

Abstract
Over 200 proteins have been identified that interact with the protein chaperone Hsp90, a recognized therapeutic target thought to participate in non-oncogene addiction in a variety of human cancers. However, defining Hsp90 clients is challenging because interactions between Hsp90 and its physiologically relevant targets involve low affinity binding and are thought to be transient. Using a chemo-proteomic strategy, we have developed a novel orthogonally cleavable Hsp90 affinity resin that allows purification of the native protein and is quite selective for Hsp90 over its immediate family members, GRP94 and TRAP 1. We show that the resin can be used under low stringency conditions for the rapid, unambiguous capture of native Hsp90 in complex with a native client. We also show that the choice of linker used to tether the ligand to the insoluble support can have a dramatic effect on the selectivity of the affinity media.
1. Introduction
Heat shock protein 90 (Hsp90) regulates cellular homeostasis by chaperoning the folding and intracellular trafficking of protein “clients” and is highly up regulated in response to stress.1, 2 The N-terminal domain contains an ATP binding site and ATPase activity is necessary for all of its cellular functions.3 To date, over 200 Hsp90 clients have been identified and many of these are involved in signal transduction.4–6 The mechanisms by which Hsp90 promotes protein folding/stability is under active investigation 7, 8 and although several solution structures of the N terminal ATP binding domain have been solved, there are no native co-solution structures of full length Hsp90 with its native clients.9, 10 The strongest evidence for its role in protein folding/stability comes from studies with selective inhibitors that bind competitively to its ATP binding domain.11–14 Typically 6–12 h exposure of a cell/xenograft tumor model to an Hsp90 inhibitor results in degradation of Hsp90 clients. To date, over ten Hsp90 inhibitors have been clinically evaluated in cancer patients for multiple indications.12, 15, 16
In vivo, Hsp90 does not function alone but acts in concert with co-chaperones such as Sba1/p23 and Cdc37.17 Interactions with co-chaperones are thought to be important to direct Hsp90 function for specific physiological processes e.g. cell cycle progression, apoptosis, or signaling cascades.5 Hsp90 is regulated at the expression level and through posttranslational modifications e.g. phosphorylation, acetylation and methylation. These processes control its ATPase activity, its ability to interact with clients and co-chaperones and its degradation.4, 18–20 Glucose-regulated protein 94 (GRP94) and tumor necrosis factor receptorassociated protein 1 (TRAP1) share homology with Hsp90 and both proteins possess ATPase activity.21, 22 Client proteins selectively interacting with these homologs have been less studied. Although there is sequence divergence between all three proteins in their N terminal domains, none of the current Hsp90 inhibitors completely discriminate between the chaperones in vitro.21, 23
Approaches to defining Hsp90 clients typically involve immunoprecipitation or affinity purification of Hsp90 with immobilized inhibitors. Client proteins are inferred by co-isolation.24, 25 However, in vivo, many clients undergoing folding are thought to be weakly associated with Hsp90, potentially complicating their identification using these approaches. Therefore, it seemed one could benefit from the use of a highly selective affinity probe that would allow for efficient Hsp90 release under mild conditions while discriminating against non-specifically associated proteins that typically bind to resin surfaces.26 Such a probe might enable isolation of native Hsp90 in quantities suitable for crystallographic studies. Also, an Hsp90 affinity resin could be used to remove the protein when its absence is desirable. We describe here the development of such a probe for Hsp90 that appears to discriminate the protein from GRP94 and TRAP1, and its use as an affinity ligand with an orthogonally cleavable linker. When incubated with cellular extracts, the ligand binds Hsp90 in complex with novel client proteins as well as established clients such as HER2. Importantly, client selectivity can be unambiguously established by prior quenching of the extracts with an Hsp90 inhibitor.
2. Results
2.1. Resin Synthesis and Evaluation
A challenge to developing an affinity resin lies in choosing an appropriate ligand and devising chemistry that allows linker attachment without disrupting binding affinity. The structure (pdb 3MNR) of the 2-aminobenzamide inhibitor 1 in complex with the N-terminal domain of human Hsp90 revealed the parts of the molecule which are solvent exposed and suggested a variety of substitutions likely to be tolerated on the aniline nitrogen (Figure. 1).27, 28 Further analyses suggested that replacing the trimethoxyphenyl group with a linker on a 3-methylpyrazole analogue would provide a good balance of potency and ease of synthesis.
Figure 1.


a) View of 2-aminobenzamide inhibitor 1, bound in Hsp90 b) Possible solvent side attachment points numbered.
Therefore, fluoro-compound 2 was initially synthesized. This intermediate provided a reactive center for the attachment of linkers and allowed for facile synthesis of a potent inhibitor 5 27 for quenching studies. Thus (Scheme 1.), dimedone was acylated with acetic anhydride to give acetyldimedone 3, and separately, 2, 4-difluorobenzonitrile reacted with hydrazine to give a mixture of arylhydrazines with the 4-substitution product 4 predominating. After aqueous workups, the crude reaction products were combined in methanol with acetic acid to give a mixture of regio-isomeric pyrazoles. The desired isomer was purified to give intermediate 2 in 50% yield from starting dimedone. For quenching experiments, the known Hsp90 inhibitor 5 was made from 2 by fluoride displacement with trans-4-aminocyclohexanol followed by peroxide mediated hydrolysis of the nitrile to the amide (91%). Constructs for affinity resins were prepared in a similar fashion except that a greater excess of diamine was used to reduce, though not eliminate, the formation of the doubly capped linkers. After hydrolysis of the nitrile to the amide, ligand-inhibitor constructs 6–9 were purified by silica gel chromatography.
Scheme 1. Synthesis of Hsp90 binding constructs.

a) Acetic anhydride, DMAP, NEt3 b) MeOH, N2H4·H2O c) MeOH, acetic acid, d) chromatography, e) i, DMSO, excess amine or diamine, 90 °C, ii, EtOH, 50% NaOH, iii, 30% H2O2 dropwise, 90 °C.
Affinity resins were prepared by reaction of the inhibitor-linker construct with activated affinity media, CNBr-activated Sepharose™ 4B according to the manufacturer’s instructions (www.gelifesciences.com). Ligand was added at 1 to 10 μmol/gram of resin in minimal methanol. We first prepared affinity resin A using the short PEG-like 8 atom linker-construct 6 and analyzed its ability to selectively capture Hsp90 from pig mammary gland extract, a tissue shown to be high in ATP binding proteins including native forms of Hsp90, GRP94 and TRAP1 (Supplementary Fig. S1 online). The resin was incubated in the protein solution then washed with a high salt buffer. The bound proteins were removed via an SDS boil procedure, separated by SDS-PAGE electrophoresis, located by silver staining and identified using mass spectrometry (MS) sequencing.29
A large number of proteins, including Hsp90, were retained (Figure 2. Lane A). However, the initial hypothesis that proteins other than Hsp90 might be clients was negated when a competition experiment, performed by pre-incubating the protein solution with known ligand 5, showed clean exclusion of Hsp90, but not the other proteins.
