolved to prevent export of faulty transcripts. In particular, if the transcript is improperly processed and/or contains a short poly(A) tail due to defective 3′-end processing and/or polyadenylation, the transcript is recognized, retained in the nucleus, and degraded by the nuclear exosome, a ring-like ribonuclease complex containing ten core subunits, including the ribonuclease, Dis3/Rrp44, and an additional nuclear riboexonuclease subunit, Rrp6 (3,5,51,52). As Nab2 binds specifically to polyadenosine RNA in vitro (10), Nab2 would be optimally placed to interface with the polyadenylation machinery and modulate polyadenylation. Given that the nuclear exosome and other nuclear ribonucleases trim the 3′-end of RNAs during normal RNA processing, Nab2 could modulate poly(A) tail length via interaction with a ribonuclease (3,5,51,52).
After correct mRNA processing, the mRNA transcript is targeted to the nuclear pore for mRNA export to the cytoplasm. Nab2 binds to the nuclear pore complex-associated protein, Mlp1, to facilitate targeting of the transcript to the pore (22,28,30). Together with the mRNA export cofactor, Yra1, Nab2 aids in recruiting the mRNA export receptor, Mex67, to promote nuclear export of the properly processed mRNA transcript (53). Following Mex67-mediated export to the cytoplasm, Gfd1 helps tether Nab2 to the cytoplasmic face of the pore, while the DEAD box RNA helicase, Dbp5, remodels the mRNP, leading to dissociation of Nab2, Mex67, and other RNA binding proteins from the transcript (29,31,37,54).
During mRNP remodeling at the cytoplasmic side of the pore, the transcript undergoes an exchange of RNA binding proteins in which cytoplasmic proteins replace nuclear proteins. The cytoplasmic RNA binding proteins target the transcript for translation or cytoplasmic processing steps, such as mRNA decay. The cytoplasmic Pab, Pab1, associates with the poly(A) tail, but how Pab1 and Nab2 interactions are coordinated is unknown. As Pab1 shuttles between the nucleus and cytoplasm and can bind the Rna15 CFIA cleavage factor (15,16,55,56), Pab1 likely initially loads onto the transcript in the nucleus. Notably, the presence of Pab1 appears to be required for the release of transcripts from the site of transcription (55). Once the transcript exits the nucleus, Pab1 stimulates translation in the cytoplasm (14). Transcripts are ultimately degraded in the cytoplasm primarily through a mechanism triggered by deadenylation, which utilizes the Ccr4-Not complex deadenylases, Ccr4 and Caf1 (57). The transcript body is then decapped by Dcp2 and degraded from the 5′-end by the 5′-3′ riboexonuclease, Xrn1, or degraded from 3′-end by the cytoplasmic exosome (4,5,58,59). In S. cerevisiae and mammals, specific transcripts can also be deadenylated and trimmed by the Pab-associated poly(A) ribonuclease (PAN) complex, Pan2 (deadenylase) and Pan3, prior to Ccr4 deadenylation (60–63).
Two simplified models for how Nab2 could control poly(A) tail length can be proposed that are not necessarily mutually exclusive (Fig. 3). In a polyadenylation limiting model, Nab2 interacts with cleavage and polyadenylation factors or potentially alters the poly(A) RNA conformation to restrict poly(A) polymerase activity and achieve the correct poly(A) tail length. Alternatively, in the poly(A) trimming model, Nab2 triggers a ribonuclease to trim the poly(A) tail to the correct length. These models for Nab2 control of poly(A) tail length can be readily tested in S. cerevisiae and thus provide a framework in which to decipher Nab2 interactions with other RNA processing factors.
Fig. 3. Models for Nab2 control of poly(A) tail length.

In a polyadenylation limiting model, Nab2 interacts with cleavage and polyadenylation factors (cleavage factor I (CFI); cleavage and polyadenylation factor (CPF)) or potentially alters the poly(A) RNA conformation to restrict poly(A) polymerase activity and limit poly(A) tail length. In the poly(A) tail trimming model, Nab2 recruits and modulates/stimulates a ribonuclease to trim the poly(A) tail to the correct length. Although, Nab2 is depicted with the cleavage and polyadenylation machinery during recruitment of a ribonuclease, Nab2 could also recruit a ribonuclease after polyadenylation at a later stage in mRNA biogenesis. These models are not necessarily mutually exclusive.
Nab2 Interactions
Nab2 regulation of poly(A) tail length suggests that Nab2 could genetically and physically interact with 3′-end processing and polyadenylation components. Indeed, NAB2 genetically interacts with genes encoding the CFIA cleavage factor, RNA15, the poly(A) polymerase, PAP1, the CPF subunit, SYC1, the cytoplasmic Pab, PAB1, the nuclear exosome subunit, RRP6, and components of the Ccr4-Not deadenylation/transcription complex (26,36,38,64) (Fig. 4). Nab2 also physically interacts with the Ccr4-Not complex deadenylases, Ccr4 and Caf1/Pop2, and the cleavage factor, Hrp1(65–67) (Fig. 4).
Fig. 4. Physical and genetic interactions of Nab2 with 3′-end processing and polyadenylation factors.
