

Abstract
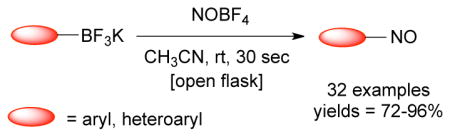
Organotrifluoroborates have emerged as an alternative to toxic and air- and moisture sensitive organometallic species for the synthesis of functionalized aryl and heteroaryl compounds. It has been shown that the trifluoroborate moiety can be easily converted into a variety of different substituents in a late synthetic stage. In this paper we disclose a mild, selective, and convenient method for the ipso-nitrosation of organotrifluoroborates using nitrosonium tetrafluoroborate (NOBF4). Aryl- and heteroaryltrifluoroborates were converted into the corresponding nitroso products in good to excellent yields. This method proved to be tolerant of a broad range of functional groups.
Keywords: Nitrosation, organotrifluoroborates, ipso-substitution
Introduction
Nitroso compounds are versatile synthetic intermediates and have been utilized in a variety of transformations1 such as nitroso aldol reactions,2 [4+2],3 [3+3],4 and [2+2]5 cycloadditions, ene reactions,6 addition of Grignard reagents,7 reactions with alkynes to yield indoles,8 coupling with amines to afford azo compounds,9 oxidation to nitro compounds10 and reduction to amines11 (Scheme 1). Additionally, aromatic nitroso species have shown some activity against HIV-1 infectivity.12 Despite their potentially wide applications, many of these reported methods utilize a single or limited subset of nitroso aromatics, presumably because of the lack of synthetic methods available to synthesize a diverse set of functionalized nitrosoarenes.
Scheme 1.

The first synthesis of nitrosobenzene was published by Baeyer over a century ago.13 Since then, various methods have been published to afford nitrosoarenes.14 Among them, the oxidation of anilines to the corresponding nitrosoarene is the most widely utilized.15 Although many protocols for this conversion are reported in the literature, their reliance on the availability of anilines makes them somewhat limited in scope. Furthermore, the use of oxidants restricts the range of functional groups allowed in this transformation. As an example, aldehyde-containing nitrosoarenes cannot be made by this method. Another problem generally associated with this method is the formation of undesired side products such as azo and azoxy compounds.14 Moreover, few heteroarylnitroso compounds have been obtained by this method, and those that have been accessed have been confined to nitrogen-containing heterocycles.16 Nitrosation of simple arenes17 and arylmetallics (e.g., organotin,18 thallium19 and silicon20 compounds) have also been reported in the literature using electrophilic nitrosonium reagents. For both of these types of transformations the reaction only works for aryl species containing electron-donating groups, which limits the breadth of nitroso products that can be accessed. Because of the limited examples using organometallic species and the drawbacks associated with oxidation reactions of aryl and heteroarylnitroso synthesis (e.g., functional group tolerance and side product formation), we were interested in finding a novel, rapid, and mild method to synthesize nitrosoarene derivatives.
The ipso-substitution of arylboron species, as previously demonstrated for halogenation21 and nitration22 of arylboronic acids, provides a potential means to accomplish this goal. Recently, our group published the chlorodeboronation of aryl and heteroaryltrifluoroborates, which most likely occurs by an ipso-substitution (Scheme 2).23
Scheme 2.

Trifluoroborates have emerged as an alternative to toxic organometallic species, such as organostannanes, and to boronic acids, which, although non-toxic, are compounds susceptible to undesired side reactions with common reagents, such as acids and bases.24 The tetracoordinate nature of the trifluoroborates makes them resistant to a variety of reaction conditions and, therefore, allows one to build complexity into a molecule while leaving the carbon-boron bond intact. This valuable bond can then be further converted in a later synthetic step into a variety of groups such as boronic acids,25 alcohols26 and halogens.21a–c,23 Moreover, trifluoroborates can be synthesized by a variety of complementary methods, including transmetalation (via metal-halogen exchange or directed metalation), Miyaura borylation, and C-H activation, all of which combine to afford an enormous diversity of available substructures (Scheme 3). Furthermore, in contrast to the corresponding aryl and heteroarylboronic acids, trifluoroborates are air and moisture stable and can be stored on the bench for months without appreciable decomposition.27
Scheme 3.

In this paper we disclose the ipso-nitrosation of a broad range of aryl and heteroaryltrifluoroborates containing both electron-donating and electron-withdrawing groups. To the best of our knowledge, this is the first nitrosation of an organoboron species, and the transformation presented is arguably the most broadly applicable approach to this underrepresented class of molecules.
RESULTS AND DISCUSSION
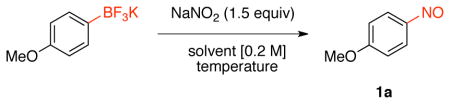
Based on the ipso-nitration of boronic acids with nitrate salts developed by Olah and co-workers,23 we began the screening for nitrosation of organotrifluoroborates with sodium nitrite in different solvents (Table 1). The choice of this nitrite salt was made by the ready availability and low cost of this reagent. After optimization, we determined that the reaction of potassium trifluoro(4-methoxyphenyl)borate with NaNO2 (1.5 equiv) in heptane: H2O at 50 °C afforded the desired nitrosated product in 89% isolated yield.
Table 1.
Optimization with Sodium Nitrite
 | ||||
|---|---|---|---|---|
| entry | solvent | temperature | reaction time (h) | 11B NMR/GC/MS |
| 1 | EtOAc | rt | 48 | S.M. |
| 2 | CH3CN | rt | 48 | S.M. |
| 3 | heptane | rt | 48 | S.M. |
| 4 | H2O | rt | 4 | 1a: protodeboronation (1:1) |
| 5 | EtOAc:H2O | rt | 4 | 1a: protodeboronation (3:1) |
| 6 | CH3CN:H2O | rt | 4 | 1a: protodeboronation (2:1) |
| 7 | heptane:H2O | rt | 4 | 1a |
| 8 | heptane:H2O | 50 °C | 2 | 1a (89% isolated yield) |
With these conditions in hand, we began to examine the nitrosation of a variety of aryltrifluoroborates (Scheme 4). Phenyltrifluoroborates bearing electron-donating groups were successfully converted into the corresponding nitrosobenzene in good yields. Unfortunately, electron-neutral aryltrifluoroborates (e.g., biphenyl) and electron-withdrawing (ester) groups inhibited this transformation, and only the protodeboronated products were obtained.
Scheme 4.

The results obtained further demonstrated that the reaction does not occur in the absence of water and that only electron-rich aryltrifluoroborates afforded the desired product. Thus, we hypothesized that aqueous conditions are necessary to form the tricoordinate boron species in situ,28 and this species, now possessing a Lewis acidic boron moiety with an empty p-orbital, could then undergo attack of sodium nitrite to form an ate-complex and a more electrophilic NO+, with subsequent ipso-substitution affording the nitroso product (Scheme 5).
Scheme 5.

To improve the scope of this reaction, the nitrosation of potassium [1,1′-biphenyl]-4-yltrifluoroborate was further optimized. A variety of solvents, additives, nitrosating agents and temperatures were investigated. As illustrated in Table 2, the use of other nitrite salts, such as KNO2 and AgNO2 (entries 1–3), were inefficient for this transformation. The use of acid additives for in situ formation of NO+ 18c,29 also did not afford the desired nitroso product, and only protodeboronation was observed (entries 4–7). Fortunately, the use of nitrosonium tetrafluoroborate (1.03 equiv) in CH3CN (0.2 M) at room temperature in an open flask proved to be efficient for this transformation, affording the nitroso product in 90% isolated yield. Importantly, the reaction can be followed visually. The slurry formed by the trifluoroborate in CH3CN becomes a bright green, homogeneous solution almost immediately. The crude reaction is then worked up by addition of water followed by dichloromethane extraction, with subsequent filtration through a plug of silica providing the product in high purity. A prolonged reaction time leads to oxidation of the formed nitroso product and affords a mixture of this compound along with the corresponding nitroaromatic. The use of more than 1.03 equivalents of nitrosonium tetrafluoroborate does not fully convert the nitroso into the nitro group. Instead, a mixture of nitroso, nitro and protodeboronation products is observed.
Table 2.
Optimization of the Nitrosation of Potassium [1,1′-Biphenyl]-4-yltrifluoroborate
 | ||||
|---|---|---|---|---|
| entry | NO agent | solvent | reaction time | GC/MS |
| 1 | NaNO2 | heptane:H2O (1:1) | 4 h | protodeboronation |
| 2 | KNO2 | heptane:H2O (1:1) | 4 h | protodeboronation |
| 3 | AgNO2 | heptane:H2O (1:1) | 2 h | protodeboronation |
| 4 | NaNO2:HCl | heptane:H2O (1:1) | 1 h | protodeboronation |
| 5 | KNO2:HCl | heptane:H2O (1:1) | 1 h | protodeboronation |
| 6 | AgNO2:HCl | heptane:H2O (1:1) | 1 h | protodeboronation |
| 7 | NaNO2:TMSCl | CH2Cl2:H2O (1:1) | 1 h | protodeboronation |
| 8 | NOBF4 | CH3CN | 30 sec | product (90% isolated yield) |
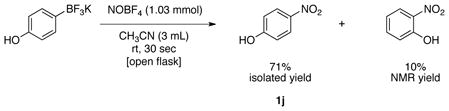
With the optimal conditions in hand, the scope of the reaction for electron-donating and electron neutral aryltrifluoroborates was investigated (Table 3). In all cases, the reaction was complete in only 30 seconds and afforded the desired product in good to excellent yields. The method proved to be selective, and aryltrifluoroborates containing ortho, meta and para substituents were readily converted to the corresponding nitrosobenzene (Table 3, entries 1–3). This regioselectivity cannot be attained by the direct nitrosation of arenes. The reaction was scaled up to 1 g, and the product was obtained in excellent yield (Table 3, entry 1). Sterically hindered substrates also afforded the desired product in good yield. Importantly, potassium (3,5-diisopropylphenyl)trifluoroborate, made by direct C-H activation of arenes30 was converted into 1,3-diisopropyl-5-nitrosobenzene in 88% yield (Table 3, entry 9). This illustrates a unique substitution pattern, because the corresponding aryl chloride (necessary for preparation of the amine utilized for the oxidation method previously mentioned) has very limited availability. Surprisingly, the reaction of potassium trifluoro(4-hydroxyphenyl)borate yielded the corresponding nitrophenol as a mixture of regioisomers (eq 1).