Figure 2. Directed chemical evolution of a selective affinity resin for Hsp90.

SDS-PAGE silver stain showing the effects of different side chain modifications on Hsp90 recovery and recovery of non-specifically bound proteins. Selectivity towards Hsp90 was demonstrated by inclusion of 1 mM 5 (+) in the tissue extract prior to mixing with affinity resin. Mass spectrometry was used to identify the bound proteins. In lane E bound proteins were eluted with 25 mM sodium dithionite in phosphate buffered saline. Numbers indicate bands that were sequenced by MS; (1) Fatty acid synthase (2) Hsp90 (3) Hsp90 (4) Hsp90 proteolytic fragments.
We then tried linker-construct 7 that has the more hydrophobic n-decane linker to prepared affinity resin B. This resin proved capable of capturing Hsp90 and was even better at capturing additional proteins, particularly in the quenching experiments with ligand 5 (Figure 2. Lane B). This may be the result of enhanced and nonspecific hydrophobic interactions of proteins with the linker.
We next examined longer, more hydrophilic linkers that allowed the ligand to extend farther away from the Sepharose surface (Supplementary Fig. S2 online). Affinity resin C was prepared using the polyethylene glycol (PEG)-like 19-atom linker-construct 8. The capture experiment was repeated and an intense band of Hsp90 was eluted with SDS (Figure 2. Lane C). Importantly, quenching with 5 showed complete blocking of Hsp90 binding as well as several N-terminal fragments. MS analysis demonstrated that all of the recovered proteins were Hsp90 or proteolytic fragments of the protein, suggesting that resin C is selective for Hsp90 over GRP94 and TRAP-1. As mentioned previously (Supplementary Fig. S2 online), the pig mammary gland tissue contained adequate levels of GRP94 and TRAP-1 to be detected by MS when eluted from an ATP resin.
Finally, we investigated another intermediate length PEG-like 13 atom linker with construct 9 and prepared affinity resin D. Although it was more selective than resins A and B, some nonspecific binding was observed (Figure 2. Lane D). All further experiments were therefore conducted with linker-construct 8.
2.2. Cleavable Linker
An important goal of our studies was to derive a probe that could be used to convincingly isolate Hsp90 under mild physiological conditions in association with potentially weakly associated client proteins. Although we can block Hsp90 recovery on our resins by quenching with 5 or other Hsp90 inhibitors (e.g. geldanamycin 30 or PU-H71 31), we were unable to elute Hsp90 bound to the affinity media without denaturing the protein. A linker that could be broken under non-denaturing conditions was needed. Recently, Verhelst et al. 32 reintroduced 33–35 an azo linker 10 (Scheme 2.), which can be cleaved under mild nondenaturing conditions with sodium dithionite solution.36 This linker was synthesized using the published procedure and coupled to our ligand-construct to give compound 11. The FMOC group was removed with neat piperidine and ligand 12 was reacted to give affinity resin E. Following exposure to pig mammary gland extract and treatment of the resin with 25 mM sodium dithionite, the eluted proteins were identified by MS as described earlier. The retained protein profile (Figure 2. Lane E) is essentially identical to resin C, except that the proteins are recovered under non-denaturing conditions.
Scheme 2. Preparation of a cleavable linker affinity resin.

a) EDC, HOBT, CH2Cl2 b) piperidine for 11, TFA for 14, c) CNBr-activated Sepharose 4B.
In our hands, synthesis of the azo-linker 10 was not without challenges. We observed production of numerous colored byproducts, possibly caused by difficulties in adequately controlling the reaction pH. Analysis of the byproducts by NMR suggested problems with reactions of the free amine. When the azo forming reaction was run with the BOC amide, using excess solid sodium bicarbonate in the reaction, we were able to obtain azo-linker 13 as the only colored product. Acidification of the reaction, followed by filtration gave clean azo-linker 13 (74%) as an orange powder. Affinity resin E was made (Scheme 2.) as before except that the amide intermediate 14 was de-protected with TFA to give the identical linker construct 12 for bead attachment.
2.3. Nonspecific Binding
One observation in these experiments (Figure 2.) is that the binding of Hsp90 to each of the resins seems to lower the level of non-specific binding, effectively protecting the resin. With each of the affinity resins (A-E), addition of compound 5 to the protein mixture completely blocked Hsp90 binding allowing recovery of other proteins, especially abundant proteins such as fatty acid synthase (240kDa). Although binding of these proteins is clearly not Hsp90 related, their recovery illustrates the challenges of using affinity media to study interactions with Hsp90. Even with a highly optimized ligand, our results illustrate the extent to which affinity based strategies in defining Hsp90 clients can lead to misleading conclusions. To define physiologically relevant associations with Hsp90, it is necessary to demonstrate competitive binding with a free ligand. Quenching experiments should be carried out using a selective inhibitor of Hsp90 in the cell/tissue extract prior to exposure to the affinity resin. Under these conditions, recovery of Hsp90 and any associated proteins should be selectively blocked. Proteins recovered in the presence of the quenching agent are likely to be non-specific.
2.4. Selectivity Evaluation
The finding that neither GRP94 nor TRAP-1 was recovered by resin C suggests that construct 8 is highly selective for Hsp90. To explore this selectivity in a somewhat reverse fashion, we tested the elution of pig mammary gland proteins from gamma phosphate linked ATP Sepharose with compound 5, known potent Hsp90 inhibitors: 17-AAG,37 PU-H71,31 and SNX2112,27 and ligand constructs 6, 8 and 9 (Supplementary Fig. S3 online). This ATP resin has been used in our lab for several years to study purine-binding proteins 38 and to discover novel Hsp90 inhibitors 27. It can be used to test ligand binding selectivity against all other purine utilizing enzymes expressed in cells.14 The ATP resin was charged with the pig mammary gland extract and aliquots were distributed into individual wells. Proteins eluted from the wells with increasing amounts of the indicated compound were analyzed as described earlier (Figure 3). Compound PU-H71 demonstrates strong potency towards both Hsp90 and GRP94, whereas SNX2112 shows weaker affinity for GRP94 and ligand 8 shows essentially no elution of GRP94. Examination of the crystal structures of Hsp90 27 and GRP94 39 with various ligands provided no obvious clue as to why ligand 8 would display a greater binding selectivity for Hsp90 over GRP94 compared to the other known Hsp90 inhibitors. It may be that the hydration of the PEG linker attached to 8 gives rise to a relatively larger, more sterically demanding structure, which precludes binding to GRP94.
