

Abstract
Effective resolution of malaria infection by avoiding pathogenesis requires regulated pro- to anti-inflammatory responses and the development of protective immunity. TLRs are known to be critical for initiating innate immune responses, but their roles in the regulation of immune responses and development of protective immunity to malaria remain poorly understood. In this study, using WT, TLR2−/−, TLR4−/−, TLR9−/−, and MyD88−/− mice infected with P. yoelii, we show that TLR9 and MyD88 regulate pro-/anti-inflammatory cytokines, Th1/Th2 development, and cellular and humoral responses. DCs from TLR9−/− and MyD88−/− mice produced significantly lower levels of pro-inflammatory cytokines and higher levels of anti-inflammatory cytokines than DCs from WT mice. NK and CD8+ T cells from TLR9−/− and MyD88−/− mice showed markedly impaired cytotoxic activity. Further, mice deficient in TLR9 and MyD88 showed higher Th2 type and lower Th1 type IgGs. Consequently, TLR9−/− and MyD88−/− mice exhibited compromised ability to control parasitemia and were susceptible to death. Our data also show that TLR9 and MyD88 distinctively regulate immune responses to malaria infection. TLR9−/− but not MyD88−/− mice produced significant levels of both pro- and anti-inflammatory cytokines, including IL-1β and IL-18, by other TLRs/inflammasome- and/or IL-1R/IL-18R-mediated signaling. Thus, while MyD88−/− mice completely lacked cell-mediated immunity, TLR9−/− mice showed low levels of cell-mediated immunity and were slightly more resistant to malaria infection than MyD88−/− mice. Overall, our findings demonstrate that TLR9 and MyD88 play central roles in the immune regulation and development of protective immunity to malaria, and have implications in understanding immune responses to other pathogens.
Introduction
Malaria caused by infection with Plasmodium species of protozoan parasites contributes substantially to the health crisis and death tolls around the world (1). Malaria infection is characterized by dominant pro-inflammatory responses with Th1 cell development during early stages of infection that decrease as infection progresses with parallel increase in production of anti-inflammatory responses (2–4). A robust production of pro-inflammatory cytokine responses at initial stages of infection is necessary for the efficient development of protective cell mediated and humoral immune responses (2–4). On the other hand, excessive and/or prolonged pro-inflammatory responses lead to the development of severe malaria clinical conditions and fatal outcomes (2, 3, 5).
To overcome the detrimental effects of inflammation, as infection progresses, the pro-inflammatory responses are down regulated (2, 3, 5). Toll-like receptors (TLRs), a family of pathogen recognition molecules that sense certain conserved structures of pathogens, play important roles in initiating innate inflammatory responses to various pathogenic infections, including malaria (6–8). In human and mice together, thirteen TLRs (TLR1 to TLR13) have been identified and their ligand recognition specificities have been studied extensively (8, 9). The signal initiated upon TLRs sensing microbial components is transmitted through their highly conserved cytoplasmic Toll/IL-1 receptor (TIR) domains, which in most TLRs recruit a common adaptor protein called MyD88 (7–9). Activation of MyD88 results in the recruitment of several other proteins forming signaling complexes, which activate MAPK and NF-κB pathways leading to the downstream cytokine production.
Several TLRs have been reported to recognize different components of malaria parasites. TLR2 and TLR4 mediate the activation of macrophages by Plasmodium falciparum glycosylphosphatidylinositols (GPIs) (10), TLR4 recognizes heme and microparticles released from parasite-infected erythrocytes (11, 12), and TLR9 is a receptor for the activation of DCs by parasite DNA (13–15). Additionally, in human and mouse malaria parasites, profilin has been reported to activate DCs through TLR11 (16, 17). In mouse malaria parasites, P. berghei, P. chabaudi chabaudi AS and P. yoelii, TLR2 and TLR9 have been reported to be involved in the activation of innate immune system (18–20). However, not much is known about the role of TLRs in the regulation of innate and adaptive cellular and humoral immunity to malaria infection.
In addition to TLRs, a family of intracellular receptors called nucleotide-binding oligomerization domain-like receptors sense microbial components or the endogenous danger signals and form multi-protein complexes known as inflammasome (21). The inflammasome is involved in the proteolytic cleavage of cell-associated pro-IL-1β and pro-IL-18 into secreted IL-1β and IL-18. The production of active IL-1β and IL-18 requires two signal components: (i) TLR-dependent activation of cells that leads to gene transcription and synthesis of pro-IL-1β and pro-IL-18, and (ii) activation of caspase-1 of the inflammasome complex, likely by a second microbial stimuli, resulting in the cleavage of pro-IL-1β and pro-IL-18 into active cytokines. In malaria, NALP3-mediated inflammasome has been reported to be involved in hemozoin- and uric acid-induced maturation of pro-IL-1β and pro-IL-18 (22, 23).