Table 3.
Nitrosation of Electron-rich and Electron-neutral Potassium Aryltrifluoroborates
 | ||
|---|---|---|
| entry | product | yield (%) |
| 1 |
 1a |
95a |
| 2 |
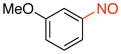 1b |
89 |
| 3 |
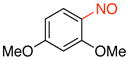 1c |
91 |
| 4 |
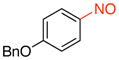 1d |
92 |
| 5 |
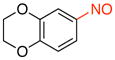 1e |
91 |
| 6 |
 1f |
90 |
| 7 |
 1g |
93 |
| 8 |
 1h |
92 |
| 9 |
 1i |
88 |
Reaction conditions: 1 mmol of aryltrifluoroborate and NOBF4 (1.03 equiv) in 3 mL of CH3CN for 30 sec at room temperature in an open flask.
5 mmol scale.
 |
(1) |
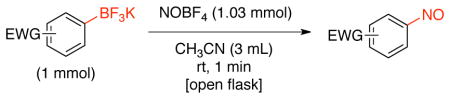
Subsequently, the reaction of aryltrifluoroborates bearing electron-withdrawing groups was investigated (Table 4). Methods such as direct nitrosation of arenes and other organometallic species have proven inefficient in the production of nitrosobenzenes with electron-poor groups.18–21 In our hands, aryltrifluoroborates containing ester, ketone, aldehyde, nitrile, amide, nitro and carboxylic acid groups (Table 4, entries 1–9) were converted into the corresponding nitroso compounds in good yields without affecting the aforementioned, embedded functional groups. The reaction was regiospecific, and ortho, meta, and para substituted nitrosobenzenes were obtained. Importantly, aldehyde-containing aryltrifluoroborates afforded the corresponding nitrosobenzaldehyde in good yields and high regioselectivity without oxidation of the aldehyde group (Table 4, entries 4–6). These aldehyde-containing nitroso products were previously obtained only by a four step procedure from the corresponding nitroarene.31 As illustrated previously with potassium (3,5-diisopropylphenyl)trifluoroborate (Table 3, entry 9), we were able to synthesize methyl 3-methyl-5-nitrosobenzoate and 1-methoxy-3-nitroso-5-(trifluoromethyl)benzene (Table 4, entries 10 and 11) from trifluoroborates made by C-H activation. Furthermore, the conversion of aryltrifluoroborates containing halogens into the corresponding nitroso product was accomplished in good yields (Table 4, entries 12–15).
Table 4.
Nitrosation of Electron-poor Potassium Aryltrifluoroborates
 | ||
|---|---|---|
| entry | product | yield (%) |
| 1 |
 2a |
96 |
| 2 |
 2b |
95 |
| 3 |
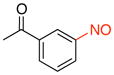 2c |
94 |
| 4 |
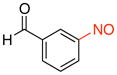 2d |
91 |
| 5 |
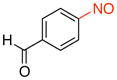 2e |
94 |
| 6 |
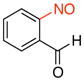 2f |
78 |
| 7 |
 2g |
89 |
| 8 |
 2h |
91 |
| 9 |
 2i |
90 |
| 10 |
 2j |
91 |
| 11 |
 2k |
81 |
| 12 |
 2l |
92 |
| 13 |
 2m |
94 |
| 14 |
 2n |
92 |
| 15 |
 2o |
79 |
Reaction conditions: 1 mmol of aryltrifluoroborate and NOBF4 (1.03 equiv) in 3 mL of CH3CN for 30 sec at room temperature in an open flask.
To expand the scope of this reaction further we turned our attention to the reaction of heteroaryltrifluoroborates. Once more, this transformation was accomplished for a variety of substrates, including dibenzofuranyl, dibenzothienyl, benzothienyl, indolyl, pyrimidinyl and pyridinyl derivatives, affording the nitrosoheteroaryl products in good yields (Table 5). Furthermore, products 3e, 3f and 3g (Table 5 entries 5– 6) were obtained with no observed nitrosation of the heterocyclic nitrogen.32 To the best of our knowledge, all compounds illustrated in Table 5 were never before synthesized by any other method. However, for 5-membered heteroaryltrifluoroborates (e.g., thienyl, furanyl, pyrrolyl, isoxazolyl and pyrazolyl) and fused system with the trifluoroborate substituent within the 5-membered heterocycle (e.g., 2- or 3-substituted dibenzofuranyl, dibenzothienyl and indolyl), the reaction was inefficient, and only protodeboronated product was recovered. Moreover, the reaction with 3-trifluoroboratopyridines containing a substituent at the 6 position afforded a mixture of nitro and dinitro products, and no nitroso derivatives were observed (eq 2). The use of more than 1 equivalent of NOBF4 did not give the dinitro product; instead a mixture of products along with protodeboronation was observed. The same pattern was observed for quinolines bearing trifluoroborates at the 2, 3 and 4 positions, where a mixture of nitro and dinitro derivatives was obtained.
Table 5.
Nitrosation of Potassium Heteroaryltrifluoroborates
 | ||
|---|---|---|
| entry | product | yield (%) |
| 1 |
 3a |
85 |
| 2 |
 3b |
80 |
| 3 |
 3c |
81 |
| 4 |
 3d |
73 |
| 5 |
 3e |
82 |
| 6 |
 3f |
76 |
| 7 |
 3g |
72 |
| 8 |
 3h |
62a |
Reaction conditions: 1 mmol of aryltrifluoroborate and NOBF4 (1.03 equiv) in 3 mL of CH 3CN for 30 sec at room temperature in an open flask.
NMR yield using EtOAc as internal standard
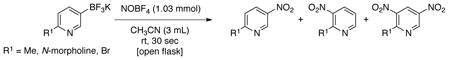 |
(2) |
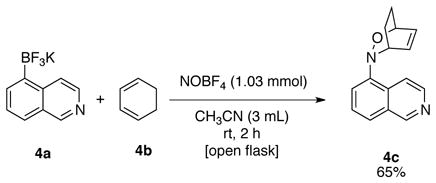
Interestingly, 5-nitrosoisoquinoline 3h (Table 5, entry 8) was obtained as a yellow solid that upon exposure to air would turn black and could not be further purified. The crude material appeared to be very pure by 1H NMR (see spectra in Supporting Information), which led to the conclusion that the nitroso product obtained is not stable. To circumvent this problem, a one-pot nitrosation of potassium trifluoro(isoquinolin-5-yl)borate, 4a, followed by Diels-Alder reaction with cyclohexa-1,3-diene 4b, was investigated (eq 3).13a The reaction afforded the Diels-Alder adduct in 65% yield over two steps.
 |
(3) |
With the success of the nitroso one-pot Diels-Alder reaction, we were interested in illustrating other reactions that potentially unstable arylnitroso compounds can undergo (Scheme 6). Nitrogen-containing compounds are found in a variety of pharmaceuticals and are also the building blocks for important synthetic transformations.33 Therefore, potassium methyl 3-trifluoroboratobenzoate was subjected to the nitrosation protocol followed by different transformations, and diverse nitrogen-containing products were obtained. The one-pot reaction of the aforementioned trifluoroborate with NOBF4 followed by addition of NaBH4 afforded the corresponding azoxy product, 5a, in 87% overall yield. Methyl 3-nitrobenzoate 5b was also obtained by a two-step procedure from the corresponding trifluoroborate. In this case, a minimal work-up of the nitrosation reaction was necessary before addition of the oxidant. Nevertheless, the desired product was obtained in 86% yield over the two steps. The one-pot nitrosation-reduction of the in situ formed methyl 3-nitrosobenzoate was performed, and the methyl 3-aminobenzoate, 5c, was obtained in 72% overall yield. The one-pot nitrosation/Diels-Alder reaction was also accomplished, with the oxazabicyclo benzoate 5d being isolated in 82% yield.
Scheme 6.

Finally, as illustrated in Scheme 7, different boron derivatives were tested under the same reaction conditions. 4-Methoxyphenylboronic acid afforded the product in nearly the same yield as the trifluoroborates, while the boronate esters were not successful in this transformation, instead providing the nitroso product in moderate yields after 1 h with starting material being recovered.
Scheme 7.

In summary, it has been demonstrated that the nitrosation of a broad range of aryl and heteroaryltrifluoroborates can be carried out under extraordinarily mild reaction conditions. Aryltrifluoroborates containing different functional groups, such as esters, ketones, aldehydes, nitriles and amides were successfully converted into the nitroso product, while leaving the aforementioned groups intact. Furthermore, nitrogen-containing heteroaryltrifluoroborates underwent nitrosation selectively, and no nitrosation of the nitrogen atom was observed. Despite their simplicity, most of the nitroso compounds prepared were previously unknown, highlighting the lack of synthetic methods available for this important class of molecules. The versatility of the nitroso products obtained has been illustrated by converting these intermediates in a variety of one-pot transformations, demonstrating that even those nitrosoarenes that may have limited stability can be employed as useful substrates for further synthetic applications.
Experimental Section
General Procedure for the Preparation of Potassium Aryl and Heteroaryltrifluoroborates from Boronic Acids
Following a published literature procedure,34 to a solution of the corresponding boronic acid in MeOH (3.5 M or enough MeOH to give a free flowing suspension) under N2 was added KHF2 (3 equiv of a 4.5 M solution in H2O) dropwise at 0° C. The ice-water bath was removed, and the reaction was stirred at rt until 11B NMR indicated completion of the reaction (~2 min). The crude mixture was concentrated and dried overnight in vacuo. The crude solid was purified using continuous Soxhlet extraction (4 h) with acetone (60 mL). The collected solvent was concentrated and then redissolved in a minimal amount of acetone (5 mL). The addition of Et2O (30 mL) led to the precipitation of the product. The product was filtered, concentrated, and dried in vacuo to afford the pure organotrifluoroborates.