Figure 3. Elution of pig mammary gland protein from ATP-Sepharose resin with SNX-2112, PU-H71 and Ligand construct 8.

ATP resin was charged with pig mammary gland extract and washed in bulk. Then, 20μl of charged resin was placed in each well of a 96-well filter plate and eluted with the indicated compounds in parallel. The eluates were characterized by SDS-PAGE, silver staining and mass spectrometry.
2.5. GRP94 Purification
It was possible to use this differential selectivity to purify GRP94. Pig mammary gland extract was passed through resin E, efficiently removing Hsp90, directly onto the ATP-resin. Elution of the ATP resin with PU-H71 gave clean GRP94 when analyzed by SDS-PAGE, silver staining and MS (Supplementary Fig. S4 online).
3. Proteomic studies
To test resin E as a proteomics tool we surveyed mouse organs to determine both Hsp90 expression levels between tissues and to define novel client proteins. Physiological conditions were used to preserve weak binding interactions with Hsp90. To demonstrate specificity, extracts were mixed with resin E +/− inhibitor 5. As seen in Figure 4A, each mouse tissue yielded a prominent protein at 90kDa of varying abundance as well as a distinct pattern of proteins of varying molecular weight. In most instances, inclusion of 5 in the extract blocked recovery of the 90 kDa protein as well as the additional proteins (Figure 4B). As expected, the 90kDa protein was identified by MS as Hsp90 (α and β isoforms). Surprisingly, many of the other proteins in the gel were either N-terminal fragments of Hsp90 or dimers of the holoenzyme (Figure 4A). Some proteins do appear to be potential clients of Hsp90 and show tissue specific associations including delta(3,5)-Delta(2,4)-dienoyl-CoA isomerase, NADPH-dependent retinol dehydrogenase/reductase, acetyl-CoA acyltransferase, and glycogen debranching enzyme. Two proteins, epoxide hydroxylase and glutaryl-CoA dehydrogenase are tentatively identified as novel clients, since recovery of these proteins was not completely quenched by 5. Their recovery may be explained by the presence of residual Hsp90 co-elution. Figure 4B also shows, as observed earlier, a few proteins were still non-specifically recovered from some tissues, even in the presence of 5, including albumin, CAZ-associated structural protein 1 and Ankrd11 protein. Their recovery underscores the necessity of including a quenching control with the free ligand to directly demonstrate Hsp90 dependence of the co-isolation.
Figure 4. Proteomic survey of mouse tissues with azo-resin E.

Tissue extracts were prepared from the indicated tissues and applied to a fixed volume of resin E (100μl) in the absence (A) and presence (B) of 5 (100 μM). Following washes at physiological ionic strength the bound proteins were eluted with 30 mM dithionite and characterized by SDS-PAGE (4–15% acrylamide), silver staining and mass spectrometry. Key: 1–22. full length Hsp90α/β or N terminal fragments of Hsp90α/β; 23. epoxide hydroxylase; 24. glutaryl-CoA dehydrogenase; 25. and 26. Hsp90α/β; 27. Hsp70; 28. epoxide hydroxylase; 29. delta(3,5)-delta(2,4)-dienoyl-CoA isomerase; 30. 3,2-trans-enoyl-CoA isomerase/NADPH-dependent retinol reductase; 31 and 32. Hsp90α/β; 33. Hsp70; 34. Hsp90α/β; 35. glycogen debranching enzyme; 36 and 37. Hsp90α/β; 38. epoxide hydroxylase; 39. 3-ketoacyl-CoA thiolase/3-ketoacyl-CoA thiolase; 40–42. Hsp90α/β; 43. Hsp70 44. epoxide hydroxylase/liver carboxylesterase 31; 45–47 Hsp90α/β; 48. epoxide hydroxylase; 49–51. Hsp90α/β; 52. Albumin; 53. Hsp90α/β; 55. Albumin; 56. CAZ-associated structural protein 1/Ankrd11 protein; 56. Hsp90α/β; 57. Albumin; 58. epoxide hydroxylase; 59. Glutaryl-CoA dehydrogenase; 60. Hsp90α/β. Combined consisted of a mixture of striated muscle, liver, testis and adipose tissue. The lane w/o dithionite was eluted with SDS. This gel is a representative example of using resin E against tissue extracts. (A larger view is shown in the supplemental material, Supplementary Fig. S5)
As a positive control, we confirmed that we could use resin E to find HER2, a known client of Hsp90. Extract from the breast cancer cell line (BTB-474) was added to the resin +/− inhibitor 5. Cleavage of the linker with dithionite and analysis by SDS-PAGE and silver staining showed only Hsp90. However, Western blot analysis clearly showed the presence of HER2 (Figure 5). Neither Hsp90 nor HER2 were seen in the sample quenched with 5.
Figure 5. Proteomic analysis for HER2 in breast cancer cell line (BTB-474) with azo-resin E.