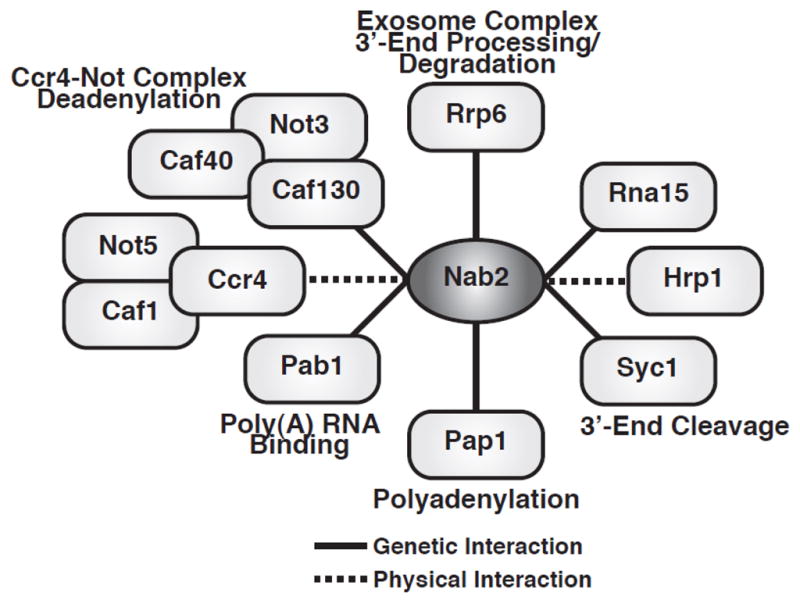
Nab2 genetically interacts with cytoplasmic poly(A) binding protein, Pab1, poly(A) polymerase, Pap1, CFIA cleavage factor, Rna15, CPF subunit, Syc1, and nuclear exosome 3′-5′ riboexonuclease, Rrp6, and Ccr4-Not complex components, Not3, Caf40, and Caf130 (26,36,38,64). Nab2 physically interacts with the CFIB cleavage factor, Hrp1, and Ccr4-Not complex deadenylases, Ccr4 and Caf1, and component, Not5 (65–67).
The Nab2 ZnF mutant, nab2-C5-7→A, exhibits negative genetic interactions with mutants of the critical CFIA cleavage factor, RNA15 (CstF-64 in mammals), (Fig. 2) and the poly(A) polymerase, PAP1 (26). Specifically, double mutants of nab2-C5-7→A and rna15-2 (rna15-L214P) or pap1-1 exhibit severe slow growth (26). As rna15-2 and pap1-1 single mutants exhibit slow growth and poly(A) RNA with shorter poly(A) tails, while the nab2-C5-7→A single mutant shows slow growth and poly(A) RNA with longer poly(A) tails, the enhanced slow growth in nab2-C5-7→A rna15-2 and nab2-C5-7→A pap1-1 double mutants could be explained if the nab2-C5-7→A protein, which shows weaker affinity for poly(A) RNA in vitro (10), binds even less efficiently to the short poly(A) tails produced by defective cleavage and polyadenylation mediated by rna15-2 and/or pap1-1 protein. Critically, the rna15-2 L214P substitution is located in an Rna15 hinge domain that interacts with the Rna14 C-terminal ‘monkeytail’ domain and disrupts the interaction with Rna14 and other CFIA subunits (41,68). Loss of Rna15 from CFIA in rna15-2 cells would therefore be predicted to reduce CFIA binding to RNA and thus impair positioning of CPF for cleavage of specific poly(A) sites (41).
Interestingly, NAB2 not only genetically interacts with the CFIA cleavage factor, RNA15, but also physically interacts with the cleavage factor, Hrp1, which contacts CFIA subunits, Rna14 and Rna15, to improve cleavage efficiency (42,66,67). In addition, NAB2 exhibits a negative genetic interaction with the gene encoding Syc1, a CPF subunit that shares sequence identity with the CPF riboendonuclease, Ysh1/Brr5, and has been proposed to negatively regulate 3′-end processing (64,69). Nab2 could thus potentially regulate poly(A) tail length by indirectly modulating cleavage efficiency.
Overexpression of Pab1, which is cytoplasmic at steady-state or a nuclear localized form of Pab1, Pab1NL, can rescue nab2Δcells (36), which would otherwise die as NAB2 is essential (21). Notably, Pab1 partially rescues the mRNA export block in nab2Δ cells, suggesting Pab1 can interact with the mRNA export machinery; however, Pab1 does not resolve the hyperadenylation defect in nab2Δ cells (36). In contrast, Pab1NL enhances the hyperadenylation defect in nab2Δ cells (36). An Arabidopsis Pab1 homologue, AtPab3, which partly rescues the mRNA export defect in nab2-ΔN cells also fails to resolve the hyperadenylation defect (70). Together with the finding that Nab2 has the capacity to restrict poly(A) tail length in pab1Δ yeast extracts, the data suggest that Nab2 interacts with factors in the nucleus to limit poly(A) tail length that cannot interface with Pab1 (15,16).
A particularly exciting discovery is that deletion of the nuclear exosome gene, RRP6, rescues the lethality of nab2Δ cells, although nab2Δ rrp6Δ double mutant cells do exhibit significant slow growth (38). As depletion of Nab2 from yeast cells results in transcripts with long poly(A) tails (36), rescue of nab2Δ cells by removal of a key nuclear exosome 3′-5′ riboexonuclease, Rrp6, could suggest that preventing Rrp6-mediated degradation of improperly processed transcripts permits nab2Δ cells to survive. This mechanism of suppression would imply that Rrp6 normally targets transcripts with long poly(A) tails. In support of this idea, in S. pombe rrp6 mutant cells, hyperadenylated native transcripts accumulate and the level of a synthetic transcript with an encoded long poly(A) tail (110 adenosines) increases relative to wildtype cells (71). Hyperadenylated transcripts could therefore be unstable due to degradation by Rrp6. Control of poly(A) tail length by Nab2 could thus involve modulation of Rrp6 activity, perhaps switching Rrp6 from a degradative to a trimming mode or binding to an Rrp6 regulator.