General Procedure for Aryl C-H borylation
Following the procedure published by Hartwig and co-workers,35 in a glove box, to an oven-dried glass vessel capable of being sealed with a Teflon cap (for microwave vials) was added B2pin2, (1 equiv), [Ir(COD)(OMe)]2 (0.1 mol %) and dtbpy (0.2 mol %). The vessel was sealed and removed from the glovebox. THF (1 M degassed) was added via syringe followed by the addition of the 1,3-substituted arene (1.5 equiv). The reaction mixture was heated in a sealed vessel at 80 °C for 16 h. The reaction was allowed to cool to rt, and then KHF2 (3 equiv of a 4.5 M solution in H2O) was added dropwise at 0° C. The ice-water bath was removed, and the reaction was stirred at rt until 11B NMR indicated completion of the reaction (~2 min). The crude mixture was concentrated and dried overnight in vacuo. The crude solid was purified using continuous Soxhlet extraction (4 h) with acetone (60 mL). The collected solvent was concentrated and then redissolved in a minimal amount of acetone (5 mL). The addition of Et2O (30 mL) led to the precipitation of the product. The product was filtered, concentrated, and dried in vacuo to afford the pure organotrifluoroborates.
General Procedure A: Nitrosation of Aryltrifluoroborates with NaNO2
To a 20 mL glass microwave vial containing a solution of potassium organotrifluoroborate (1 mmol) in heptane: H2O (1:1, 5 mL, 0.2 M) was added NaNO2 (104 mg, 1.5 mmol, 1.5 equiv) in one portion. The reaction was stirred open to air at 50 °C until the trifluoroborate was consumed (as indicated by 11B NMR). To the crude mixture was added H2O (20 mL) and CH2Cl2 (10 mL). The layers were separated and the aqueous layer was extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were dried (Na2SO4), filtered, and concentrated under reduced pressure. The products were obtained in high purity after filtration through a small plug of silica topped with Celite, eluting with CH2Cl2.
General Procedure B: Nitrosation of Aryl and Heteroaryltrifluoroborates with NOBF4
To a 20 mL glass microwave vial containing a solution of potassium organotrifluoroborate (1 mmol) in CH3CN (3 mL, 0.33 M) was added NOBF4 (120 mg, 1.03 mmol, 1.03 equiv) in one portion. The reaction was stirred open to air at rt until the reaction became homogeneous. The reaction changed from a white slurry to a green or black solution. To the crude mixture was added H2O (20 mL) and CH2Cl2 (10 mL). The layers were separated, and the aqueous layer was extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were dried (Na2SO4), filtered, and concentrated under reduced pressure. In general the product was obtained in high purity after filtration through a small plug of silica topped with Celite, eluting with CH2Cl2/hexanes. In specific cases trace impurities were removed by column chromatography using CH2Cl2/hexanes or EtOAc/hexanes to afford the desired pure product.
1-Methoxy-4-nitrosobenzene (1a).8a
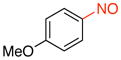
General procedure B was employed using potassium 4-methoxyphenyltrifluoroborate (214 mg, 1 mmol) and NOBF4 (120 mg, 1.03 mmol). The reaction was complete in 30 seconds. The desired pure product was obtained in 95% yield (130 mg, 0.95 mmol) as a green oil after filtration through a short plug of silica topped with Celite using hexanes/CH2Cl2 (3:1). 1H NMR (500 MHz, CDCl3) δ 7.91 (d, J = 6.5 Hz, 2H), 7.02 (d, J = 8.0 Hz, 2H), 3.94 (s, 3H). 13C NMR (125.8 MHz, CDCl3) δ 165.6, 163.9, 124.3, 113.8, 55.9. IR (neat) 1598, 1504, 1411, 1263, 1020, 837 cm−1. HRMS (ESI) m/z calcd. for C7H8NO2 (M+H)+ 138.0555, found 138.0558.
1-Methoxy-3-nitrosobenzene (1b)
General procedure B was employed using potassium 3-methoxyphenyltrifluoroborate (214 mg, 1 mmol) and NOBF4 (120 mg, 1.03 mmol). The reaction was complete in 30 seconds. The desired pure product was obtained in 89% yield (122 mg, 0.89 mmol) as a green oil after column chromatography with hexanes/CH2Cl2 (3:1) as eluent. 1H NMR (500 MHz, CDCl3) δ 8.02 (m, 1H), 7.60 (t, J = 8 Hz, 1H), 7.28 (m, 1H), 6.89 (t, J = 2 Hz, 1H), 3.86 (s, 3H). 13C NMR (125.8 MHz, CDCl3) δ 166.9, 160.5, 130.5, 122.9, 119.8, 99.8, 55.8. IR (neat) 1604, 1483, 1384, 1041, 789 cm−1. HRMS (ESI) m/z calcd. for C7H8NO2 (M+H)+ 138.0555, found 138.0558.
2,4-Dimethoxy-1-nitrosobenzene (1c)
General procedure B was employed using potassium (2,4-dimethoxyphenyl)trifluoroborate (244 mg, 1 mmol) and NOBF4 (120 mg, 1.03 mmol). The reaction was complete in 30 seconds. The desired pure product was obtained in 91% yield (152 mg, 0.91 mmol) as a green solid, mp 93–95 °C, after filtration through a short plug of silica topped with Celite using hexanes/CH2Cl2 (3:1). 1H NMR (500 MHz, CDCl3) δ 6.65 (d, J = 2.5 Hz, 1H), 6.50 (d, J = 9 Hz, 1H) 6.34 (m, 1H), 4.22 (s, 3H), 3.92 (s, 3H). 13C NMR (125.8 MHz, CDCl3) δ 168.5, 164.4, 157.1, 112.1, 105.8, 98.5, 56.9, 56.2. IR (neat) 1600, 1397, 1246, 1014, 837 cm−1. HRMS (ESI) m/z calcd. for C8H10NO3 (M+H)+ 168.0661, found 168.0664.
1-(Benzyloxy)-4-nitrosobenzene (1d)
General procedure B was employed using potassium (4-(benzyloxy)phenyl)trifluoroborate (290 mg, 1 mmol) and NOBF4 (120 mg, 1.03 mmol). The reaction was complete in 30 seconds. The desired pure product was obtained in 92% yield (196 mg, 0.92 mmol) as a blue solid, mp 81–83 °C, after filtration through a short plug of silica topped with Celite using hexanes/CH2Cl2 (3:1). 1H NMR (500 MHz, CDCl3) δ 7.93 (brs, 2H), 7.45 – 7.37 (m, 5H), 7.10 (t, J = 8 Hz, 2H), 5.21 (s, 2H). 13C NMR (125.8 MHz, CDCl3) δ 164.9, 164.1, 135.6, 129.0, 128.7, 127.7, 114.9, 70.8. IR (neat) 1598, 1502, 1262, 1117, 844, 730 cm−1. HRMS (ESI) m/z calcd. for C13H11NO2 (M)+ 213.0790, found 213.0797.
6-Nitroso-2,3-dihydrobenzo[b][1,4]dioxine (1e)
General procedure B was employed using potassium (2,3-dihydrobenzo[b][1,4]dioxin-6-yl)trifluoroborate (242 mg, 1 mmol) and NOBF4 (120 mg, 1.03 mmol). The reaction was complete in 30 seconds. The desired pure product was obtained in 91% yield (150 mg, 0.91 mmol) as a green solid, mp 88–90 °C, after column chromatography with hexanes/CH2Cl2 (3:1) as eluent. 1H NMR (500 MHz, CDCl3) δ 7.84 (d, J = 7.5 Hz, 1H), 7.13 (s, 1H), 7.08 (d, J = 8.5 Hz, 1H), 4.38 – 4.36 (m, 2H), 4.33 – 4.31 (m, 2H). 13C NMR (125.8 MHz, CDCl3) δ 163.6, 150.9, 143.9, 120.8, 117.6, 107.7, 65.1, 64.2. IR (neat) 1591, 1495, 1280, 1054, 913 cm−1. HRMS (ESI) m/z calcd. for C8H8NO3 (M+H)+ 166.0504, found 166.0504.
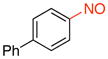
4-Nitrosobiphenyl (1f).36
General procedure B was employed using potassium biphenyl-4-yltrifluoroborate (260 mg, 1 mmol) and NOBF4 (120 mg, 1.03 mmol). The reaction was complete in 30 seconds. The desired pure product was obtained in 90% yield (165 mg, 0.90 mmol) as an orange solid, mp 72–74 °C (lit.34 73–74 °C), after filtration through a short plug of silica topped with Celite using hexanes/CH2Cl2 (3:1). 1H NMR (500 MHz, CDCl3) δ 7.98 (d, J = 8.5 Hz, 2H), 7.83 (d, J = 8 Hz, 2H), 7.68 – 7.66 (m, 2H), 7.52 – 7.49 (m, 2H), 7.45 (m, 1H). 13C NMR (125.8 MHz, CDCl3) δ 165.12, 148.2, 139.3, 129.3, 129.1, 128.0, 127.6, 121.8. IR (neat) 1483, 1249, 760, 695 cm−1. HRMS (ESI) m/z calcd. for C12H10NO (M+H)+ 184.0762, found 184.0758.
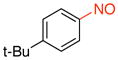
1-tert-Butyl-4-nitrosobenzene (1g)
General procedure B was employed using potassium (4-tert-butylphenyl)trifluoroborate (240 mg, 1 mmol) and NOBF4 (120 mg, 1.03 mmol). The reaction was complete in 30 seconds. The desired pure product was obtained in 93% yield (152 mg, 0.93 mmol) as a green oil after filtration through a short plug of silica topped with Celite using hexanes/CH2Cl2 (3:1). 1H NMR (500 MHz, CDCl3) δ 7.83 (d, J = 8.5 Hz, 2H), 7.60 (d, J = 9 Hz, 2H), 1.37 (s, 9H). 13C NMR (125.8 MHz, CDCl3) δ 165.3, 159.9, 126.2, 121.1, 35.7, 31.1. IR (neat) 1601, 1509, 1453, 1124, 1099, 840, 710 cm−1. HRMS (ESI) m/z calcd. for C10H14NO (M+H)+ 164.1075, found 164.1082.