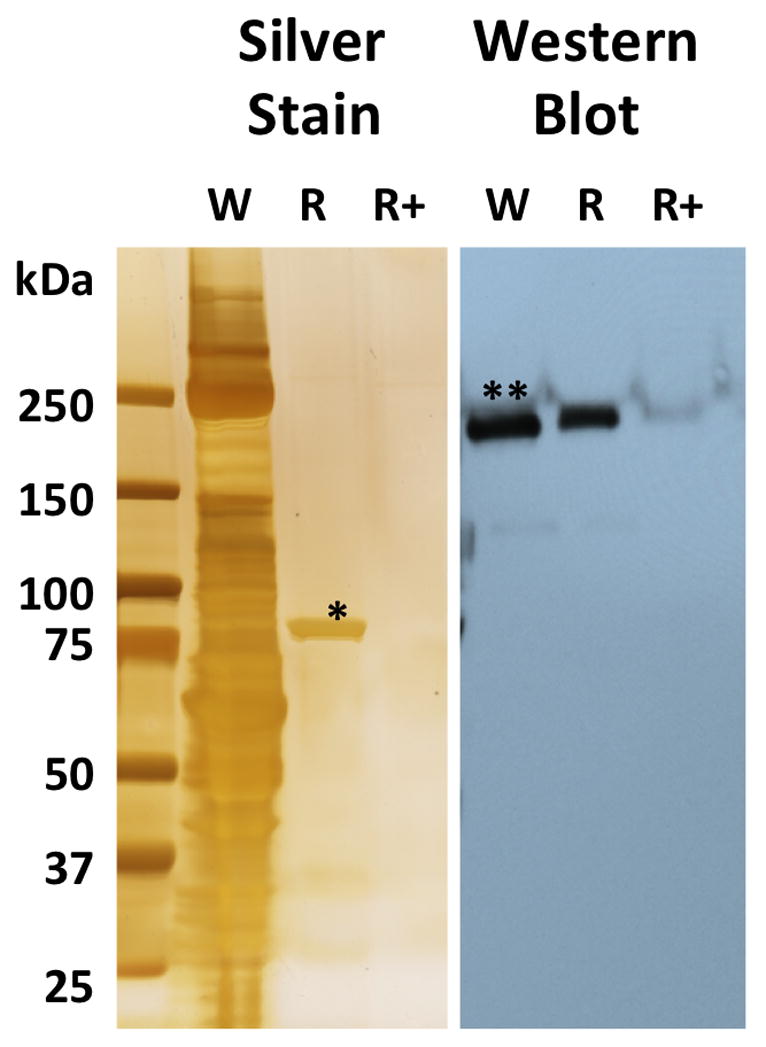
W – whole cell lysate (BTB474); R – BTB474 lysate on Hsp90 Resin (resin E); R+ – BTB474 lysate pretreated with free drug, compound 5 (1 mM), before passing over Hsp90 resin (resin E). * Hsp90, ** HER2.
4. Discussion and Conclusions
We report the development of a novel affinity probe, resin E, for the selective recovery of Hsp90 in native complex with its physiologically relevant client proteins. Our ligand is the first reported affinity reagent that only binds Hsp90 and shows no affinity for GRP94 and TRAP1 at the limits of silver staining and MS analysis. Therefore, resin E enables the study of native client proteins selective for Hsp90 independent of other chaperones. The development of resin E illustrates the complexities of utilizing affinity approaches to study native protein-protein interactions. Clearly, linker choice is critical to avoid artifacts. Study of Hsp90 is particularly challenging because interactions with its clients may be weak and readily disrupted under stringent, non-physiological conditions. Before assigning a particular protein as a client or co-chaperone of Hsp90, it is essential to also perform a co-isolation experiment with the appropriate quenching control. Any proteins recovered under quenching conditions are probably artifacts and unlikely to be a client or co-chaperone of Hsp90.
In proteomic studies of mouse tissues using cleavable resin E, Hsp90 was recovered either alone or in association with novel client proteins. Recovery of the putative clients was authenticated by quenching control experiments with 5. Interestingly, the majority of the recovered clients are associated with the metabolism of various lipids namely delta(3,5)-delta(2,4)-dienoyl-CoA isomerase, acetyl-CoA acyltransferase, NADPH-dependent retinol reductase and epoxide hydroxylase 2. Delta(3,5)-delta(2,4)-dienoyl-CoA isomerase and acetyl-CoA acyltransferase are peroximal enzymes participating in β oxidation of long chain fatty acids. Delta(3,5)-delta(2,4)-dienoyl-CoA isomerase isomerizes 3-trans, 5-cis-dienoyl- CoA to 2-trans,4-trans-dienoyl-CoA functioning as a auxiliary step of the fatty acid beta-oxidation pathway, enabling the metabolism of unsaturated fatty acids in mammals. NADPH-dependent retinol reductase (RDH12) has activity toward 9-cis and all-trans-retinol and is involved in the metabolism of short-chain aldehydes. The enzyme forms 11-cis-retinal from 11-cis-retinol during regeneration of the cone visual pigments. Mutations in the RDH12 are associated with retinitis pigmentosa type 53.40, 41 Retinitis pigmentosa is characterized by retinal pigment deposits as well as loss of rod photoreceptor cells with some secondary cone photoreceptor loss. The condition is typified by night vision blindness and reduced peripheral visual field. If RDH12 is a client for Hsp90, loss of expression of the protein in the eye could explain some of the idiosyncratic visual side effects anecdotally reported in some patients treated with a variety of Hsp90 inhibitors in clinical trials.42, 43 This would require that these drugs cross the blood-brain barrier since the retina is contiguous with the central nervous system. Epoxide hydroylase 2 and its immediate family members have roles in the metabolism of arachidonic and linoleic acid epoxides, as well as, xenobiotics.44 The finding that epoxide hydroylase 2 is a client of Hsp90 could explain some of the anti-inflammatory effects of Hsp90 inhibitors in addition to their indications in cancer.45, 46
The mouse study was used primarily to demonstration the use of the affinity resin for initial identification of potential client proteins. Further studies are needed to confirm these proteins are true clients of Hsp90. As shown with the breast cancer cell line, actual client proteins may be present below the detection limits of silver staining or MS detection but still be detectable by western blot analysis.
A final observation relates to the impact of the linker choice on the performance of an affinity resin. There is a radical difference in the amount of nonspecific binding between using the n-decane linker (resin B) and PEG-6 linker (resin C). We were initially disappointed with the number of clients identified with our affinity media, although we now believe the approach of using a “clean” cleavable linker along with a quenching agent gives rise to fewer experimental artifacts. It may be worthwhile to re-examine other affinity experiments where hydrophobic linkers are used, beyond the field of chaperone proteins. Judicious linker replacement may show some affinity experimental conclusions to be premature and may yet yield positive results in other previously failed experiments. We continue to explore this interesting observation.
5. Experimental Section
5.1. General
Reagents were obtained from commercial sources and used without further purification. 1,19-Diamino-4,7,10,13,16-pentaoxanonadecane was obtained from Berry and Associates. Proton NMR spectra were obtained on Varian 400 and 500 MHz spectrometers. LC/MS were obtained on an Agilent ion-trap LC/MS system. HRMS results were obtained on an Agilent 6224 LCMS-TOF and are reported as an average of four runs. PU-H71 and 17-AAG were purchased from commercial sources. SNX2112 was synthesized using published methods. Spectral data (NMR etc.), descriptions of tissue collection, cell culture, tissue extraction and proteomic analysis are included in the supplementary material.