A final intriguing genetic interaction is that deletion of NOT3, CAF40, or CAF130 genes encoding components of the Ccr4-Not deadenylation complex can improve the growth of Nab2-depleted cells (64). Notably, Nab2 also physically interacts with the Ccr4-Not complex deadenylases, Ccr4 and Caf1/Pop2, and subunit, Not5, by co-immunoprecipitation (65). These interactions suggest that Nab2 could modulate the Ccr4-Not complex Ccr4/Caf1 deadenylases to affect poly(A) tail length in the cytoplasm. Removal of Ccr4-Not subunits from Nab2-depleted cells could conceivably improve growth by impairing Ccr4/Caf1 deadenylation and increasing the poly(A) tail length of transcripts in the cytoplasm. Alternatively, as the Ccr4-Not complex physically and genetically interacts with the nuclear exosome, impairment of the Ccr4-Not complex in Nab2-depleted cells could improve growth by disrupting exosome function in the nucleus (72,73).
Control of Poly(A) Tail Length By Nab2 and PABPN1
How does Nab2 actually control poly(A) tail length? As proposed, in a polyadenylation limiting model, Nab2 could interact with cleavage and polyadenylation factors or alter the poly(A) RNA conformation to restrict poly(A) polymerase activity and poly(A) tail length and/or, in a poly(A) tail trimming model, Nab2 could recruit a ribonuclease to trim the poly(A) tail (Fig. 3). Recombinant Nab2 can limit the poly(A) tail length of CYC1 pre-mRNA in a coupled cleavage and polyadenylation reaction or pre-cleaved CYC1 pre-mRNA in a polyadenylation reaction in vitro containing purified CFIA and CPF from yeast and recombinant Hrp1 (36,67). These in vitro data strongly suggest that Nab2 restriction of poly(A) tail partly or wholly involves Nab2 interaction with cleavage and polyadenylation factors and/or Nab2 interaction with the RNA, supporting the polyadenylation limiting model (Fig. 3). However, conceivably, Nab2 could also interact with other RNA processing components that associate and purify with the CFIA and CPF complexes from yeast to limit poly(A) tail length.
Recognition of polyadenosine RNA by the Nab2 CCCH-type zinc finger (ZnF) domain is correlated with control of poly(A) tail length (26,36). In particular, amino acid substitutions in Nab2 ZnF5-7, which reduce binding to polyadenosine RNA in vitro, give rise to bulk poly(A) RNA with extended poly(A) tails in vivo (26,36). These data strongly suggest that Nab2 ZnF5-7 binding to the poly(A) tail facilitates control of poly(A) tail length. As mentioned, Nab2 binding to polyadenosine RNA could regulate poly(A) tail length by interacting with polyadenylation and/or ribonuclease proteins. Alternatively, the novel polyadenosine binding ZnF domain of Nab2 could alter the conformation of the RNA to impact polyadenylation and poly(A) tail length. In support of this idea, recombinant Nab2 ZnF1-7 alone, but not the Nab2-N+Q-rich domains alone, can limit the poly(A) tail length of a CYC1 pre-mRNA using purified CFIA, CPF, and Hrp1 in vitro (74). In addition, recombinant nab2 ZnF6-7 mutant, nab2-21, which lacks part of ZnF6 and all of ZnF7, and a nab2Δ ZnF1-7 mutant, both fail to reduce the poly(A) tail length of this transcript in vitro (74). With the caveat that these recombinant Nab2 domain mutants could be misfolded, these results suggest that the Nab2 ZnF domain, especially ZnF6-7, helps restrict poly(A) tail length. Notably, a recombinant nab2 RGG-ZnK1-7 mutant, lacking the Nab2-N and Q-rich domains, does not efficiently limit the poly(A) tail length of a transcript in vitro (74). This observation could suggest that the N-terminal domain of Nab2 also plays a role in control of poly(A) tail length, which correlates with the fact that nab2-ΔN mutant cells exhibit long poly(A) tails (26).
Recombinant Nab2 restricts the poly(A) tail length of pre-cleaved CYC1 pre-mRNA in pab1Δ yeast extract (36). Transcripts become hyperadenylated and exhibit long poly(A) tails in pab1Δ cell extract because Pab1 normally limits poly(A) tail length, but addition of recombinant Nab2 to this pab1Δ extract rapidly and efficiently limits the length of a transcript poly(A) tail (15,16,36). In yeast cells and pab1Δ extract, in addition to cleavage and polyadenylation factors, Nab2 could also interact with RNA processing factors, such as ribonucleases, after poly(A) tail synthesis to restrict poly(A) tail length, consistent with the poly(A) trimming model (Fig. 3). Over time, the poly(A) tail of precleaved CYC1 pre-mRNA is not appreciably trimmed in the presence of recombinant Nab2 (36). This result could be construed to suggest that Nab2 does not promote trimming of the poly(A) tail and therefore does not recruit a ribonuclease. However, Nab2 could still recruit and finely regulate a ribonuclease to distributively trim the poly(A) tail without allowing this ribonuclease to processively shorten the poly(A) tail. In support of Nab2 poly(A) tail trimming, NAB2 genetically interacts with genes encoding the nuclear exosome riboexonuclease, RRP6, and physically interacts with the Ccr4-Not complex deadenylases, Ccr4 and Caf1 (38,65).
How could Nab2 restrict the poly(A) tail length to an average length of 60–80 adenosines in S. cerevisiae? Nab2 could play a role similar to the mammalian nuclear Pab, PABPN1, or the 3′-end processing factor, nucleophosmin (NPM1), which do not have orthologues in S. cerevisiae. In mammalian cells, in conjunction with CPSF, PABPN1 directly binds to the distributive poly(A) polymerase (PAP), increasing the affinity of the polymerase for RNA and stimulating PAP to processively add approximately 250 adenosines without dissociating (44–46). After reaching a poly(A) tail length of ~250 adenosines, synthesis terminates and the polyadenylation complex becomes distributive and incompetent for processive elongation (18,75). The mechanism for this termination of processive polyadenylation is currently unknown, but must involve disruption of CPSF and/or PABPN1 interaction with PAP (18,75). Notably, in mammalian cells, nucleophosmin is deposited only on mRNA transcripts that have completed polyadenylation, interacts with polyadenylation factors (e.g. PABPN1, CPSF, and PAP), and causes hyperadenylation of transcripts upon depletion, suggesting that nucleophosmin plays a critical role in the termination of polyadenylation (47).