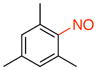
1,3,5-Trimethyl-2-nitrosobenzene (1h).17a
General procedure B was employed using potassium trifluoro(mesityl)borate (260 mg, 1 mmol) and NOBF4 (120 mg, 1.03 mmol). The reaction was complete in 30 seconds. The desired pure product was obtained in 92% yield (137 mg, 0.90 mmol) as a white solid, mp 120–122 °C (lit.17a 121–122 °C), after filtration column chromatography with hexanes/CH2Cl2 (3:1) as eluent. 1H NMR (500 MHz, CDCl3) δ 6.99 (s, 2H), 2.62 (s, 2H), 2.41 (s, 4H), 2.34 (s, 1H), 2.33 (s, 2H). 13C NMR (125.8 MHz, CDCl3) δ 140.7, 139.3, 132.7, 129.9, 21.2, 18.7. IR (neat) 1603, 1475, 1245, 807 cm−1. HRMS (ESI) m/z calcd. for C9H12NO (M+H)+ 150.0919, found 150.0919.
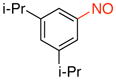
1,3-Diisopropyl-5-nitrosobenzene (1i)
General procedure B was employed using potassium (3,5-diisopropylphenyl)trifluoroborate (268 mg, 1 mmol) and NOBF4 (120 mg, 1.03 mmol). The reaction was complete in 30 seconds. The desired pure product was obtained in 88% yield (168 mg, 0.88 mmol) as a green oil after filtration through a short plug of silica topped with Celite using hexanes/CH2Cl2 (3:1). 1H NMR (500 MHz, CDCl3) δ 7.61 (s, 2H), 7.46 (s, 1H), 3.07 – 3.01 (m, 2H), 1.32 (d, J = 7 Hz, 12 H). 13C NMR (125.8 MHz, CDCl3) δ 167.4, 150.5, 132.7, 117.0, 34.1, 24.0. IR (neat) 1608, 1493, 1096, 886, 694 cm−1. HRMS (ESI) m/z calcd. for C12H18NO (M+H)+ 192.1388, found 192.1384.
4-Nitrophenol (1j).37
General procedure B was employed using potassium trifluoro(4-hydroxyphenyl)borate (200 mg, 1 mmol) and NOBF4 (120 mg, 1.03 mmol). The reaction was complete in 30 seconds. In this case no nitroso product was observed and a mixture of 4-nitrophenol and 2-nitrophenol (10% NMR yield) was obtained. The pure 4-nitrophenol product was obtained in 71% yield (99 mg, 0.71 mmol) as a yellow solid, mp 108–110 °C (lit.38 109–110 °C), after column chromatography with hexanes/CH2Cl2 (2:1) as eluent. 1H NMR (500 MHz, CDCl3) δ 8.18 (d, J = 9.5 Hz, 2H), 6.92 (d, J = 9 Hz, 2H), 5.72 (s, 1H). 13C NMR (125.8 MHz, CDCl3) δ 161.6, 141.7, 126.5, 115.9. IR (neat) 3359, 1592, 1488, 1331, 1113, 844 cm−1.
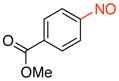
Methyl 4-Nitrosobenzoate (2a).39
General procedure B was employed using potassium trifluoro(4-(methoxycarbonyl)phenyl)borate (242 mg, 1 mmol) and NOBF4 (120 mg, 1.03 mmol). The reaction was complete in 1 min. The desired pure product was obtained in 95% yield (157 mg, 0.95 mmol) as a light yellow solid, mp 123–125 °C (lit.40 129.5 °C), after filtration through a short plug of silica topped with Celite using CH2Cl2. 1H NMR (500 MHz, CDCl3) δ 8.28 (d, J = 8 Hz, 2H), 7.92 (d, J = 8 Hz, 2H), 3.97 (s, 3H). 13C NMR (125.8 MHz, CDCl3) δ 165.9, 164.5, 135.3, 131.2, 120.5, 52.9. IR (neat) 1727, 1441, 1266, 766, 694 cm−1. HRMS (ESI) m/z calcd. for C8H8NO3 (M+H)+ 166.0504, found 166.0510.
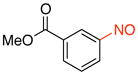
Methyl 3-Nitrosobenzoate (2b).15b
General procedure B was employed using potassium trifluoro(3-(methoxycarbonyl)phenyl)borate (242 mg, 1 mmol) and NOBF4 (120 mg, 1.03 mmol). The reaction was complete in 1 min. The desired pure product was obtained in 96% yield (158 mg, 0.96 mmol) as a light yellow solid, mp 91–93 °C (lit.41 93 °C), after filtration through a short plug of silica topped with Celite using CH2Cl2. 1H NMR (500 MHz, CDCl3) δ 8.60 (t, J = 1.5 Hz, 1H), 8.39 (m, 1H), 8.01 (m, 1H), 7.71 (t, J = 7.5 Hz, 1H), 4.01 (s, 3H). 13C NMR (125.8 MHz, CDCl3) δ 165.8, 164.9, 135.8, 131.9, 129.7, 123.9, 122.6, 52.8. IR (neat) 1727, 1433, 1259, 754, 685 cm−1. HRMS (ESI) m/z calcd. for C8H8NO3 (M+H)+ 166.0504, found 166.0510.
1-(3-Nitrosophenyl)ethanone (2c)
General procedure B was employed using potassium (3-acetylphenyl)trifluoroborate (226 mg, 1 mmol) and NOBF4 (120 mg, 1.03 mmol). The reaction was complete in 1 min. The desired pure product was obtained in 94% yield (140 mg, 0.94 mmol) as a light yellow solid, mp 78–80 °C (lit.38 81.5 °C), after filtration through a short plug of silica topped with Celite using CH2Cl2. 1H NMR (500 MHz, CDCl3) δ 8.47 (t, J = 1.5 Hz, 1H), 8.33 (m, 1H), 8.05 (m, 1H), 7.75 (t, J = 8 Hz, 1H), 2.73 (s, 3H). 13C NMR (125.8 MHz, CDCl3) δ 196.8, 165.0, 138.2, 134.3, 130.0, 124.3, 121.0, 26.9. IR (neat) 1691, 1248, 800, 676 cm−1. HRMS (CI) m/z calcd. for C8H8NO2 (M+H)+ 150.0555, found 150.0557.
3-Nitrosobenzaldehyde (2d)
General procedure B was employed using potassium trifluoro(3-formylphenyl)borate (212 mg, 1 mmol) and NOBF4 (120 mg, 1.03 mmol). The reaction was complete in 1 min. The desired pure product was obtained in 88% yield (119 mg, 0.88 mmol) as a light yellow solid, mp 106–108 °C (lit.42 106.5–107 °C), after filtration through a short plug of silica topped with Celite using CH2Cl2. 1H NMR (500 MHz, CDCl3) δ 10.20 (s, 1H), 8.38 (s, 1H), 8.26 (m, 1H), 8.15 (m, 1H), 7.83 (t, J = 8 Hz, 1H). 13C NMR (125.8 MHz, CDCl3) δ 190.9, 164.7, 137.4, 135.0, 130.5, 125.7, 121.7. IR (neat) 1689, 1257, 1121, 678 cm−1. HRMS (ESI) m/z calcd. for C7H6NO2 (M+H)+ 136.0399, found 136.0402.
4-Nitrosobenzaldehyde (2e).31a
General procedure B was employed using potassium trifluoro(4-formylphenyl)borate (212 mg, 1 mmol) and NOBF4 (120 mg, 1.03 mmol). The reaction was complete in 1 min. The desired pure product was obtained in 94% yield (127 mg, 0.94 mmol) as a light yellow solid, mp 135–137 °C (lit.41 135–136 °C), after filtration through a short plug of silica topped with Celite using CH2Cl2. 1H NMR (500 MHz, CDCl3) δ 10.20 (s, 1H), 8.17 (d, J = 8.5 Hz, 2H), 8.04 (d, J = 8 Hz, 2H). 13C NMR (125.8 MHz, CDCl3) δ 191.4, 163.9, 139.6, 131.2, 121.2. IR (neat) 1691, 1259, 789 cm−1. HRMS (CI) m/z calcd. for C7H5NO2 (M)+ 135.0320, found 135.0322.
2-Nitrosobenzaldehyde (2f).43
General procedure B was employed using potassium trifluoro(2-formylphenyl)borate (212 mg, 1 mmol) and NOBF4 (120 mg, 1.03 mmol). The reaction was complete in 1 min. The desired pure product was obtained in 78% yield (105 mg, 0.78 mmol) as a light yellow solid, mp 110–112 °C (lit.12 110 °C), after filtration through a short plug of silica topped with Celite using CH2Cl2. 1H NMR (500 MHz, CDCl3) δ 12.1 (s, 1H), 8.22 (m, 1H), 7.91 (t, J = 7.5 Hz, 1H), 7.69 (m, 1H), 6.44 (m, 1H). 13C NMR (125.8 MHz, CDCl3) δ 193.5, 162.2, 136.6, 134.2, 132.8, 127.8, 106.7. IR (neat) 1702, 1248, 1196, 768 cm−1. HRMS (ESI) m/z calcd. for C7H6NO2 (M+H)+ 136.0399, found 136.0404.
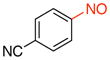
4-Nitrosobenzonitrile (2g).44
General procedure B was employed using potassium (4-cyanophenyl)trifluoroborate (209 mg, 1 mmol) and NOBF4 (120 mg, 1.03 mmol). The reaction was complete in 1 min. The desired pure product was obtained in 89% yield (117 mg, 0.94 mmol) as a light yellow solid, mp 128–130 °C (lit.42 128–129 °C), after filtration through a short plug of silica topped with Celite using CH2Cl2. 1H NMR (500 MHz, CDCl3) δ 7.97 (s, 4 H). 13C NMR (125.8 MHz, CDCl3) δ 162.3, 134.1, 120.9, 118.5, 117.6. IR (neat) 2239, 1499, 1252, 868 cm−1. HRMS (ESI) m/z calcd. for C7H4N2O (M)+ 132.0324, found 132.032.