Among cells of the innate immune system, DCs play crucial roles in TLR- and other pathogen specific receptor-mediated recognition, and initiation of innate immune responses and development of adaptive immunity (24–26). At early stages of malaria infection, DCs efficiently produce pro-inflammatory cytokines, and as the infection progresses, their ability to produce pro-inflammatory responses becomes low but acquire increased capacity to produce anti-inflammatory responses (27). Additionally, DCs activate NK cells, instruct T cells to induce programmed Th1/Th2 responses, and initiate the development of cell-mediated and humoral adaptive immunity (24, 25, 28, 29). Thus, DCs provide a critical link between the innate and adaptive immune responses and help shaping the pathogen-specific adaptive immune responses. TLRs and MyD88 are also expressed by T and B cells and play important roles in the function of these cells (30–32). For example, MyD88-mediated signaling is essential for T cell-mediated resistance to Toxoplasma gondii (33), and TLR2-MyD88-mediated signaling is necessary for CD8+ T cell clonal expansion and memory cell formation (34, 35). MyD88 has also been shown to regulate virus-specific CD4+ T-cell responses (36), virus-induced B cell activation and antibody production and Ig class switching to IgG2c (37, 38).
Of different immunostimulatory components of malaria parasites that activate TLRs (10–17), parasite protein-DNA complex/nucleosome is the major factor that activates DCs through TLR9/MyD88-mediated signaling and induces the production of pro-inflammatory responses (15, 39). Given that the cytokine milieu of the initial immune responses determines the effectiveness of adaptive immune responses, we hypothesized that TLR9 and MyD88 play crucial roles in the regulation of Th1/Th2 development and cellular and humoral adaptive immunity to malaria. Here, we tested this hypothesis by studying innate and adaptive immune responses to the blood stage P. yoelii mouse malaria infection. The results show that TLR9 and MyD88 are critical for the robust pro-inflammatory responses, Th1 development, and efficient cell-mediated and humoral immunity to malaria infection. Hence, the deficiency in TLR9 and MyD88 resulted in the decreased capacity of DCs to produce pro-inflammatory cytokines with the increased ability to elicit anti-inflammatory cytokine responses, impaired NK and CD8+ T cell cytotoxic activity, and increased Th2 type antibody responses. Consequently, TLR9−/− and MyD88−/− mice harbored significantly higher parasitemia and exhibit lower survival rates than wild type (WT) mice.
Materials and Methods
Reagents
DMEM and RPMI 1640 medium were purchased from Mediatech, Inc. (Manassas, VA). Penicillin/streptomycin solution was from Invitrogen Corp. (Carlsbad, CA). Fetal bovine serum (FBS) was purchased from Atlanta Biologicals (Lawrenceville, GA). Collagenase D was from Roche Applied Science (Mannheim, Germany). Non-essential amino acids and 2-mercaptoethanol for cell culturing, LPS from Salmonella minnesota Re595strain and percoll were from Sigma-Aldrich (St. Louis, MO). CpG ODN1826 was from Coley Pharmaceuticals (Kanata, ON, Canada). Pam3CSK4 was from Microcollections (Tübingen, Germany). Poly (I:C), a synthetic analog of dsRNA (TLR3 ligand), was from InvivoGen (San Diego, CA). Chicken ovalbumin peptide323–339 (OVA323–339) was from Peptides International, Inc. (Louisville, KY). Isolymph was from CTL Scientific Supply (Deer Park, NY). CytoTox 96® non-radioactive cytotoxicity assay kit was from Promega Corp. (Madison, WI). Mouse immunoglobulin isotyping ELISA kit and Cy5-conjugated annexin V were from BD Biosciences (San Jose, CA). Target cells, YAC-1 (a murine lymphoma cell line) and EL-4 (a mouse thymocyte cell line), for the measurement of NK cell and CD8+ T cell cytotoxic activity were provided, respectively, by Drs. Nikki Keasey and Todd Schell, Penn State University College of Medicine, Hershey, PA.
Anti-mouse CD11c antibody (clone N418)-conjugated MicroBeads (cat # 130-052-001) for the isolation of DCs, anti-mouse CD90.2 antibody-conjugated MicroBeads (cat # 130-049-101) for total T cell isolation, mouse CD8α T cell isolation kit (cat # 130-090-859), and mouse NK cell isolation kit (cat # 130-090-864) were purchased from Miltenyi Biotec Inc. (Auburn, CA).