As CPSF, PABPN1, and PAP processively elongate short poly(A) tails to ~250 adenosines, but only slowly elongate long poly(A) tails in vitro, without showing sensitivity to changes in elongation rate, the CPSF/PABPN1 mechanism of poly(A) tail length control seems to truly measure the length of the poly(A) tail (75). In the current model, the CPSF/PABPN1/PAP complex may count the number of PABPN1 molecules on the tail to measure its length (18,75). Notably, PABPN1 forms a spherical particle in complex with poly(A) RNA of 200–300 nucleotides, suggesting a specific PABPN1 structure could signal the poly(A) tail length (76). As recombinant Nab2 protects a poly(A) RNA length of 23–25 nucleotides from micrococcal nuclease digestion, a single Nab2 molecule may bind approximately 20 adenosines (74). Three or four Nab2 molecules could therefore bind to the average poly(A) tail of 60–80 adenosines and serve as a molecular ruler of poly(A) tail length (74). By analogy to PABPN1, Nab2 molecules could form a specific structure in complex with poly(A) RNA to signal the poly(A) tail length to the polyadenylation machinery. As recombinant Nab2 can inhibit adenylation of a polyadenylated transcript by Pap1 and inhibit Pap1 activity in vitro, Nab2 could conceivably limit poly(A) tail length by impairing access of Pap1 to the 3′-end of the mature poly(A) tail (74). A major challenge will be integrating the function of the human Nab2 counterpart, ZC3H14, into the already complicated mammalian poly(A) tail length control machinery.
Nab2/ZC3H14 and Disease
Mammalian nuclear poly(A) RNA binding proteins (Pabs), such as the Nab2 counterpart, ZC3H14, and PABPN1, are ubiquitously expressed in mammalian tissues (11,77) and likely regulate the expression of numerous mRNA transcripts. Intriguingly, mutations in genes encoding ubiquitous RNA binding proteins that recognize a broad range of target mRNAs lead to a variety of tissue-specific diseases, including muscular dystrophy, neurological disorders, and cancer (78,79). At present, there are two diseases known to be caused by mutations in Pab genes. Oculopharyngeal muscular dystrophy (OPMD), an adult onset disease characterized by eyelid drooping, difficulty in swallowing, and weakness in proximal limb muscles, is caused by mutations in the PABPN1 gene (13). In contrast to the muscle disease caused by mutations in the PABPN1 gene, mutations in the ZC3H14 gene, which encodes the human Nab2 counterpart, causes a form of non-syndromic autosomal recessive intellectual disability (ARID) (12). We speculate on how the ubiquitously expressed ZC3H14 leads to a neurological disorder like intellectual disability
Intellectual disability is characterized by a limitation in learning function or adaptive behavior (80). A large scale study identified ZC3H14 as the sole gene responsible for a non-syndromic form of ARID (12). Many forms of intellectual disability are syndromic, meaning that intellectual disability is accompanied by additional symptoms. The fact that the intellectual disability caused by ZC3H14 mutation is non-syndromic strongly suggests that ZC3H14 performs a critical function specifically in neurons. The best characterized mutation in ZC3H14 produces a premature stop codon, which eliminates the expression of nuclear ZC3H14 protein (12). Consistent with a role for ZC3H14 in neurons, ZC3H14 mRNA transcripts are readily detected in human hippocampal and temporal lobe brain samples and ZC3H14 protein is expressed in murine hippocampal neurons where it colocalizes with poly(A) RNA in neuronal cell bodies (12). Studies of the Drosophila orthologue of ZC3H14, dNab2, in Drosophila melanogaster show that dNab2 is important for proper poly(A) tail length of bulk poly(A) RNA and indicate that dNab2 is required for normal neuronal function in flies (12). The analysis of dNab2 in flies supports a role for dNab2/ZC3H14 in poly(A) tail length control and neuronal function.
Mammalian RNA binding proteins bind specifically to the same target transcript in different tissues, but this transcript specificity of RNA binding proteins can be modulated by interactions with other proteins of the ribonucleoprotein complex in each tissue. Although most poly(A) binding proteins bind specifically to polyadenosine RNA, suggesting that they can interact with all polyadenylated transcripts, Pabs often exhibit transcript specificity. Transcript specificity can be conferred to Pabs by post-translationally modifying the Pab, recruiting additional protein factors to the Pab, or altering the cell-type specific expression or subcellular localization of the Pab. The C-terminal domain of yeast Nab2 possesses seven tandem zinc fingers, but only zinc fingers 5–7 are required for high affinity binding to polyadenosine RNA (26). As zinc finger domains can bind nucleic acid, proteins, or even lipids (81), Nab2 zinc fingers 1–4 could potentially bind to another region of the mRNA transcript or interact with other RNA binding proteins that show specificity for specific mRNA transcripts. Alternatively, the Nab2 RGG domain (82), which is also a putative RNA binding domain, could facilitate binding to specific transcripts. The range of RNA binding proteins and the modularity of RNA binding elements in transcripts permits mRNA biogenesis to be fine-tuned in a tissue specific manner.