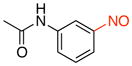
N-(3-Nitrosophenyl)acetamide (2h)
General procedure B was employed using potassium (3-acetamidophenyl)trifluoroborate (241 mg, 1 mmol) and NOBF4 (120 mg, 1.03 mmol). The reaction was complete in 1 min. The desired pure product was obtained in 91% yield (149 mg, 0.91 mmol) as a light yellow solid, mp 118–120 °C, after filtration through a short plug of silica topped with Celite using CH2Cl2. 1H NMR (500 MHz, CDCl3) δ 7.97 (d, J = 8 Hz, 1H), 7.84 (d, J = 8 Hz, 1H), 7.79 (s, 1H), 7.61 (t, J = 8 Hz, 1H), 7.55 (s, 1H), 2.24 (s, 3H). 13C NMR (125.8 MHz, CDCl3) δ 168.9, 165.9, 139.2, 130.2, 126.5, 118.8, 110.3, 24.8. IR (neat) 1672, 1598, 1492, 1076, 800 cm−1. HRMS (ESI) m/z calcd. for C8H9N2O2 (M+H)+ 165.0664, found 165.0659.
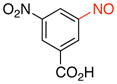
3-Nitro-5-nitrosobenzoic acid (2i)
General procedure B was employed using potassium (3-carboxy-5-nitrophenyl)trifluoroborate (273 mg, 1 mmol) and NOBF4 (120 mg, 1.03 mmol). The reaction was complete in 1 min. The desired pure product was obtained in 92% yield (180 mg, 0.92 mmol) as a green solid, mp 148–150 °C, after filtration through a short plug of silica topped with Celite using CH2Cl2. 1H NMR (500 MHz, CDCl3) δ 9.30 (t, J = 2 Hz, 1H), 9.10 (s, 1H), 8.78 (t, J = 1.5 Hz, 1H). 13C NMR (125.8 MHz, CDCl3) δ 168.6, 162.5, 149.5, 132.8, 129.6, 128.0, 118.2. IR (neat) 3095, 1700, 1545, 1294, 1177, 918, 736 cm−1. HRMS (ESI) m/z calcd. for C7H3N2O5 (M–H)− 195.0042, found 195.0045.
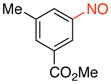
Methyl 3-Methyl-5-nitrosobenzoate (2j)
General procedure B was employed using potassium trifluoro(3-(methoxycarbonyl)-5-methylphenyl)borate (256 mg, 1 mmol) and NOBF4 (120 mg, 1.03 mmol). The reaction was complete in 1 min. The desired pure product was obtained in 91% yield (163 mg, 0.91 mmol) as a yellow solid, mp 68–70 °C, after filtration through a short plug of silica topped with Celite using CH2Cl2. 1H NMR (500 MHz, CDCl3) δ 8.44 (s, 1H), 8.20 (s, 1H), 7.73 (s, 1H), 3.99 (s, 3H), 2.55 (s, 3H). 13C NMR (125.8 MHz, CDCl3) δ 165.9, 165.3, 140.1, 136.3, 131.6, 123.9, 120.6, 52.7, 21.2. IR (neat) 1726, 1445, 1253, 1134, 760 cm−1. HRMS (ESI) m/z calcd. for C9H10NO3 (M+H)+ 180.0661, found 180.0667.
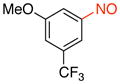
1-Methoxy-3-nitroso-5-(trifluoromethyl)benzene (2k)
General procedure B was employed using potassium trifluoro(3-methoxy-5-(trifluoromethyl)phenyl)borate (282 mg, 1 mmol) and NOBF4 (120 mg, 1.03 mmol). The reaction was complete in 10 min. The desired pure product was obtained in 81% yield (166 mg, 0.81 mmol) as a green oil after filtration through a short plug of silica topped with Celite using CH2Cl2. 1H NMR (500 MHz, CDCl3) δ 8.11 (s, 1H), 7.50 (s, 1H), 7.21 (m, 1H), 3.93 (s, 3H). 13C NMR (125.8 MHz, CDCl3) δ 165.2, 161.0, 133.4 (d, J = 34 Hz), 123.3 (m), 118.4 (d, J = 3.5 Hz), 114.0 (d, J = 3.5 Hz), 104.8, 56.3. 19F NMR (470.8 MHz, CDCl3) δ −62.9. IR (neat) 1507, 1325, 1131, 1046, 873, 688 cm−1. HRMS (ESI) m/z calcd. for C8H7NO2F3 (M+H)+ 206.0429, found 206.0431.
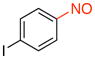
1-Iodo-4-nitrosobenzene (2l).45
General procedure B was employed using potassium trifluoro(4-iodophenyl)borate (310 mg, 1 mmol) and NOBF4 (120 mg, 1.03 mmol). The reaction was complete in 1 min. The desired pure product was obtained in 92% yield (214 mg, 0.92 mmol) as a green solid, mp 100–102 °C (lit.46 104 –106 °C), after filtration through a short plug of silica topped with Celite using CH2Cl2. 1H NMR (500 MHz, CDCl3) δ 8.03 (d, J = 8.5 Hz, 2H), 7.60 (d, J = 8.5 Hz, 2H). 13C NMR (125.8 MHz, CDCl3) δ 164.3, 138.9, 122.0, 105.6. IR (neat) 1579, 1481, 1113, 822 cm−1.
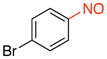
1-Bromo-4-nitrosobenzene (2m).8a
General procedure B was employed using potassium (4-bromophenyl)trifluoroborate (263 mg, 1 mmol) and NOBF4 (120 mg, 1.03 mmol). The reaction was complete in 1 min. The desired pure product was obtained in 94% yield (175 mg, 0.94 mmol) as a light yellow solid, mp 92–94 °C (lit.8a 99–101 °C), after filtration through a short plug of silica topped with Celite using CH2Cl2. 1H NMR (500 MHz, CDCl3) δ 7.78 (s, 4H). 13C NMR (125.8 MHz, CDCl3) δ 164.0, 132.9, 131.8, 122.3. IR (neat) 1478, 1257, 1011, 856 cm−1. HRMS (ESI) m/z calcd. for C6H5NOBr (M+H)+ 185.9554, found 185.9555.
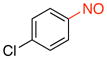
1-Chloro-4-nitrosobenzene (2n).15d
General procedure B was employed using potassium (4-chlorophenyl)trifluoroborate (219 mg, 1 mmol) and NOBF4 (120 mg, 1.03 mmol). The reaction was complete in 1 min. The desired pure product was obtained in 92% yield (130 mg, 0.92 mmol) as a light yellow solid, mp 87–89 °C (lit.47 88–89 °C), after filtration through a short plug of silica topped with Celite using CH2Cl2. 1H NMR (500 MHz, CDCl3) δ 7.86 (d, J = 9 Hz, 2H), 7.60 (d, J = 9 Hz, 2H). 13C NMR (125.8 MHz, CDCl3) δ 164.0, 142.6, 129.8, 122.3. IR (neat) 1481, 1256, 1089, 857 cm−1. HRMS (CI) m/z calcd. for C6H5NOCl (M+H)+ 142.0060, found 142.0056.
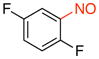
1,4-Difluoro-2-nitrosobenzene (2o)
General procedure B was employed using potassium (2,5-difluorophenyl)trifluoroborate (220 mg, 1 mmol) and NOBF4 (120 mg, 1.03 mmol). The reaction was complete in 10 min. The desired pure product was obtained in 81% yield (116 mg, 0.81 mmol) as a white solid, mp 35–37 °C, after filtration through a short plug of silica topped with Celite using CH2Cl2. 1H NMR (500 MHz, CDCl3) δ 7.22 (m, 1H), 6.87 (m, 1H), 6.61 (m, 1H). 13C NMR (125.8 MHz, CDCl3) δ 168.9 (d, J = 12 Hz), 166.9 (m), 164.8 (d, J = 13 Hz), 153.3 (d, J = 4 Hz), 112.0 (m), 106.4 (m). 19F NMR (470.8 MHz, CDCl3) δ –94.4, –123.7. IR (neat) 1613, 1501, 1241, 845 cm−1. HRMS (ESI) m/z calcd. for C6H4NOF2 (M+H)+ 144.0261, found 144.0260.
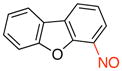
4-Nitrosodibenzo[b,d]furan (3a)
General procedure B was employed using potassium dibenzo[b,d]furan-4-yltrifluoroborate (274 mg, 1 mmol) and NOBF4 (120 mg, 1.03 mmol). The reaction was complete in 30 sec. The desired pure product was obtained in 85% yield (168 mg, 0.85 mmol) as a green solid, mp 84–86 °C, after filtration through a short plug of silica topped with Celite using CH2Cl2. 1H NMR (500 MHz, CDCl3) δ 8.26 (m, 1H), 8.01 (m, 1H), 7.78 (d, J = 8 Hz, 1H), 7.59 – 7.56 (m, 2H), 7.49 – 7.44 (m, 2H). 13C NMR (125.8 MHz, CDCl3) δ 157.4, 153.3, 148.8, 128.7, 128.5, 128.4, 124.1, 122.6, 122.5, 121.0, 116.3, 112.7. IR (neat) 1456, 1417, 1174, 1107, 830, 744 cm−1. HRMS (CI) m/z calcd. for C12H8NO2 (M+H)+ 198.0555, found 198.0553.
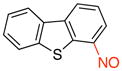
4-Nitrosodibenzo[b,d]thiophene (3b)
General procedure B was employed using potassium dibenzo[b,d]thiophen-4-yltrifluoroborate (290 mg, 1 mmol) and NOBF4 (120 mg, 1.03 mmol). The reaction was complete in 30 sec. The desired pure product was obtained in 80% yield (171 mg, 0.80 mmol) as a green solid, mp 115–117 °C, after filtration through a short plug of silica topped with Celite using CH2Cl2. 1H NMR (500 MHz, CDCl3) δ 9.56 (m, 1H), 8.46 (m, 1H), 8.15 (m, 1H), 7.93 – 7.89 (m, 2H), 7.53 – 7.50 (m, 2H). 13C NMR (125.8 MHz, CDCl3) δ 162.9, 142.1, 138.1, 137.9, 132.4, 128.2, 127.8, 125.9, 125.5, 123.7, 121.7, 120.1. IR (neat) 1421, 1190, 1086, 920, 750 cm−1. HRMS (ESI) m/z calcd. for C12H8NOS (M+H)+ 214.0327, found 214.0336.