Duoset ELISA kits for measuring mouse TNF-α, IL-12p40, IL-1β, IL-4, IL-10, and IFN-γ were from R&D Systems (Minneapolis, MN). Coating and biotin-labeled detecting antibodies against mouse IL-18 (clones 74 and 93–10C) and mouse IL-18 standard for the measurement of IL-18 by ELISA were purchased from MBL International Corp. (Woburn, MA).
Antibodies
The antibodies used in this study were as follows: Purified anti-mouse CD3ε antibody (clone 17A2), anti-mouse CD16/32 monoclonal antibody (clone 93), FITC-conjugated monoclonal antibodies against mouse CD3ε (clone 145-2C11), CD11c (N418), CD19 (eBio1D3), pan NK cell (DX5) and TNF-α (MP6-XT22), PE-conjugated anti-mouse NK1.1 antibody (PK136), PE-Cy5-conjugated antibodies against mouse CD80 (16-10A1) and hamster IgG (eBio299Arm) isotype control, CD86 (GL1) and rat IgG2aκ isotype control, PerCP-Cy5.5-conjugated anti-CD8α monoclonal antibody (53-6.7), and allophycocyanin (APC)-conjugated antibodies against mouse CD3ε (clone 145-2C11), mouse CD40 (1C10) and mouse IFN-γ (XMG1.2), and rat IgG1κ isotype control were from eBioscience (San Diego, CA). PerCP-Cy5.5-conjugated anti-mouse IL-4 (11B11) and PE-conjugated anti-mouse IL-12 antibodies (clone 15.6) were from BD Biosciences (San Jose, CA).
Ethics statement
The Institutional Animal Care and Use Committee of the Pennsylvania State University College of Medicine, Hershey, has reviewed and approved the protocols for use of animals in this study. Theanimal care was according to the institutional guidelinesof the Pennsylvania State University College of Medicine.
Mice
WT, and TLR2, TLR4, TLR9 and MyD88 knockout mice, and OT-II transgenic mice expressing TCR for OVA323–339 peptide on CD4 T cells were housed in a pathogen-free environment. All mice used in this study were in C57BL/6J background.
Parasite infection, and parasitemia and survival rate measurements
WT C57BL/6 mice were infected with cryopreserved nonlethal P. yoelii (Py17XNL strain) parasites and blood from these mice was used for infecting experimental mice. Experimental mice were infected by intraperitoneal injection of 1 × 106 infected erythrocytes from infected donor mice in 100 μl of saline. Parasitemia was monitored on alternative days after infection by examining Giemsa-stained thin smears of tail blood on glass slides, and results were expressed as percentage of parasite infected erythrocytes. Mice were monitored for survival twice a day. Blood was collected, sera prepared, and stored at −80 °C until used for cytokine and antibody analyses by ELISA (15). Mice were also infected as above with P. berghei NK65 strain and IRBCs isolated and used for testing the antigen specificity of cytotoxic CD8+ T cells.
Preparation of FLT3- and GM-CSF-differentiated DCs
FLT3 (Fms-like tyrosine kinase 3)-differentiated DCs (FL-DCs) were prepared by culturing mouse bone marrow cells in complete DMEM supplemented with 15% of FLT3 ligand-containing conditioned medium, which is obtained by culturing B16 cells expressing retrovirus-coded FLT3 ligand (15, 40).
GM-CSF differentiated DCs (GM-DCs) were obtained by culturing bone marrow cells from WT mice for 7 or 8 days in complete DMEM containing 10% conditioned DMEM from the cultured GM-CSF producing cells (41).
Isolation of spleen and liver cells
Single cell suspensions of mouse spleens were prepared as described previously (15) and used for the isolation of total T cells and CD8+ T cells by magnetic column separation (MACS). Total T cells from mouse spleens were isolated using anti-mouse CD90.2 antibody-conjugated magnetic beads; the purity of cells as assessed by flow cytometry after staining with anti-CD3ε antibody was ~90%. CD8+ T cells were isolated using CD8α T cell isolation kit; the purity of cells as analyzed by flow cytometry by staining with anti-CD3ε and anti-CD8α antibodies was ~92%.
DCs were isolated from the single cell suspensions of mouse spleens, prepared by digesting with 1 mg/ml of collagenase D (15), by MACS after staining with anti-mouse CD11c antibody-conjugated microbeads. The purity of cells as assessed by flow cytometry after staining with anti-CD11c antibody was ~90%.