Processing and localization of transcripts in neurons is highly regulated. Neuronal transcripts undergo a large amount of alternative splicing and polyadenylation and the transcripts are also actively transported through neurites to the termini of neurons (83). Given such post-transcriptional regulation of neuronal transcripts, any change in the expression level of RNA binding proteins could significantly alter neuronal function. For example, in response to external cues, the level of an RNA binding protein could be modulated to generate an alternatively spliced transcript that encodes a protein isoform critical for synaptic navigation (84). Expression of ZC3H14 protein could be critical for proper neuronal function, such that loss of ZC3H14 could lead to impaired axon or synaptic function and thus disruption in the normal neuronal circuitry required for learning and memory. Notably, depletion of ZC3H14 could also alter the stoichiometry or complement of RNA binding proteins on the transcripts normally bound by ZC3H14. Given the genetic links between Nab2 and the polyadenylation and deadenylation machinery in S. cerevisiae, ZC3H14 could make similar interactions with polyadenylation factors and deadenylases in mammalian cells. In support of this idea, select subunits of human CCR4-NOT are expressed solely in neuronal tissue and are temporally regulated during neuronal development (85). Furthermore, there are a few examples of human cleavage and polyadenylation factors that are regulated in a tissue specific manner (86). In particular, mRNA transcripts of a splice variant of CstF-64 (S. cerevisiae Rna15) are enriched in neuronal tissue (87). These results suggest that ZC3H14 could control poly(A) tail length in neuronal tissue by interacting with polyadenylation and deadenylation components. If the poly(A) tails of transcripts are indeed longer in the neurons of people with mutations in ZC3H14, as observed in nab2 mutant yeast cells and dNab2 mutant fly tissue, ZC3H14 and the regulation of poly(A) tail length could play significant roles in shaping neuronal function and development.
Concluding Remarks
The growing array of proteins implicated in control of poly(A) tail length suggests that the regulation of poly(A) tail length plays a critical role in fine-tuning gene expression. Proper poly(A) tail length depends upon the right balance of polyadenosine synthesis by poly(A) polymerase and polyadenosine trimming by ribonucleases. RNA binding proteins, in particular the poly(A) RNA binding proteins, facilitate control of poly(A) tail length by modulating Pap or ribonuclease activity. As Nab2 makes numerous contacts with cleavage and polyadenylation factors, deadenylases, and ribonucleases, and causes transcript hyperadenylation upon depletion, Nab2 may likely sit at the epicenter of poly(A) tail regulation.
An important challenge in the future will be to determine if Nab2 directly modulates the poly(A) tail by inhibiting poly(A) polymerase, stimulating ribonucleases, or both. Genetic experiments indicate that Nab2 interacts with the 3′-end processing machinery, but physical interactions with these 3′-end processing factors must be tested to determine which cleavage and polyadenylation components directly contact Nab2. Upon identifying these physical interactors, biochemical and structural analysis of Nab2 interactions with polyadenylation factors could help to determine how and where Nab2 interfaces with the polyadenylation machinery. Unbiased identification of all the proteins that physically interact with Nab2 by co-precipitation and mass spectroscopy is also required. Knowledge of these Nab2 physical interactors would shed light on how Nab2 influences the poly(A) tail length of transcripts. At present, co-precipitation of 3′-end processing components from mammalian cell lysates and identification of the protein interactors by mass spectrometry has not identified ZC3H14. However, Pabs such as ZC3H14 may only interact transiently with the 3′-end processing machinery, making it difficult to identify the ZC3H14 interactors without further optimization of binding conditions. Future biochemical experiments using recombinant Nab2 in yeast lysates from polyadenylation factor mutant strains or in vitro reconstituted assays with purified polyadenylation components would provide insight into the role of Nab2 in limiting or trimming poly(A) tails. Importantly, analysis of the function of Nab2 in polyadenylation control must be performed in the presence of additional RNA binding proteins in order to decipher the complete mechanism of poly(A) tail control and the dynamic interplay between different RNA binding proteins.
To gain a more comprehensive view of the regulation of 3′-end processing of an mRNA transcript, the consequences of altered poly(A) tail length will need to be addressed. Steps in mRNA processing upstream of cleavage and polyadenylation, such as transcription or splicing, and events downstream, such as nuclear export, could have a dramatic impact on the quality of the 3′-end of an mRNA transcript. Changes in the poly(A) tail length of the transcript could act as a signal to the RNA quality control machinery, in which Nab2 assists in the recognition of a faulty transcript and communicates this information to nuclear pore components to block the export of the transcript or trigger ribonucleases to degrade the transcript.
Significant strides in our understanding of 3′-end processing in metazoans have been achieved, but the mechanisms that regulate poly(A) tail length in vivo still remain poorly characterized. Unraveling the composition of mRNP complexes by analyzing the protein levels of 3′-end processing factors in specific tissues will be important. To elucidate the mechanisms underlying human disease caused by mutations in genes encoding RNA binding proteins, a major goal is to identify the target mRNA transcripts of these RNA binding proteins in specific tissues. What are the target mRNA transcripts of ZC3H14 in neurons? If there is a broad range of target mRNA transcripts, does another RNA binding protein modulate the interaction of ZC3H14 with these mRNA transcripts? If there are specific target mRNA transcripts, does ZC3H14 directly modulate the length of the poly(A) tails on these transcripts? By deciphering the nuts and bolts of poly(A) tail length control, we will gain novel insights into how RNA binding proteins like the ubiquitously expressed ZC3H14 give rise to neuronal-specific defects that underlie intellectual disability.
Highlight.