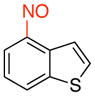
4-Nitrosobenzo[b]thiophene (3c)
General procedure B was employed using potassium benzo[b]thiophen-4-yltrifluoroborate (240 mg, 1 mmol) and NOBF4 (120 mg, 1.03 mmol). The reaction was complete in 30 sec. The desired pure product was obtained in 81% yield (132 mg, 0.81 mmol) as a yellow solid, mp 76–78 °C, after filtration through a short plug of silica topped with Celite using CH2Cl2. 1H NMR (500 MHz, CDCl3) δ 8.48 (d, J = 8 Hz, 1H), 8.31 (m, 1H), 8.22 (d, J = 8 Hz, 1H), 7.86 (d, J = 5.5 Hz, 1H), 7.72 (t, J = 7.5 Hz, 1H). 13C NMR (125.8 MHz, CDCl3) δ 160.6, 142.8, 134.1, 130.2, 127.0, 126.8, 124.2, 121.7. IR (neat) 1450, 1265, 861, 746 cm−1. HRMS (ESI) m/z calcd. for C8H6NOS (M+H)+ 164.0170, found 164.0168.
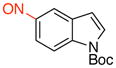
tert-Butyl 5-Nitroso-1H-indole-1-carboxylate (3d)
General procedure B was employed using potassium (1-(tert-butoxycarbonyl)-1H-indol-5-yl)trifluoroborate (323 mg, 1 mmol) and NOBF4 (120 mg, 1.03 mmol). The reaction was complete in 30 sec. The desired pure product was obtained in 73% yield (180 mg, 0.73 mmol) as a green solid, mp 101–103 °C, after filtration through a short plug of silica topped with Celite using CH2Cl2. 1H NMR (500 MHz, CDCl3) δ 8.43 (s, 1H), 8.26 (d, J = 8.5 Hz, 1H), 7.72 (d, J = 4 Hz, 1H), 7.61 (d, J = 8.5 Hz, 1H), 6.83 (d, J = 3.5 Hz, 1H), 1.70 (s, 9H). 13C NMR (125.8 MHz, CDCl3) δ 164.8, 149.2, 139.0, 130.6, 128.8, 119.7, 115.3, 115.1, 109.3, 85.2, 28.2. IR (neat) 1743, 1467, 1325, 1155, 1071, 721 cm−1. HRMS (ESI) m/z calcd. for C13H13N2O3 (M-H)− 245.0926, found 245.0932.
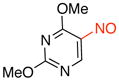
2,4-Dimethoxy-5-nitrosopyrimidine (3e)
General procedure B was employed using potassium (2,4-dimethoxypyrimidin-5-yl)trifluoroborate (246 mg, 1 mmol) and NOBF4 (120 mg, 1.03 mmol). The reaction was complete in 30 sec. The desired pure product was obtained in 82% yield (139 mg, 0.82 mmol) as a green solid, mp 78–80 °C, after filtration through a short plug of silica topped with Celite using CH2Cl2. 1H NMR (500 MHz, CDCl3) δ 8.18 (s, 1H), 4.33 (s, 3H), 4.16 (s, 3H). 13C NMR (125.8 MHz, CDCl3) δ 166.6, 166.3, 149.6, 149.2, 56.5, 55.4. IR (neat) 1594, 1547, 1474, 1314, 1054, 796 cm−1. HRMS (ESI) m/z calcd. for C6H8N3O3 (M+H)+ 170.0566, found 170.0570.
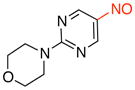
4-(5-Nitrosopyrimidin-2-yl)morpholine (3f)
General procedure B was employed using potassium trifluoro(2-morpholinopyrimidin-5-yl)borate (271 mg, 1 mmol) and NOBF4 (120 mg, 1.03 mmol). The reaction was complete in 30 sec. The desired pure product was obtained in 76% yield (148 mg, 0.76 mmol) as a green solid, mp 151–153 °C, after column chromatography with CH2Cl2 as eluent. 1H NMR (500 MHz, CDCl3) δ 9.07 (s, 2H), 4.00 (t, J = 5 Hz, 4H), 3.79 (t, J = 5 Hz, 4H). 13C NMR (125.8 MHz, CDCl3) δ 161.5, 154.8, 133.6, 66.6, 44.8. IR (neat) 2359, 1601, 1548, 1329, 1109, 790 cm−1. HRMS (ESI) m/z calcd. for C8H11N4O2 (M+H)+ 195.0882, found 195.0884.
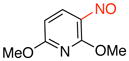
2,6-Dimethoxy-3-nitrosopyridine (3g)
General procedure B was employed using potassium (2,6-dimethoxypyridin-3-yl)trifluoroborate (245 mg, 1 mmol) and NOBF4 (120 mg, 1.03 mmol). The reaction was complete in 30 sec. The desired pure product was obtained in 72% yield (121 mg, 0.72 mmol) as a green solid, mp 95–97 °C, after column chromatography with CH2Cl2 as eluent. 1H NMR (500 MHz, CDCl3) δ 6.87 (d, J = 8.5 Hz, 1H), 6.23 (d, J = 8.5 Hz, 1H), 4.37 (s, 3H), 4.11 (s, 3H). 13C NMR (125.8 MHz, CDCl3) δ 168.3, 165.6, 151.2, 122.1, 103.3, 54.8. IR (neat) 1588, 1384, 1286, 1001, 825 cm−1. HRMS (ESI) m/z calcd. for C7H9N2O3 (M+H)+ 169.0613, found 169.0618.
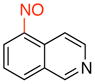
5-Nitrosoisoquinoline (3h)
General procedure B was employed using potassium trifluoro(isoquinolin-5-yl)borate (235 mg, 1 mmol) and NOBF4 (120 mg, 1.03 mmol). The reaction was complete in 30 sec. The desired pure product was obtained in 62% yield (NMR yield) as a yellow solid that upon exposure to air becomes black after column chromatography with EtOAc/CH2Cl2 (1:1) as eluent. 1H NMR (500 MHz, CDCl3) δ 9.49 (s, 1H), 9.41 (d, J = 6 Hz, 1H), 8.97 (d, J = 6 Hz, 1H), 8.40 (d, J = 8 Hz, 1H), 7.73 (t, J = 8 Hz, 1H), 7.14 (q, J = 1 and 8 Hz, 1H). 13C NMR (125.8 MHz, CDCl3) δ 157.7, 152.6, 147.2, 136.4, 131.5, 129.6, 126.2, 116.2, 114.0.
General Procedure C: One-pot Nitrosation/Diels-Alder
Adapted from a previously reported method.15a To a 20 mL glass microwave vial containing a solution of potassium organotrifluoroborate (1 mmol) in CH3CN (3 mL, 0.33 M) was added 1,3-cyclohexadiene (114 μL, 1.2 mmol, 1.2 equiv). To the mixture was added NOBF4 (120 mg, 1.03 mmol, 1.03 equiv) in one portion. The flask was then capped (exothermic reaction) and stirred for 2 h. To the crude mixture was added H2O (20 mL) and EtOAc (10 mL). The layers were separated and the aqueous layer was extracted with EtOAc (3 × 10 mL). The combined organic layers were dried (Na2SO4), filtered, and concentrated under reduced pressure. The product was purified by column chromatography using EtOAc/hexanes.
3-(Isoquinolin-5-yl)-2-Oxa-3-azabicyclo[2.2.2]oct-5-ene (4c)
General procedure C was employed using potassium trifluoro(isoquinolin-5-yl)borate (235 mg, 1 mmol), 1,3-cyclohexadiene (114 μL, 1.2 mmol, 1.2 equiv), and NOBF4 (120 mg, 1.03 mmol). The desired pure product was obtained in 65% yield (155 mg, 0.65 mmol) as a yellow solid, mp 68–70 °C, after column chromatography with EtOAc/hexanes (1:1) as eluent. 1H NMR (500 MHz, CDCl3) δ 9.19 (s, 1H), 8.51 (d, J = 6 Hz, 1H), 7.91 (d, J = 6 Hz, 1H), 7.64 (d, J = 8 Hz, 1H), 7.45 (t, J = 8 Hz, 1H), 7.37 (m, 1H), 6.75 (d, J = 2 and 6.5 Hz, 1H), 5.96 (t, J = 6.5 Hz, 1H), 4.82 (m, 1H), 4.34 (m, 1H), 2.51 (m, 1H), 2.36 (m, 1H), 1.60 (m, 1H), 1.50 (m, 1H). 13C NMR (125.8 MHz, CDCl3) δ 152.6, 146.1, 142.5, 132.1, 129.1, 129.1, 128.9, 126.8, 123.1, 120.5, 116.0, 69.5, 56.9, 23.7, 22.0. IR (neat) 1579, 1371, 1271, 932, 838, 768 cm−1. HRMS (ESI) m/z calcd. for C15H15N2O (M+H)+ 239.1184, found 239.1183.
Methyl 3-(-2-Oxa-3-azabicyclo[2.2.2]oct-5-en-3-yl)benzoate (5d)
General procedure C was employed using potassium trifluoro(3-(methoxycarbonyl)phenyl)borate (242 mg, 1 mmol), 1,3-cyclohexadiene (114 μL, 1.2 mmol, 1.2 equiv), and NOBF4 (120 mg, 1.03 mmol). The desired pure product was obtained in 82% yield (201 mg, 0.82 mmol) as a yellow solid, mp 87–89 °C, after column chromatography with EtOAc/hexanes (5:1) as eluent. 1H NMR (500 MHz, CDCl3) δ 7.66 (s, 1H), 7.58 (m, 1H), 7.26 (t, J = 8 Hz, 1H), 7.19 (m, 1H), 6.57 (m, 1H), 6.12 (m, 1H), 4.73 (m, 1H), 4.48 (m, 1H), 3.88 (s, 3H), 2.29 – 2.22 (m, 2H), 1.57 (m, 1H), 1.38 (m, 1H). 13C NMR (125.8 MHz, CDCl3) δ 167.3, 152.7, 131.9, 130.5, 129.9, 128.6, 123.3, 122.1, 118.4, 69.5, 56.6, 52.2, 24.0, 21.4. IR (neat) 1719, 1439, 1270, 944, 759 cm−1. HRMS (ESI) m/z calcd. for C14H16NO3 (M+H)+ 246.1130, found 246.1130.