5.1.1. 2-Acetyl-5,5-dimethylcyclohexane-1,3-dione (3)
Dimedone (10 g, 71.3 mmol) was dissolved in methylene chloride (200 mL) and treated with Hunig’s base (9.7 g, 74.9 mmol) and DMAP (440 mg, 3.6 mmol) followed by slow addition of acetic anhydride (7.65 g, 74.9 mmol). After 24 h, the mixture was concentrated, partitioned between hexanes (150 mL) and 1 N HCl (70 mL), then washed with brine (50 mL), treated with Norit A and then dried (MgSO4), filtered and concentrated to give 2-acetyl-5,5-dimethylcyclohexane-1,3-dione 3 (~11+ g) as a yellow oil. The entire product was used in the next step.
5.1.2. 2-Fluoro-4-hydrazinylbenzonitrile (4)
2,4-difluorobenzonitrile (10 g, 72 mmol) was dissolved in methanol (100 mL) and treated drop wise with hydrazine hydrate (18 g, 0.36 mol) and stirred at RT. After 16 h, the reaction mixture, containing a mixture of isomers, was concentrated then partitioned between ethyl acetate (100 mL), water (70 mL) and 1 N NaOH (30 mL). The organic layer was washed with brine (40 mL) then concentrated to give 4 in a mixture of isomers as a white solid.
5.1.3. 2-Fluoro-4-(3,6,6-trimethyl-4-oxo-4,5,6,7-tetrahydro-1H-indazol-1-yl)benzonitrile (2)
All of 3 (assume 13 g, 71.8 mmol) and 4 (assume 10.9 g, 71.8 mmol) were combined, then dissolved in methanol (40 mL) and treated with acetic acid (1 mL) and stirred at RT for 3 days. The mixture was concentrated and then dissolved in methylene chloride (20 ml). The mixture was chromatographed in batches, with the best approach to load a DCE solution onto a dry column, wait (15 to 20 min) then elute with 10% ethyl acetate in hexanes to remove the yellow (isomeric product) followed by elution with 20% to get the product. The chromatographed products were recrystallized from ethyl acetate/hexanes to give 2 (10.4 g, 49%). TLC (hexane/EtOAc: 60/40) Rf = 0.47; 1H NMR (CDCl3) δ 7.73 (dd, J = 7, 8.5 Hz, 1H), 7.49 (dd, J = 2, 9.8 Hz, 1H), 7.45 (dd, J = 2, 8.5 Hz, 1H), 2.90 (s, 2H) 2.54 (s, 3H) 2.43 (s, 2H), 1.14 (s, 6H); MS (ESI): m/z 298.2 [M+H]+.
5.1.4. 2-(((1r,4r)-4-Hydroxycyclohexyl)amino)-4-(3,6,6-trimethyl-4-oxo-4,5,6,7-tetrahydro-1H-indazol-1-yl)benzamide (5)
A mixture of nitrile 2 (200mg, 0.67 mmol) and trans-4-aminocyclohexanol (232 g, 2.0 mmol), Hunig’s base (117 μL) and DMSO (300 μL) were heated to 90 °C for 30 m. The mixture diluted with ethanol (2 mL) and treated with 50% NaOH (5 drops) and then, very slowly, a drop at a time, with hydrogen peroxide. After each drop, the reaction foamed up a bit. After adding 5 drops over 10 m, the mixture was diluted with water (18 mL) and allowed to cool slowly with rapid stirring. After stirring overnight, the solid was filtered off to give the product 5 (251 mg, 91%) as a white powder. TLC (EtOAc) Rf = 0.25; 1H NMR (CDCl3) δ 8.07 (d, J = 7.2 Hz), 7.44 (d, J = 8.6 Hz, 1H), 6.75 (d, J = 2 Hz, 1H), 6.60 (dd, J = 2, 8.6 Hz, 1H), 5.6 (br s, 2H), 3.71 (m, 1H), 3.35 (m, 1H), 2.80 (s, 2H) 2.53 (s, 3H) 2.38 (s, 2H), 2.13 (m,2H), 2.02 (m, 2H), 1.64 (br s, 1H), 1.41 (m, 4H), 1.09 (s, 6H); MS (ESI): m/z 411.3 [M+H]+.
5.1.5. 2-((2-(2-(2-aminoethoxy)ethoxy)ethyl)amino)-4-(3,6,6-trimethyl-4-oxo-4,5,6,7-tetrahydro-1H-indazol-1-yl)benzamide (6)
A mixture of 2 (500 mg, 1.7 mmol) and 2,2′-(ethylenedioxy) bis(ethylamine) (1.2 g, 8.4 mmol) in DMSO (1 mL) were and heated to 90 °C for 30 minutes. The mixture was diluted with ethanol (2 mL) and, still at 90 °C, treated with 50% NaOH (20 drops) and very slowly with 30% hydrogen peroxide (40 drops). The reaction mixture was diluted with methylene chloride and methanol and adsorbed onto silica gel. The mixture was chromatographed (silica gel, 1.5 cm × 20 cm) and eluted with 9/1 CH2Cl2/MeOH, followed by 9/1/0.1 CH2Cl2/MeOH/NH3 to give 6 (670 mg, 89%) as a clear glass. TLC (4/1/0.1 CH2Cl2/MeOH/NH3) Rf = 0.14; 1H NMR (CDCl3) δ 8.17 (t, J = 5 Hz), 7.47 (d, J = 8.4 Hz, 1H), 6.77 (d, J = 2 Hz, 1H), 6.60 (dd, J = 2, 8.4 Hz, 1H), 6.1 (br s, 2H), 3.73 (t, 3H), 3.65 (m, 4H), 3.50 (t, 2H), 3.36 (m, 2H), 2.85 (t, 2H), 2.77 (s, 2H) 2.51 (s, 3H) 2.36 (s, 2H), 1.07 (s, 6H); HRMS (ESI) [M+H]+ calcd for C23H34N5O4, 444.2605; found 444.2607.