For NK cell isolation, livers were flushed with 10 ml of PBS, pH 7.2, crushed and filtered through a 70-mm strainer to obtain single cell suspensions. The cell suspensions were centrifuged on Isolymph cushions at 1200 g at 4°C for 15 min, Buffy coat on the top of Isolymph were collected, washed, NK cells were isolated by MACS using the mouse NK cell isolation kit. The purity of cells by flow cytometry analysis after staining with anti-mouse NK1.1 antibody was ~60%.
Isolation of P. yoelii- and P. berghei-infected red blood cells and preparation of cell lysates
Erythrocyte pellets from blood samples of parasite infected mice were suspended in two volumes of PBS, pH 7.2, centrifuged on Isolymph cushions at 1200 g at 4°C for 15 min, and Buffy coat removed. The erythrocyte pellets were resuspended in two volumes of PBS and, centrifuged on 75% percoll cushions at 1200 g at 4°C for 15 min. The infected red blood cells (IRBCs) on the top of percoll were collected, and washed.
Lysates of P. yoelii IRBCs were prepared by alternative freezing and thawing of IRBC suspensions three times followed by sonication in water-bath sonicator for 5 min. After centrifugation at 13, 000 rpm in microcentrifuge for 10 min, the protein contents in the clear supernatants were measured using Pierce micro BCA protein estimation kit (Thermo Scientific, Rockford, IL), and used for antibody analysis in mice sera.
Stimulation of DCs and co-cultures of DCs plus OT-II T cells
Mouse spleen DCs (1 × 105/well) from infected mice were seeded into 96-well plates. Cells in 200 μl of complete DMEM were stimulated with standard TLR ligands: Pam3CSK4 (TLR2 ligand, 10 ng/ml), Poly I:C (TLR3 ligand, 2 μg/ml), LPS (TLR4 ligand, 100 ng/ml) or CpG ODN (TLR9 ligand, 2 μg/ml). After 24 h, the culture supernatants were analyzed for cytokines by ELISA (15). In coculturing experiments, spleen DCs (1 × 105/well) from infected mice were either untreated or treated with 2 μg/ml OVA323–339 peptide and cocultured with spleen T cells (0.5 × 105/well) from the naïve OT-II transgenic mice that expresses OVA-specific T cell receptor in 96 well U-bottomed plates containing 200 μl of complete medium. OT-II T cells alone were similarly stimulated with OVA323–339 peptide as controls. After 72 h, the culture supernatants were collected and cytokines analyzed by ELISA (15).
Flow cytometry analysis of cytokine expression and costimulatory molecules
To determine the cell types that produce cytokines in coculture experiments, cells were stimulated, treated with GolgiPlug (BD Biosciences) for 6 h, treated with Fc block (anti-mouse CD16/32 antibody), and stained for surface markers followed by intracellular staining with anti-cytokine antibodies (15). The survival of DCs was assessed by flow cytometry using FACSCalibur (BD Biosciences) after staining with annexin V.
For the analysis of costimulatory molecules, total spleen cells from WT, TLR9−/− and MyD88−/− mice at 3 days postinfection were stained with dye-conjugated antibodies against costimulatory molecules. After washing, cells were analyzed by flow cytometry and the results were analyzed using CellQuest software (BD Biosciences).
Restimulation of T cells from P. yoelii-infected mice
Total spleen T cells isolated from the parasite-infected mice were allowed to rest overnight in complete DMEM and plated (1 × 105 cells/well) into 96-well U-bottomed plates. To each well was added FL-DCs (5 × 104/well) and P. yoelii IRBCs (1.5 × 105/well) in 200 μl of complete DMEM. After 72 h, the culture supernatants were collected and cytokines measured by ELISA. Supernatants of T cells cocultured with WT FL-DCs in the absence of IRBCs or T cells alone cultured in the presence of IRBCs were used as controls. Cytokine production by DCs and T cells were also analyzed by intracellular staining as described above.
Cell cytotoxicity assay
The cytotoxic activity of spleen CD8+ T cells was determined by measuring the lysis of P. yoelii IRBC-pulsed GM-DCs and the anti-mouse CD3ε antibody-redirected lysis of EL-4 cells (H-2b background, 42). The cytotoxic activity of NK cells was measured by assaying the lysis of YAC-1 target cells (43). Briefly, to different doses of spleen CD8+ T cells in 96-well U-bottomed plates were added GM-DCs (1 × 104/well) that were previously pulsed with P. yoelii or P. berghei IRBC lysates at DC to IRBC equivalent of 1:5 for 4 h. In separate sets of experiments, EL-4 cells were treated with 10 μg/ml anti-CD3ε antibody, washed with incomplete RPMI 1640 medium. To different doses of spleen CD8+ T cells plated in 96-well U-bottomed plates were added antibody-treated EL-4 target cells (1 × 104/well) in 100 μl of RPMI 1640 medium. Similarly, to liver NK cells plated in 96-well U-bottomed plates were added YAC-1 cells (1X104/well) in 100 μl of RPMI 1640 medium. In each case, plates were centrifuged for 3 min at 300 g to facilitate cell-cell contact and incubated at 37 °C for 4 h. Target cell alone to which control lysis solution was added served as positive controls. Fifty-μl of culture supernatants were transferred into 96-well opaque plates, the extent of target cell lysis was measured using CytoTox 96 non-radioactive cytotoxicity assay reagents according to the manufacturer’s instructions.