One-pot Procedure for Formation of Azoxy Compound: Synthesis of 1,2-Bis(3-(methoxycarbonyl)phenyl)diazene Oxide (5a):48
Adapted from a previously reported method.49 To a 20 mL glass microwave vial containing potassium trifluoro(3-(methoxycarbonyl)phenyl)borate (242 mg, 1 mmol) in CH3CN (3 mL, 0.33 M) was added NOBF4 (120 mg, 1.03 mmol, 1.03 equiv) in one portion. After 30 sec the solvent was removed and EtOH was added (3.5 mL, 0.3M), followed by (NH4)2SO4 (1 g, 5 mmol, 5 equiv) and NaBH4 (661 mg, 3 mmol, 3 equiv). The flask was then capped (exothermic reaction) and stirred at rt for 30 min. The reaction mixture changed from green to yellow. To the crude mixture was added H2O (20 mL) and EtOAc (10 mL). The layers were separated, and the aqueous layer was extracted with EtOAc (3 × 10 mL). The combined organic layers were dried (Na2SO4), filtered, and concentrated under reduced pressure. Purification by filtration through a short plug of silica topped with Celite using EtOAc afforded the pure product in 87% yield (137 mg, 0.44 mmol) as a yellow solid, mp 135–137 °C (lit.46 135 °C). 1H NMR (500 MHz, CDCl3) δ 8.96 (d, J = 2 Hz, 1H), 8.77 (t, J = 2 Hz, 1H), 8.51 (m, 1H), 8.42 (m, 1H), 8.25 (d, J = 7.5 Hz, 1H), 8.07 (d, J = 7.5 Hz, 1H), 7.61 (t, J = 7.5 Hz, 1H), 7.57 (t, J = 7.5 Hz, 1H), 3.99 (s, 3H), 3.96 (s, 3H). 13C NMR (125.8 MHz, CDCl3) δ 166.5, 165.8, 148.4, 143.9, 132.9, 131.4, 131.1, 130.8, 129.6, 129.3, 129.0, 127.2, 126.7, 123.7, 52.7, 52.5. IR (neat) 1726, 1466, 1301, 1267, 1083, 753 cm−1. HRMS (ESI) m/z calcd. for C16H15N2O5 (M+H)+ 315.0981, found 315.0981.
Procedure for Formation of Nitro Compounds: Synthesis of Methyl 3-Nitrobenzoate (5b):50
Adapted from a previously reported method.15b To a 20 mL glass microwave vial containing potassium trifluoro(3-(methoxycarbonyl)phenyl)borate (242 mg, 1 mmol), in CH3CN (3 mL, 0.33 M) was added NOBF4 (120 mg, 1.03 mmol, 1.03 equiv) in one portion. After 30 sec the crude mixture was filtered through a plug of silica topped with Celite using CH2Cl2, the solvent was removed, and acetone: H2O (1:1, 5 mL) was added. To the solution was then added Oxone (461 mg, 1.5 mmol, 1.5 equiv). The flask was then capped and stirred at 60 °C for 2 h. The reaction solution changed from green to yellow. To the crude mixture was added H2O (20 mL) and EtOAc (10 mL). The layers were separated and the aqueous layer was extracted with EtOAc (3 × 10 mL). The combined organic layers were dried (Na2SO4), filtered, and concentrated under reduced pressure. Purification by filtration through a short plug of silica topped with Celite using EtOAc afforded the pure product in 86% yield (156 mg, 0.86 mmol) as a yellow solid, mp 75–77 °C (lit.48 77–79 °C). 1H NMR (500 MHz, CDCl3) δ 8.87 (t, J = 1.5 Hz, 1H), 8.42 (m, 1H), 8.37 (m, 1H), 7.67 (t, J = 8.5 Hz, 1H), 3.99 (s, 3H). 13C NMR (125.8 MHz, CDCl3) δ 164.9, 148.3, 135.2, 131.9, 129.6, 127.4, 124.6, 52.8. IR (neat) 3098, 1718, 1527, 1350, 1290, 1269, 1134, 720 cm−1.
One-pot Procedure for Formation of Aniline Compounds: Synthesis of Methyl 3-Aminobenzoate (5c):51
Adapted from a previously reported method.52 To a 20 mL glass microwave vial containing potassium trifluoro(3-(methoxycarbonyl)phenyl)borate (242 mg, 1 mmol) in CH3CN (3 mL, 0.33 M) was added nitrosonium tetrafluoroborate (120 mg, 1.03 mmol, 1.03 equiv) in one portion. EtOH (2 mL) and SnCl2•2H2O (1.13 g, 5 mmol, 5 equiv) were added after 30 sec. The flask was then capped, and the reaction was stirred at 80 °C for 2 h. To the crude mixture was added 5% NaHCO3 (3 mL, or enough to make the pH slightly basic, 7–9). The resulting emulsion was filtered, and the solid was washed with H2O (10 mL) and EtOAc (10 mL). The layers of the filtrate were separated, and the aqueous layer was extracted with EtOAc (3 × 10 mL). The combined organic layers were dried (Na2SO4), filtered, and concentrated under reduced pressure. Purification by filtration through a short plug of silica topped with Celite using EtOAc afforded the pure product as a yellow oil in 72% yield (109 mg, 0.72 mmol). 1H NMR (500 MHz, CDCl3) δ 7.42 (m, 1H), 7.35 (t, J = 2 Hz, 1H), 7.21 (t, J = 8 Hz, 1H), 6.85 (m, 1H), 3.89 (s, 3H), 3.79 (brs, 2H). 13C NMR (125.8 MHz, CDCl3) δ 167.4, 146.6, 131.3, 129.4, 119.8, 119.5, 115.9, 52.2. IR (neat) 3372, 2951, 1710, 1603, 1239, 753 cm−1. HRMS (ESI) m/z calcd. for C8H10NO2 (M+H)+ 152.0712, found 152.0713.
Supplementary Material
Acknowledgments
This work was generously supported by the NIGMS (R01 GM086209, GM035249). We acknowledge Conselho Nacional de Desenvolvimento Cientifico e Tecnologico (CNPq) for Graduate Research Fellowship to L.N.C. We also acknowledge Aldrich, BoroChem, and Frontier Scientific for their donation of the boronic acids used to prepare the organotrifluoroborates. Dr. Rakesh Kohli (University of Pennsylvania) is acknowledged for obtaining the HRMS data.
Footnotes
Supporting Information Available: Copies of 1H, 13C, and 19F spectra for all compounds prepared by the method described. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.(a) Patai S. The Chemistry of Amino, Nitroso, Nitro and Related Groups. Wiley-VCH; Weinheim: 1996. [Google Scholar]; (b) Momiyama N, Yamamoto H. Chem Commun. 2005:3514. doi: 10.1039/b503212c. [DOI] [PubMed] [Google Scholar]
- 2.For selected examples see: Momiyama N, Yamamoto H. J Am Chem Soc. 2005;127:1080. doi: 10.1021/ja0444637.Momiyama N, Yamamoto H. J Am Chem Soc. 2003;125:6038. doi: 10.1021/ja0298702.Momiyama N, Yamamoto H. J Am Chem Soc. 2004;126:5360. doi: 10.1021/ja039103i.
- 3.For selected examples see: Yamamoto Y, Yamamoto H. J Am Chem Soc. 2004;126:4128. doi: 10.1021/ja049849w.Stephenson GR, Balfe AM, Hughes DL, Kelsey RD. Tetrahedron Lett. 2010;51:6806.Sakai H, Ding X, Yoshida T, Fujinami S, Ukaji Y, Inomata K. Heterocycles. 2008;76:1285.Jana CK, Studer A. Chem Eur J. 2008;14:6326. doi: 10.1002/chem.200800903.Jana CK, Grimme S, Studer A. Chem Eur J. 2009;15:9078. doi: 10.1002/chem.200901331.Calvet G, Coote SC, Blanchard N, Kouklovsky C. Tetrahedron. 2010;66:2969.Jana CK, Studer A. Angew Chem Int Ed. 2007;46:6542. doi: 10.1002/anie.200701631.Yamamoto Y, Yamamoto H. Angew Chem Int Ed. 2005;44:7082. doi: 10.1002/anie.200501345.
- 4.Pagar VV, Jadhav AM, Liu R-S. J Am Chem Soc. 2011;133:20728. doi: 10.1021/ja209980d. [DOI] [PubMed] [Google Scholar]
- 5.For selected examples see: Ginsburg VA. J Org Chem USSR (Engl Trans) 1974;10:1427.Barr A, Hazeldine RN. J Chem Soc. 1955:1881.Dochnahl M, Fu GC. Angew Chem Int Ed. 2009;48:2391. doi: 10.1002/anie.200805805.Wang T, Huang X-L, Ye Org Biomol Chem. 2010;8:5007. doi: 10.1039/c0ob00249f.
- 6.Adam W, Krebs O. Chem Rev. 2003;103:4131. doi: 10.1021/cr030004x. and references therein. [DOI] [PubMed] [Google Scholar]
- 7.(a) Aston A, Menard M. J Am Chem Soc. 1935;57:1922. [Google Scholar]; (b) Forrester AR, Hepburn SP. J Chem Soc C. 1971:3322. [Google Scholar]; (c) Goldman J. Tetrahedron. 1973;29:3833. [Google Scholar]
- 8.For selected examples see: Tibiletti F, Simonetti M, Nicholas KM, Palmisano G, Parravicini M, Imbesi F, Tollari S, Penoni A. Tetrahedron. 2010;66:1280.Penoni A, Volkman J, Nicholas KM. Org Lett. 2002;4:699. doi: 10.1021/ol017139e.Penoni A, Palmisano G, Broggini G, Kadowaki A, Nicholas KM. J Org Chem. 2006;71:823. doi: 10.1021/jo051609r.Penoni A, Palmisano G, Zhao YL, Houk KN, Volkman J, Nicholas KM. J Am Chem Soc. 2009;131:653. doi: 10.1021/ja806715u.Lamar AA, Nicholas KM. Tetrahedron. 2009;65:3829.
- 9.Goelitz P, Meijere A. Angew Chem. 1977;89:892. [Google Scholar]
- 10.For selected examples see: McKillop A, Tarbin JA. Tetrahedron. 1987;43:1753.Astolfi P, Carloni P, Damiani E, Greci L, Marini M, Rizzoli C, Stipa P. Eur J of Org Chem. 2008:3279.Ibne-Rasa KM, Lauro CG, Edwards JO. J Am Chem Soc. 1963;85:1165.Johnson NA, Guld ES. J Am Chem Soc. 1973;95:5198.