5.1.6. 2-((19-Amino-4,7,10,13,16-pentaoxanonadecyl)amino)-4-(3,6,6-trimethyl-4-oxo-4,5,6,7-tetrahydro-1H-indazol-1-yl)benzamide (8)
A mixture of 2 (482 mg, 1.62 mmol) and 1,19-diamino-4,7,10,13,16-pentaoxanonadecane (1 g, 3.24mol), diisopropylethylamine (628 mg, 4.8 mmol) and DMSO (1 mL) were heated to 90 °C for 20 m. Still at 90 °C, the mixture diluted with ethanol (2 mL) and treated with 50% NaOH (10 drops) and then, very slowly, a drop at a time, with hydrogen peroxide. After each drop, the reaction foamed up substantially. After about 10 drops over 10 m, the reaction mixture was diluted with ethanol and added to silica gel (6 g), concentrated to a powder, added to a silica gel column (2.5 × 20 cm) and chromatographed with CH2Cl2 (300 mL), CH2Cl2/MeOH/NH3 19/0.9/0.1 (300 mL), 9/0.9/0.1 (300 mL) and 4/0.9/0.1 (500 mL). The cleanest fractions were combined to give 8 (600 mg, 61%) as a lightly yellow glass. TLC (4/1/0.1 CH2Cl2/MeOH/NH3) Rf = 0.30; 1H NMR (CDCl3) δ 7.98 (t, J = 4 Hz), 7.47 (d, J = 8.4 Hz, 1H), 6.77 (d, J = 2 Hz, 1H), 6.60 (dd, J = 2, 8.4 Hz, 1H), 6.0 (br s, 2H), 3.61 (m, 16H), 3.28 (m, 2H), 2.85 (t, 2H), 2.79 (s, 2H), 2.52 (s, 3H) 2.37 (s, 2H), 2.28 (br s, 2H), 1.94 (m, 2H), 1.76 (m, 2H), 1.07 (s, 6H); MS (ESI): m/z 604.4 [M+H]+; HRMS (ESI) [M+H]+ calcd for C31H50N5O7, 604.3705; found 604.37155.
5.1.7. 2-((3-(2-(2-(3-aminopropoxy)ethoxy)ethoxy)propyl)amino)-4-(3,6,6-trimethyl-4-oxo-4,5,6,7-tetrahydro-1H-indazol-1-yl)benzamide (9)
Compound 9 was prepared in the same way as compound 6. TLC (4/1/0.1 CH2Cl2/MeOH/NH3) Rf = 0.39; 1H NMR (CDCl3) δ 8.00 (t, J = 5 Hz, 1H), 7.46 (d, J = 8.2 Hz, 1H), 6.77 (s, 1H), 6.60 (d, J = 8.4 Hz, 1H), 6.1 (br s, 2H), 3.59 (m, 8H), 3.29 (m, 2H), 2.82 (t, 2H), 2.79 (s, 2H) 2.59 (br s, 2H), 2.51 (s, 3H), 2.37 (s, 2h), 1.93 (M, 2h), 1.73 (m, 2H), 1.07 (s, 6H); MS (ESI): m/z 516.4 [M+H]+; HRMS (ESI) [M+H]+ calcd for C27H42N5O5, 516.3180; found 516.3191.
5.1.8. (E)-4-((5-(2-((tert-butoxycarbonyl)amino)ethyl)-2- hydroxyphenyl)diazenyl)benzoic acid (13)
4-Aminobenzoate (500 mg, 3.6 mmol) was slurried in 6N HCl (10 mL), cooled to 0 °C and treated slowly with sodium nitrite (629 mg, 9.11 mmol). After stirring for 20 m, the mixture was added slowly to an ice-cooled solution of N-Boc tyramine (865 mg, 3.6 mmol) in saturated sodium bicarbonate solution (40 mL) with added sodium bicarbonate (4 g) and a bit of acetone (~5 mL). The orange reaction slurry was left to stir overnight. The reaction mixture was treated 1N HCl (100 mL) until acidic and then stirred an additional 2 h. The solids were filtered off, washed with water and air dried, then dried under vacuum to give 13 (1.03 g, 73%) as a reddish-orange solid. The product was used as is though it contained a minor impurity by NMR. TLC (9/1 CH2Cl2/MeOH) Rf = 0.20; 1H NMR (DMSO-d6) δ 13.19 (br s, 1H), 10.82 (s, 1H), 8.11 (d, J = 8.5 Hz, 2H), 8.05 (d, J = 8.5 Hz, 2H), 7.55 (s, 1H), 7.28 (d, J = 8.3 Hz, 1H), 7.00 (d, J = 8.3 Hz, 1H), 6.88 (t, 1H), 3.12 (m, 2H), 2.67 (t, J = 6.8 Hz, 2H), 1.33 (s, 9H); MS (ESI): m/z 384.2 [M]−, 791.4 [2M+Na]−; HRMS (ESI) [M+Na]+ calcd for C20H23N3O5Na, 408.1536; found 408.1518.
5.1.9. (E)-tert-butyl 3-((4-((19-((2-carbamoyl-5-(3,6,6-trimethyl-4-oxo-4,5,6,7-tetrahydro-1H-indazol-1-yl)phenyl)amino)-4,7,10,13,16-pentaoxanonadecyl)carbamoyl)phenyl)diazenyl)-4-hydroxyphenethylcarbamate (14)
Amine 8 (483 mg, 800 μmol), acid 13 (308 mg, 800 μmol), EDC (161 mg, 840 μmol) and HOBT (113 mg, 840 μmol) and 2 chips of DMAP and were dissolved in CH2Cl2 (10 mL) and stirred at RT for 2 h. The reaction mixture was added to a column and chromatographed (2.5 × 20 cm, silica gel, CH2Cl2 (300 mL), 9/1 CH2Cl2/MeOH (300 mL), 4/1 CH2Cl2/MeOH (300 mL). The active fractions were combined and concentrated to a frothy glass to give 14 (560 mg, 72%). The hard glass was scraped out to give an orange powder (460 mg). TLC (9/0.9/0.1 CH2Cl2/MeOH/NH3) Rf = 0.44; 1H NMR (DMSO-d6) δ 10.85 (s, 1H), 8.60 (br t, 1H), 8.40 (br t, 1H), 8.02 (br s, 4H), 7.92 (b s, 1H), 7.73 (d, J = 8.1 Hz, 1H), 7.55 (s, 1H), 7.27 (d, J = 8.1 Hz, 1H), 7.00 (d, J = 8.1 Hz, 1H), 6.88 (br t, 1H), 6.76 (s, 1H), 6.66 (d, J = 8.1 Hz, 1H), 3.47 (m, 20H), 3.19 (m, 1H), 3.12 (m, 1H), 2.90 (s, 2H), 2.67 (t, 2H), 2.52 (m under DMSO, 2H), 2.38 (s, 3H), 2.31 (s, 2H), 1.77 (m, 4H), 1.33 (s, 9H), 0.99 (s, 6H); HRMS (ESI) [M+H]+ calcd for C51H71N8O11, 971.5237; found 971.523575.