Transcript poly(A) tails of correct length are critical for proper gene expression
Nab2 is an essential nuclear polyadenosine binding protein in S. cerevisiae
Nab2 is important for control of poly(A) tail length of RNA transcripts
The Drosophila Nab2 homologue, dNab2, is critical for neuronal function in flies
Mutations in the gene encoding human Nab2, ZC3H14, causes intellectual disability
Acknowledgments
We are most grateful to members of the Corbett, Moberg and Pavlath laboratories for their helpful discussions and comments.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Moore MJ, Proudfoot NJ. Cell. 2009;136:688–700. doi: 10.1016/j.cell.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 2.Proudfoot NJ, Furger A, Dye MJ. Cell. 2002;108:501–512. doi: 10.1016/s0092-8674(02)00617-7. [DOI] [PubMed] [Google Scholar]
- 3.Schmid M, Jensen TH. Wiley Interdiscip Rev RNA. 2010;1:474–485. doi: 10.1002/wrna.24. [DOI] [PubMed] [Google Scholar]
- 4.Doma MK, Parker R. Cell. 2007;131:660–668. doi: 10.1016/j.cell.2007.10.041. [DOI] [PubMed] [Google Scholar]
- 5.Lebreton A, Seraphin B. Biochim Biophys Acta. 2008;1779:558–565. doi: 10.1016/j.bbagrm.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 6.Kelly SM, Corbett AH. Traffic. 2009;10:1199–1208. doi: 10.1111/j.1600-0854.2009.00944.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rodriguez-Navarro S, Hurt E. Curr Opin Cell Biol. 2011;23:302–309. doi: 10.1016/j.ceb.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 8.Stewart M. Trends Biochem Sci. 2010;35:609–617. doi: 10.1016/j.tibs.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 9.Eckmann CR, Rammelt C, Wahle E. Wiley Interdiscip Rev RNA. 2011;2:348–361. doi: 10.1002/wrna.56. [DOI] [PubMed] [Google Scholar]
- 10.Kelly SM, Pabit SA, Kitchen CM, Guo P, Marfatia KA, Murphy TJ, Corbett AH, Berland KM. Proc Natl Acad Sci U S A. 2007;104:12306–12311. doi: 10.1073/pnas.0701244104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leung SW, Apponi LH, Cornejo OE, Kitchen CM, Valentini SR, Pavlath GK, Dunham CM, Corbett AH. Gene. 2009;439:71–78. doi: 10.1016/j.gene.2009.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pak C, Garshasbi M, Kahrizi K, Gross C, Apponi LH, Noto JJ, Kelly SM, Leung SW, Tzschach A, Behjati F, Abedini SS, Mohseni M, Jensen LR, Hu H, Huang B, Stahley SN, Liu G, Williams KR, Burdick S, Feng Y, Sanyal S, Bassell GJ, Ropers HH, Najmabadi H, Corbett AH, Moberg KH, Kuss AW. Proc Natl Acad Sci U S A. 2011;108:12390–12395. doi: 10.1073/pnas.1107103108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brais B, Bouchard JP, Xie YG, Rochefort DL, Chretien N, Tome FM, Lafreniere RG, Rommens JM, Uyama E, Nohira O, Blumen S, Korczyn AD, Heutink P, Mathieu J, Duranceau A, Codere F, Fardeau M, Rouleau GA. Nat Genet. 1998;18:164–167. doi: 10.1038/ng0298-164. [DOI] [PubMed] [Google Scholar]
- 14.Sachs AB, Davis RW. Cell. 1989;58:857–867. doi: 10.1016/0092-8674(89)90938-0. [DOI] [PubMed] [Google Scholar]
- 15.Amrani N, Minet M, Le Gouar M, Lacroute F, Wyers F. Mol Cell Biol. 1997;17:3694–3701. doi: 10.1128/mcb.17.7.3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Minvielle-Sebastia L, Preker PJ, Wiederkehr T, Strahm Y, Keller W. Proc Natl Acad Sci U S A. 1997;94:7897–7902. doi: 10.1073/pnas.94.15.7897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Caponigro G, Parker R. Genes Dev. 1995;9:2421–2432. doi: 10.1101/gad.9.19.2421. [DOI] [PubMed] [Google Scholar]
- 18.Kuhn U, Wahle E. Biochim Biophys Acta. 2004;1678:67–84. doi: 10.1016/j.bbaexp.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 19.Zhang X, Virtanen A, Kleiman FE. Cell Cycle. 2010;9:4437–4449. doi: 10.4161/cc.9.22.13887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marfatia KA, Crafton EB, Green DM, Corbett AH. J Biol Chem. 2003;278:6731–6740. doi: 10.1074/jbc.M207571200. [DOI] [PubMed] [Google Scholar]
- 21.Anderson JT, Wilson SM, Datar KV, Swanson MS. Mol Cell Biol. 1993;13:2730–2741. doi: 10.1128/mcb.13.5.2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grant RP, Marshall NJ, Yang JC, Fasken MB, Kelly SM, Harreman MT, Neuhaus D, Corbett AH, Stewart M. J Mol Biol. 2008;376:1048–1059. doi: 10.1016/j.jmb.2007.11.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aitchison JD, Blobel G, Rout MP. Science. 1996;274:624–627. doi: 10.1126/science.274.5287.624. [DOI] [PubMed] [Google Scholar]
- 24.Lee DC, Aitchison JD. J Biol Chem. 1999;274:29031–29037. doi: 10.1074/jbc.274.41.29031. [DOI] [PubMed] [Google Scholar]
- 25.Truant R, Fridell RA, Benson RE, Bogerd H, Cullen BR. Mol Cell Biol. 1998;18:1449–1458. doi: 10.1128/mcb.18.3.1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kelly SM, Leung SW, Apponi LH, Bramley AM, Tran EJ, Chekanova JA, Wente SR, Corbett AH. J Biol Chem. 2010;285:26022–26032. doi: 10.1074/jbc.M110.141127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Green DM, Marfatia KA, Crafton EB, Zhang X, Cheng X, Corbett AH. J Biol Chem. 2002;277:7752–7760. doi: 10.1074/jbc.M110053200. [DOI] [PubMed] [Google Scholar]