- 11.For selected examples see: Feuer H, Braunstein DM. J Org Chem. 1969;34:2024.Fischer B, Sheihet L. J Org Chem. 1998;63:393.Baik W, Rhee JU, Lee SH, Lee NH, Kim BH, Kim KS. Tetrahedron Lett. 1995;36:2793.
- 12.Rice WG, Schaeffer CA, Graham L, Bu M, McDougal JS, Orloff SL, Villinger F, Young M, Oroszlan S, Fesen MR, Pommier Y, Mendeleyev J, Kun E. Nature. 1993;361:473. doi: 10.1073/pnas.90.20.9721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baeyer A. Chem Ber. 1874;7:1638. [Google Scholar]
- 14.For a review on the synthesis of nitroso compunds see: Gowenlock BG, Richter-Addo GB. Chem Rev. 2004;104:3315. doi: 10.1021/cr030450k.
- 15.For selected examples see: Zhao D, Johansson M, Backvall JE. Eur J Org Chem. 2007:4431.Priewisch B, Ruck-Braun K. J Org Chem. 2005;70:2350. doi: 10.1021/jo048544x.Bordoloi A, Halligudi SB. Adv Synth Catal. 2007:2085.Defoin A. Synthesis. 2004:706.Gowenlock BG, Maidment MJ, Orrell KG, Prokes I, Roberts JR. J Chem Soc. 2001:1904.
- 16.For selected examples see: Lin W, Gupta A, Kim KH, Mendel D, Miller MJ. Org Lett. 2009;11:449. doi: 10.1021/ol802553g.Rogers MAT. J Chem Soc. 1943:590.Tedder JM, Webster B. J Chem Soc. 1960:3270.Alkorta I, Garcia-Gomez C, Paz JLG, Jimeno ML, Aran VJ. J Chem Soc. 1996:293.
- 17.For selected examples see: Bosch E, Kochi JK. J Org Chem. 1994;59:5573.Zyk NV, Nesterov EE, Khiobystov AN, Zefirov NS. Russ Chem Bull. 1999;48:506.Atherton JH, Moodie RB, Noble DR. J Chem Soc, Perkin Trans. 1999;2:699.D’Amicoc JJ, Tung CC, Walker LAJ. Am Chem Soc. 1959;81:5957.
- 18.Bartlett EH, Eaborn C, Walton DRM. J Chem Soc C. 1970:1717. [Google Scholar]
- 19.Taylor EC, Danforth RH, McKillop A. J Org Chem. 1973;38:2088. [Google Scholar]
- 20.Birkofer L, Franz M. Chem Ber. 1971;104:3062. [Google Scholar]
- 21.For selected examples see: Yao M-L, Reddy MS, Yong L, Walsh I, Blevins DW, Kabalka GW. Org Lett. 2010;12:700. doi: 10.1021/ol9027144.Kabalka GW, Mereddy AR. Organometallics. 2004;23:4519.Kabalka GW, Mereddy AR. Tetrahedron Lett. 2004;45:343.Thiebes C, Prakash GK, Petasis NA, Olah GA. Synlett. 1998:141.Szumigala RH, Devine PN, Gauthier DR, Jr, Volante RP. J Org Chem. 2004;69:566. doi: 10.1021/jo035184p.
- 22.(a) Salzbrunn S, Simon J, Prakash GKS, Petasis NA, Olah GA. Synlett. 2000:1485. [Google Scholar]; (b) Prakash GKS, Panja C, Mathew T, Surampudi V, Petasis NA, Olah GA. Org Lett. 2004;6:2205. doi: 10.1021/ol0493249. [DOI] [PubMed] [Google Scholar]
- 23.Molander GA, Cavalcanti LN. J Org Chem. 2011;76:7195. doi: 10.1021/jo201313a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.For reviews, see: Darses S, Genet JP. Chem Rev. 2008;108:288. doi: 10.1021/cr0509758.Doucet H. Eur J Org Chem. 2008:2013.Molander GA, Ellis NM. Acc Chem Res. 2007;40:275. doi: 10.1021/ar050199q.Stefani HA, Cella R, Vieira AS. Tetrahedron. 2007;63:3623.Molander GA, Figueroa R. Aldrichim Acta. 2005;38:49.Darses S, Genet JP. Eur J Org Chem. 2003:4313.
- 25.(a) Kabalka GW, Coltuclu V. Tetrahedron Lett. 2009;50:6271. [Google Scholar]; (b) Molander GA, Cavalcanti LN, Canturk B, Pan PS, Kennedy LE. J Org Chem. 2009;74:7364. doi: 10.1021/jo901441u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu J, Wang X, Shao C, Su D, Cheng G, Hu Y. Org Lett. 2010;12:1964. doi: 10.1021/ol1003884. [DOI] [PubMed] [Google Scholar]; (b) Molander GA, Cavalcanti LN. J Org Chem. 2011;76:623. doi: 10.1021/jo102208d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gillis EP, Burke MD. J Am Chem Soc. 2007;129:6716. doi: 10.1021/ja0716204. [DOI] [PubMed] [Google Scholar]
- 28.Ting R, Harwig CW, Lo J, Li Y, Adam MJ, Ruth TJ, Perrin DM. J Org Chem. 2008;73:4662. doi: 10.1021/jo800681d. [DOI] [PubMed] [Google Scholar]
- 29.For selected examples see: Baeyer A, Caro H. Ber. 1874;7:963.Radner F, Wall A, Loncar M. Acta Chem Scand. 1990;44:152.
- 30.Ishiyama T, Takagi J, Hartwig JF, Miyaura N. Angew Chem Int Ed. 2002;41:3056. doi: 10.1002/1521-3773(20020816)41:16<3056::AID-ANIE3056>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 31.(a) Creary X, Engel PS, Kavaluskas N, Pan L, Wolf A. J Org Chem. 1999;64:5634. doi: 10.1021/jo990732d. [DOI] [PubMed] [Google Scholar]; (b) Morrison J, Wan P, Corrie JET, Munasinghe VRN. Can J Chem. 2003;81:586. [Google Scholar]
- 32.Olah GA, Olah JA, Overchuck NA. J Org Chem. 1965;30:3373. [Google Scholar]
- 33.For selected examples see: Hamon F, Djedaini-Pilard F, Barbot F, Len C. Tetrahedron. 2009;65:10105.Burkhardt ER, Matos K. Chem Rev. 2006;106:2617. doi: 10.1021/cr0406918.Adams JP. J Chem Soc, Perkin Trans. 2002;1:2586.Tafesh AM, Weiguny J. Chem Rev. 1996;96:2035. doi: 10.1021/cr950083f.Kumar GS, Neckers DC. Chem Rev. 1989;89:1915.Ikeda T, Tsutumi O. Science. 1995;268:1873. doi: 10.1126/science.268.5219.1873.Campbell D, Dix LR, Rostron P. Dyes Pigm. 1995;29:77.Waghmode SB, Sabne SM, Sivasanker S. Green Chem. 2001;3:285.Sakaue S, Tsubakino T, Nishiyama Y, Ishii Y. J Org Chem. 1993;58:3633.Muller WE. The Benzodiazepine Receptor. Cambridge University Press; New York: 1988. Belciug M, Ananthanarayanan VS. J Med Chem. 1994;37:4392. doi: 10.1021/jm00051a017.Zollinger H. Color Chemistry. Wiley-VCH; New York: 1987. p. 161.Fan F-RF, Yao Y, Cai L, Cheng L, Tour JM, Bard AJ. J Am Chem Soc. 2004;126:4035. doi: 10.1021/ja0359815.
- 34.Molander GA, Canturk B, Kennedy LE. J Org Chem. 2009;74:973. doi: 10.1021/jo802590b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murphy JM, Tzschucke CC, Hartwig JF. Org Lett. 2007;9:757. doi: 10.1021/ol062903o. [DOI] [PubMed] [Google Scholar]
- 36.Pace G, Ferri V, Grave C, Elbing M, von Hanisch C, Zharnikov M, Mayor M, Rampi MA, Samori P. Proc Natl Acad Sci USA. 2007;104:9937. doi: 10.1073/pnas.0703748104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zarchi MAK, Rahmani F. J Appl Polym Sci. 2011;120:2830. [Google Scholar]
- 38.Marshall LJ, Cable KM, Botting NP. Tetrahedron. 2009;65:8165. [Google Scholar]
- 39.Krakert S, Terfort A. Aust J Chem. 2010;63:303. [Google Scholar]
- 40.Gowenlock BG, Pfab J, Young VM. J Chem Soc, Perkin Trans. 1997;2:1793. [Google Scholar]
- 41.Alway W. Chem Ber. 1903;36:2312. [Google Scholar]
- 42.Bamberger E. Chem Ber. 1895;28:248. [Google Scholar]
- 43.Il’ichev YV, Schwoerer MA, Wirz J. J Am Chem Soc. 2004;126:4581. doi: 10.1021/ja039071z. [DOI] [PubMed] [Google Scholar]
- 44.Zarwell S, Rueck-Braun K. Tetrahedron Lett. 2008;49:4020. [Google Scholar]
- 45.Biljan I, Cvjetojevic G, Novak P, Mihalic Z, Vancik H, Smrecki V, Babic D, Mali G, Plavec J. J Mol Struct. 2010;979:22. [Google Scholar]
- 46.Tsuzuki U, Hirasawa Chem Ber. 1941;74:616. [Google Scholar]
- 47.Knight GT, Loadman MJR. J Chem Soc, Perkin Trans. 1973;2:1550. [Google Scholar]
- 48.Gebhardt C, Priewisch B, Irran E, Rück-Braun K. Synthesis. 2008:1889. [Google Scholar]
- 49.Gohain S, Prajapati D, Sandhu JS. Chem Lett. 1995:725. [Google Scholar]
- 50.Aridoss G, Laali KK. J Org Chem. 2011;76:8088. doi: 10.1021/jo201374a. [DOI] [PubMed] [Google Scholar]
- 51.Monguchi Y, Maejima T, Mori S, Maegawa T, Sajiki H. Chem Eur J. 2010;16:7372. doi: 10.1002/chem.200903511. [DOI] [PubMed] [Google Scholar]
- 52.Bellamy FD, Ou K. Tetrahedron Lett. 1984;25:839. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.