5.2. Resin Synthesis (GE Healthcare Instructions 71-7086-00 AFA)
Buffers and solutions
| Swelling solution | 1 mM HCl |
| Coupling buffer | 0.1 M NaHCO3, 0.5 M NaCl, pH = 8.3 |
| Capping solution | 1 M ethanolamine |
| Low buffer | 0.1 M AcOH/NaAcOH, 0.5 M NaCl pH = 4 |
| High buffer | 0.1 M TRIS-HCl, 0.5 M NaCl pH = 8 |
| Storage buffer | 0.1M KH2PO4, pH = 7.4 w/200 mg NaN3/L |
Ligand 14 (25 mg 25.74 μmol) was dissolved in trifluoroacetic acid (1 mL). TLC (9/1/0.1: CH2Cl2/MeOH/NH3) showed loss of starting material and formation of a lower product 12. The mixture was concentrated, then dissolved in ethanol (5 mL) and concentrated again. The residue was then dissolved in ethanol (5 mL) for addition to the resin.
In a big 275 mL column, CNBr-activated Sepharose™ 4B (25 g) was swelled in 1 mM HCl (450 ml) and then washed with 1 mM HCl (5 L). The resin was washed with coupling buffer (125 mL) and then slurried with coupling buffer (125 mL). The mixture was then treated with the linker-PEG-6 compound described above. The mixture was tumbled at RT for 4 h. The resin was then drained and washed with coupling buffer (5 × 125 mL), diluted with more coupling buffer (~125 mL) and treated with capping solution (2 mL) and rotated at RT for 2 h. The solution was drained and washed with 3 rounds of high buffer/low buffer (250 mL ea.) and finally washed with water (250 mL) and transferred in storage buffer (125 mL) to a bottle and stored at 4 °C.
Supplementary Material
Acknowledgments
This work was funded by 1R01-AI089526-01 and 1R01-AI090644-01 to TAJH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.De Maio A. Shock. 1999;11:1. doi: 10.1097/00024382-199901000-00001. [DOI] [PubMed] [Google Scholar]
- 2.Csermely P, Schnaider T, Soti C, Prohaszka Z, Nardai G. Pharmacology & Therapeutics. 1998;79:129. doi: 10.1016/s0163-7258(98)00013-8. [DOI] [PubMed] [Google Scholar]
- 3.Hessling M, Richter K, Buchner J. Nature Structural & Molecular Biology. 2009;16:287. doi: 10.1038/nsmb.1565. [DOI] [PubMed] [Google Scholar]
- 4.Mollapour M, Tsutsumi S, Truman AW, Xu WP, Vaughan CK, Beebe K, Konstantinova A, Vourganti S, Panaretou B, Piper PW, Trepel JB, Prodromou C, Pearl LH, Neckers L. Molecular Cell. 2011;41:672. doi: 10.1016/j.molcel.2011.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vaughan CK, Neckers L, Piper PW. Nature Structural & Molecular Biology. 2010;17:1400. doi: 10.1038/nsmb1210-1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moulick K, Ahn JH, Zong HL, Rodina A, Cerchietti L, DaGama EMG, Caldas-Lopes E, Beebe K, Perna F, Hatzi K, Vu LP, Zhao XY, Zatorska D, Taldone T, Smith-Jones P, Alpaugh M, Gross SS, Pillarsetty N, Ku T, Lewis JS, Larson SM, Levine R, Erdjument-Bromage H, Guzman ML, Nimer SD, Melnick A, Neckers L, Chiosis G. Nature Chemical Biology. 2011;7:818. doi: 10.1038/nchembio.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taipale M, Jarosz DF, Lindquist S. Nature Reviews Molecular Cell Biology. 2010;11:515. doi: 10.1038/nrm2918. [DOI] [PubMed] [Google Scholar]
- 8.Ratzke C, Berkemeier F, Hugel T. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:161. doi: 10.1073/pnas.1107930108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grenert JP, Sullivan WP, Fadden P, Haystead TAJ, Clark J, Mimnaugh E, Krutzsch H, Ochel HJ, Schulte TW, Sausville E, Neckers LM, Toft DO. Journal of Biological Chemistry. 1997;272:23843. doi: 10.1074/jbc.272.38.23843. [DOI] [PubMed] [Google Scholar]
- 10.Prodromou C, Roe SM, Obrien R, Ladbury JE, Piper PW, Pearl LH. Cell. 1997;90:65. doi: 10.1016/s0092-8674(00)80314-1. [DOI] [PubMed] [Google Scholar]
- 11.Hadden MK, Lubbers DJ, Blagg BSJ. Current Topics in Medicinal Chemistry. 2006;6:1173. doi: 10.2174/156802606777812031. [DOI] [PubMed] [Google Scholar]
- 12.Trepel J, Mollapour M, Giaccone G, Neckers L. Nature Reviews Cancer. 2010;10:537. doi: 10.1038/nrc2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chiosis G, Huezo H, Rosen N, Mimnaugh E, Whitesell L, Neckers L. Molecular Cancer Therapeutics. 2003;2:123. [PubMed] [Google Scholar]
- 14.Fadden P, Huang KH, Veal JM, Steed PM, Barabasz AF, Foley B, Hu M, Partridge JM, Rice J, Scott A, Dubois LG, Freed TA, Silinski MAR, Barta TE, Hughes PF, Ommen A, Ma W, Smith ED, Spangenberg AW, Eaves J, Hanson GJ, Hinkley L, Jenks M, Lewis M, Otto J, Pronk GJ, Verleysen K, Haystead TA, Hall SE. Chemistry & Biology. 2010;17:686. doi: 10.1016/j.chembiol.2010.04.015. [DOI] [PubMed] [Google Scholar]
- 15.Biamonte MA, Van de Water R, Arndt JW, Scannevin RH, Perret D, Lee WC. Journal of Medicinal Chemistry. 2010;53:3. doi: 10.1021/jm9004708. [DOI] [PubMed] [Google Scholar]
- 16.Solit DB, Chiosis G. Drug Discovery Today. 2008;13:38. doi: 10.1016/j.drudis.2007.10.007. [DOI] [PubMed] [Google Scholar]
- 17.Ali MMU, Roe SM, Vaughan CK, Meyer P, Panaretou B, Piper PW, Prodromou C, Pearl LH. Nature. 2006;440:1013. doi: 10.1038/nature04716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scroggins BT, Robzyk K, Wang DX, Marcu MG, Tsutsumi S, Beebe K, Cotter RJ, Felts S, Toft D, Karnitz L, Rosen N, Neckers L. Molecular Cell. 2007;25:151. doi: 10.1016/j.molcel.2006.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mollapour M, Tsutsumi S, Donnelly AC, Beebe K, Tokita MJ, Lee MJ, Lee S, Morra G, Bourboulia D, Scroggins BT, Colombo G, Blagg BS, Panaretou B, Stetler-Stevenson WG, Trepel JB, Piper PW, Prodromou C, Pearl LH, Neckers L. Molecular Cell. 2010;37:333. doi: 10.1016/j.molcel.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scroggins BT, Neckers L. Expert Opinion on Drug Discovery. 2007;2:1403. doi: 10.1517/17460441.2.10.1403. [DOI] [PubMed] [Google Scholar]