- 28.Green DM, Johnson CP, Hagan H, Corbett AH. Proc Natl Acad Sci U S A. 2003;100:1010–1015. doi: 10.1073/pnas.0336594100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Suntharalingam M, Alcazar-Roman AR, Wente SR. J Biol Chem. 2004;279:35384–35391. doi: 10.1074/jbc.M402044200. [DOI] [PubMed] [Google Scholar]
- 30.Fasken MB, Stewart M, Corbett AH. J Biol Chem. 2008;283:27130–27143. doi: 10.1074/jbc.M803649200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zheng C, Fasken MB, Marshall NJ, Brockmann C, Rubinson ME, Wente SR, Corbett AH, Stewart M. J Biol Chem. 2010;285:20704–20715. doi: 10.1074/jbc.M110.107276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Galy V, Gadal O, Fromont-Racine M, Romano A, Jacquier A, Nehrbass U. Cell. 2004;116:63–73. doi: 10.1016/s0092-8674(03)01026-2. [DOI] [PubMed] [Google Scholar]
- 33.Strambio-de-Castillia C, Blobel G, Rout MP. J Cell Biol. 1999;144:839–855. doi: 10.1083/jcb.144.5.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kolling R, Nguyen T, Chen EY, Botstein D. Mol Gen Genet. 1993;237:359–369. doi: 10.1007/BF00279439. [DOI] [PubMed] [Google Scholar]
- 35.Hodge CA, Colot HV, Stafford P, Cole CN. EMBO J. 1999;18:5778–5788. doi: 10.1093/emboj/18.20.5778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hector RE, Nykamp KR, Dheur S, Anderson JT, Non PJ, Urbinati CR, Wilson SM, Minvielle-Sebastia L, Swanson MS. EMBO J. 2002;21:1800–1810. doi: 10.1093/emboj/21.7.1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tran EJ, Zhou Y, Corbett AH, Wente SR. Mol Cell. 2007;28:850–859. doi: 10.1016/j.molcel.2007.09.019. [DOI] [PubMed] [Google Scholar]
- 38.Gonzalez-Aguilera C, Tous C, Babiano R, de la Cruz J, Luna R, Aguilera A. Mol Biol Cell. 2011;22:2729–2740. doi: 10.1091/mbc.E11-01-0055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tian B, Graber JH. Wiley Interdiscip Rev RNA. 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gordon JM, Shikov S, Kuehner JN, Liriano M, Lee E, Stafford W, Poulsen MB, Harrison C, Moore C, Bohm A. Biochemistry. 2011;50:10203–10214. doi: 10.1021/bi200964p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moreno-Morcillo M, Minvielle-Sebastia L, Fribourg S, Mackereth CD. Structure. 2011;19:534–545. doi: 10.1016/j.str.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 42.Leeper TC, Qu X, Lu C, Moore C, Varani G. J Mol Biol. 2010;401:334–349. doi: 10.1016/j.jmb.2010.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kessler MM, Henry MF, Shen E, Zhao J, Gross S, Silver PA, Moore CL. Genes Dev. 1997;11:2545–2556. doi: 10.1101/gad.11.19.2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wahle E. Cell. 1991;66:759–768. doi: 10.1016/0092-8674(91)90119-j. [DOI] [PubMed] [Google Scholar]
- 45.Wahle E, Lustig A, Jeno P, Maurer P. J Biol Chem. 1993;268:2937–2945. [PubMed] [Google Scholar]
- 46.Bienroth S, Keller W, Wahle E. EMBO J. 1993;12:585–594. doi: 10.1002/j.1460-2075.1993.tb05690.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sagawa F, Ibrahim H, Morrison AL, Wilusz CJ, Wilusz J. EMBO J. 2011;30:3994–4005. doi: 10.1038/emboj.2011.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Preker PJ, Lingner J, Minvielle-Sebastia L, Keller W. Cell. 1995;81:379–389. doi: 10.1016/0092-8674(95)90391-7. [DOI] [PubMed] [Google Scholar]
- 49.Helmling S, Zhelkovsky A, Moore CL. Mol Cell Biol. 2001;21:2026–2037. doi: 10.1128/MCB.21.6.2026-2037.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kaufmann I, Martin G, Friedlein A, Langen H, Keller W. EMBO J. 2004;23:616–626. doi: 10.1038/sj.emboj.7600070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lykke-Andersen S, Brodersen DE, Jensen TH. J Cell Sci. 2009;122:1487–1494. doi: 10.1242/jcs.047399. [DOI] [PubMed] [Google Scholar]
- 52.Tomecki R, Drazkowska K, Dziembowski A. Chembiochem. 2010;11:938–945. doi: 10.1002/cbic.201000025. [DOI] [PubMed] [Google Scholar]
- 53.Iglesias N, Tutucci E, Gwizdek C, Vinciguerra P, Von Dach E, Corbett AH, Dargemont C, Stutz F. Genes Dev. 2010;24:1927–1938. doi: 10.1101/gad.583310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lund MK, Guthrie C. Mol Cell. 2005;20:645–651. doi: 10.1016/j.molcel.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 55.Dunn EF, Hammell CM, Hodge CA, Cole CN. Genes Dev. 2005;19:90–103. doi: 10.1101/gad.1267005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brune C, Munchel SE, Fischer N, Podtelejnikov AV, Weis K. RNA. 2005;11:517–531. doi: 10.1261/rna.7291205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tucker M, Valencia-Sanchez MA, Staples RR, Chen J, Denis CL, Parker R. Cell. 2001;104:377–386. doi: 10.1016/s0092-8674(01)00225-2. [DOI] [PubMed] [Google Scholar]
- 58.Wiederhold K, Passmore LA. Biochem Soc Trans. 2010;38:1531–1536. doi: 10.1042/BST0381531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Garneau NL, Wilusz J, Wilusz CJ. Nat Rev Mol Cell Biol. 2007;8:113–126. doi: 10.1038/nrm2104. [DOI] [PubMed] [Google Scholar]
- 60.Brown CE, Tarun SZ, Jr, Boeck R, Sachs AB. Mol Cell Biol. 1996;16:5744–5753. doi: 10.1128/mcb.16.10.5744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yamashita A, Chang TC, Yamashita Y, Zhu W, Zhong Z, Chen CY, Shyu AB. Nat Struct Mol Biol. 2005;12:1054–1063. doi: 10.1038/nsmb1016. [DOI] [PubMed] [Google Scholar]
- 62.Uchida N, Hoshino S, Katada T. J Biol Chem. 2004;279:1383–1391. doi: 10.1074/jbc.M309125200. [DOI] [PubMed] [Google Scholar]
- 63.Brown CE, Sachs AB. Mol Cell Biol. 1998;18:6548–6559. doi: 10.1128/mcb.18.11.6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wilmes GM, Bergkessel M, Bandyopadhyay S, Shales M, Braberg H, Cagney G, Collins SR, Whitworth GB, Kress TL, Weissman JS, Ideker T, Guthrie C, Krogan NJ. Mol Cell. 2008;32:735–746. doi: 10.1016/j.molcel.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kerr SC, Azzouz N, Fuchs SM, Collart MA, Strahl BD, Corbett AH, Laribee RN. PLoS One. 2011;6:e18302. doi: 10.1371/journal.pone.0018302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yu H, Braun P, Yildirim MA, Lemmens I, Venkatesan K, Sahalie J, Hirozane-Kishikawa T, Gebreab F, Li N, Simonis N, Hao T, Rual JF, Dricot A, Vazquez A, Murray RR, Simon C, Tardivo L, Tam S, Svrzikapa N, Fan C, de Smet AS, Motyl A, Hudson ME, Park J, Xin X, Cusick ME, Moore T, Boone C, Snyder M, Roth FP, Barabasi AL, Tavernier J, Hill DE, Vidal M. Science. 2008;322:104–110. doi: 10.1126/science.1158684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dheur S, Nykamp KR, Viphakone N, Swanson MS, Minvielle-Sebastia L. J Biol Chem. 2005;280:24532–24538. doi: 10.1074/jbc.M504720200. [DOI] [PubMed] [Google Scholar]
- 68.Qu X, Perez-Canadillas JM, Agrawal S, De Baecke J, Cheng H, Varani G, Moore C. J Biol Chem. 2007;282:2101–2115. doi: 10.1074/jbc.M609981200. [DOI] [PubMed] [Google Scholar]
- 69.Zhelkovsky A, Tacahashi Y, Nasser T, He X, Sterzer U, Jensen TH, Domdey H, Moore C. RNA. 2006;12:435–445. doi: 10.1261/rna.2267606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chekanova JA, Belostotsky DA. RNA. 2003;9:1476–1490. doi: 10.1261/rna.5128903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen HM, Futcher B, Leatherwood J. PLoS One. 2011;6:e26804. doi: 10.1371/journal.pone.0026804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Assenholt J, Mouaikel J, Saguez C, Rougemaille M, Libri D, Jensen TH. RNA. 2011;17:1788–1794. doi: 10.1261/rna.2919911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Azzouz N, Panasenko OO, Colau G, Collart MA. PLoS One. 2009;4:e6760. doi: 10.1371/journal.pone.0006760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Viphakone N, Voisinet-Hakil F, Minvielle-Sebastia L. Nucleic Acids Res. 2008;36:2418–2433. doi: 10.1093/nar/gkn080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wahle E. J Biol Chem. 1995;270:2800–2808. doi: 10.1074/jbc.270.6.2800. [DOI] [PubMed] [Google Scholar]
- 76.Keller RW, Kuhn U, Aragon M, Bornikova L, Wahle E, Bear DG. J Mol Biol. 2000;297:569–583. doi: 10.1006/jmbi.2000.3572. [DOI] [PubMed] [Google Scholar]
- 77.Hino H, Araki K, Uyama E, Takeya M, Araki M, Yoshinobu K, Miike K, Kawazoe Y, Maeda Y, Uchino M, Yamamura K. Hum Mol Genet. 2004;13:181–190. doi: 10.1093/hmg/ddh017. [DOI] [PubMed] [Google Scholar]
- 78.Lukong KE, Chang KW, Khandjian EW, Richard S. Trends Genet. 2008;24:416–425. doi: 10.1016/j.tig.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 79.Danckwardt S, Hentze MW, Kulozik AE. EMBO J. 2008;27:482–498. doi: 10.1038/sj.emboj.7601932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ropers HH. Curr Opin Genet Dev. 2008;18:241–250. doi: 10.1016/j.gde.2008.07.008. [DOI] [PubMed] [Google Scholar]
- 81.Matthews JM, Sunde M. IUBMB Life. 2002;54:351–355. doi: 10.1080/15216540216035. [DOI] [PubMed] [Google Scholar]
- 82.Godin KS, Varani G. RNA Biol. 2007;4:69–75. doi: 10.4161/rna.4.2.4869. [DOI] [PubMed] [Google Scholar]
- 83.Glisovic T, Bachorik JL, Yong J, Dreyfuss G. FEBS Lett. 2008;582:1977–1986. doi: 10.1016/j.febslet.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kang H, Schuman EM. Science. 1996;273:1402–1406. doi: 10.1126/science.273.5280.1402. [DOI] [PubMed] [Google Scholar]
- 85.Chen C, Ito K, Takahashi A, Wang G, Suzuki T, Nakazawa T, Yamamoto T, Yokoyama K. Biochem Biophys Res Commun. 2011;411:360–364. doi: 10.1016/j.bbrc.2011.06.148. [DOI] [PubMed] [Google Scholar]
- 86.MacDonald CC, McMahon KW. Wiley Interdiscip Rev RNA. 2010;1:494–501. doi: 10.1002/wrna.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shankarling GS, Coates PW, Dass B, Macdonald CC. BMC Mol Biol. 2009;10:22. doi: 10.1186/1471-2199-10-22. [DOI] [PMC free article] [PubMed] [Google Scholar]