- 21.Schulte TW, Akinaga S, Murakata T, Agatsuma T, Sugimoto S, Nakano H, Lee YS, Simen BB, Argon Y, Felts S, Toft DO, Neckers LM, Sharma SV. Molecular Endocrinology. 1999;13:1435. doi: 10.1210/mend.13.9.0339. [DOI] [PubMed] [Google Scholar]
- 22.Felts SJ, Owen BAL, Nguyen P, Trepel J, Donner DB, Toft DO. Journal of Biological Chemistry. 2000;275:3305. doi: 10.1074/jbc.275.5.3305. [DOI] [PubMed] [Google Scholar]
- 23.Kang BH, Plescia J, Song HY, Meli M, Colombo G, Beebe K, Scroggins B, Neckers L, Altieri DC. Journal of Clinical Investigation. 2009;119:454. doi: 10.1172/JCI37613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taldone T, Zatorska D, Patel PD, Zong HL, Rodina A, Ahn JH, Moulick K, Guzman ML, Chiosis G. Bioorganic & Medicinal Chemistry. 2011;19:2603. doi: 10.1016/j.bmc.2011.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Citri A, Gan J, Mosesson Y, Vereb G, Szollosi J, Yarden Y. Embo Reports. 2004;5:1165. doi: 10.1038/sj.embor.7400300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vasko RC, Rodriguez RA, Cunningham CN, Ardi VC, Agard DA, McAlpine SR. Acs Medicinal Chemistry Letters. 2010;1:4. doi: 10.1021/ml900003t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang KH, Veal JM, Fadden RP, Rice JW, Eaves J, Strachan JP, Barabasz AF, Foley BE, Barta TE, Ma W, Silinski MA, Hu M, Partridge JM, Scott A, DuBois LG, Freed T, Steed PM, Ommen AJ, Smith ED, Hughes PF, Woodward AR, Hanson GJ, McCall WS, Markworth CJ, Hinkley L, Jenks M, Geng LF, Lewis M, Otto J, Pronk B, Verleysen K, Hall SE. Journal of Medicinal Chemistry. 2009;52:4288. doi: 10.1021/jm900230j. [DOI] [PubMed] [Google Scholar]
- 28.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. Nucleic Acids Research. 2000;28:235. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Graves PR, Haystead TAJ. Microbiology and Molecular Biology Reviews. 2002;66:39. doi: 10.1128/MMBR.66.1.39-63.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Whitesell L, Mimnaugh EG, Decosta B, Myers CE, Neckers LM. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:8324. doi: 10.1073/pnas.91.18.8324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.He HZ, Zatorska D, Kim J, Aguirre J, Llauger L, She YH, Wu N, Immormino RM, Gewirth DT, Chiosis G. Journal of Medicinal Chemistry. 2006;49:381. doi: 10.1021/jm0508078. [DOI] [PubMed] [Google Scholar]
- 32.Verhelst SHL, Fonovic M, Bogyo M. Angewandte Chemie-International Edition. 2007;46:1284. doi: 10.1002/anie.200603811. [DOI] [PubMed] [Google Scholar]
- 33.Joshi VK, Shahani KM, Kilara A, Wagner FW. Journal of Chromatography. 1979;176:11. [Google Scholar]
- 34.Gray GR. Analytical Chemistry. 1980;52:R9. [Google Scholar]
- 35.Cuatreca P. Journal of Biological Chemistry. 1970;245:3059. [PubMed] [Google Scholar]
- 36.Yang YY, Grammel M, Raghavan AS, Charron G, Hang HC. Chemistry & Biology. 2010;17:1212. doi: 10.1016/j.chembiol.2010.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schulte TW, Neckers LM. Cancer Chemotherapy and Pharmacology. 1998;42:273. doi: 10.1007/s002800050817. [DOI] [PubMed] [Google Scholar]
- 38.Graves PR, Kwiek JJ, Fadden P, Ray R, Hardeman K, Coley AM, Foley M, Haystead TAJ. Molecular Pharmacology. 2002;62:1364. doi: 10.1124/mol.62.6.1364. [DOI] [PubMed] [Google Scholar]
- 39.Immormino RM, Metzger LE, Reardon PN, Dollins DE, Blagg BSJ, Gewirth DT. Journal of Molecular Biology. 2009;388:1033. doi: 10.1016/j.jmb.2009.03.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Janecke AR, Thompson DA, Utermann G, Becker C, Hubner CA, Schmid E, McHenry CL, Nair AR, Ruschendorf F, Heckenlively J, Wissinger B, Nurnberg P, Gal A. Nature Genetics. 2004;36:850. doi: 10.1038/ng1394. [DOI] [PubMed] [Google Scholar]
- 41.Thompson DA, Janecke AR, Lange J, Feathers KL, Hubner CA, McHenry CL, Stockton DW, Rammesmayer G, Lupski JR, Antinolo G, Ayuso C, Baiget M, Gouras P, Heckenlively JR, den Hollander A, Jacobson SG, Lewis RA, Sieving PA, Wissinger B, Yzer S, Zrenner E, Utermann G, Gal A. Human Molecular Genetics. 2005;14:3865. doi: 10.1093/hmg/ddi411. [DOI] [PubMed] [Google Scholar]
- 42.Giaccone G, Rajan A, Crandon S, Gutierrez M, Jain L, Figg WD, Houk BE, Shnaidman M, Brega N. Annals of Oncology. 2010;21:23. [Google Scholar]
- 43.Infante J, Weiss G, Jones S, Tibes R, Bendell J, Brega N, Torti V, Von Hoff D, Burris H, Ramanathan R. Ejc Supplements. 2010;8:119. [Google Scholar]
- 44.Morisseau C, Hammock BD. Annual Review of Pharmacology and Toxicology. 2005;45:311. doi: 10.1146/annurev.pharmtox.45.120403.095920. [DOI] [PubMed] [Google Scholar]
- 45.Bucci M, Roviezzo F, Cicala C, Sessa WC, Cirino G. British Journal of Pharmacology. 2000;131:13. doi: 10.1038/sj.bjp.0703549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rice JW, Veal JM, Fadden RP, Barabasz AF, Partridge JM, Barta TE, Dubois LG, Huang KH, Mabbett SR, Silinski MA, Steed PM, Hall SE. Arthritis and Rheumatism. 2008;58:3765. doi: 10.1002/art.24047. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.