EL-4 and YAC-1 cells used in the above assays were cultured in RPMI 1640 medium containing 10% FBS, 1% penicillin-streptomycin, 1% non-essential amino acids, 1 mM sodium pyruvate, 50 μM 2-mercaptoethanol, and harvested at exponential growth phase.
Analysis of antibody responses in infected mice
The levels of total IgG in the sera of P. yoelii infected mice were analyzed by ELISA. Ninety-six well microtiter plates were coated with IRBC lysates (20 μg protein per ml) in 50 mM sodium bicarbonate buffer, pH 9.6, overnight at 4 °C, and blocked with 1% BSA in PBS. The plates were incubated with serially diluted mouse sera at room temperature for 1 h. After washing, the plates were incubated with 1:3000 diluted horseradish peroxidase-conjugated goat anti-mouse IgG for 1 h followed by SureBlue TMB peroxidase substrate solution (KPL, Gaithersburg, MD). The reaction was stopped by adding 2 N H2SO4, and absorbance at 450 nm measured.
The antibody isotypes in the sera from P. yoelii infected mice were analyzed using the mouse immunoglobulin isotyping ELISA kit according to the manufacturer’s instructions.
Statistical Analysis
The data were plotted as mean values ± SD or SEM. Statistical analysis of data was performed by one-way ANOVA followed by the Newman-Keuls test. GraphPad prism software version 3.0 was used for the analysis. p values <0.05 were considered statistically significant.
Results
TLR9 and MyD88 regulate malaria-induced DC functions
Malaria infection is characterized by a robust pro-inflammatory cytokine production during the early stages of infection that declines as infection progresses with parallel gradual increase in the production of anti-inflammatory cytokines (2–4). The initial cytokine responses play important roles in the regulation and development of immunity to malaria (2–5). Although several TLRs, including TLR9, TLR2 and TLR4, have been shown to recognize various components of malaria parasites and induce pro-inflammatory cytokines (10–17), which of these TLRs play key roles in the generation of protective immunity to malaria remains unknown. Since DCs are central to the development of innate and adaptive immunity to malaria (2–4), we first studied the roles of TLRs and their key shared adaptor protein MyD88 in the regulation of pro- and anti-inflammatory cytokine responses by DCs isolated from the spleen of WT, TLR2−/−, TLR4−/−, TLR9−/− or MyD88−/− mice infected with P. yoelii. At the early stages of infection (5 days postinfection), when the parasite is establishing a stable infection (see Fig. 8A and 8B later), DCs obtained from WT, TLR2−/− and TLR4−/− mice, but not those from TLR9−/− and MyD88−/− mice efficiently produced TNF-α and IL-12 (Fig. 1). At the later stages (10 and 17 days) of infection, i.e., when parasites grew exponentially, the levels of these cytokines produced by DCs from the WT, TLR2−/− and TLR4−/− mice were markedly declined. In parallel, however, DCs from these mice at 10 days and 17 days postinfection produced increased levels of IL-4 as compared to DCs from the infected mice at 5 days postinfection. The expression of IL-10 was also modestly increased in DCs from mice at 10 days postinfection compared to cells from mice at 5 days postinfection, but decreased considerably in DCs from mice at 17 days postinfection when parasitemia was high (Fig. 1). Further, DCs produced high levels of cytokines in response to standard immunostimulatory ligands in TLR-specific manner (Fig. S1). Together these results indicated that at the early stages of infection, DCs produce mainly pro-inflammatory responses, which are known to instruct the immune system to develop effective Th1 adaptive immunity for parasite clearance, whereas at later stages of infection these cells produce primarily anti-inflammatory cytokines, presumably leading to Th2 responses.
FIGURE 8. P. yoelii infected TLR9 and MyD88 deficient mice exhibit higher parasitemia and mortality than infected WT, TLR2−/−, and TLR4−/− mice.
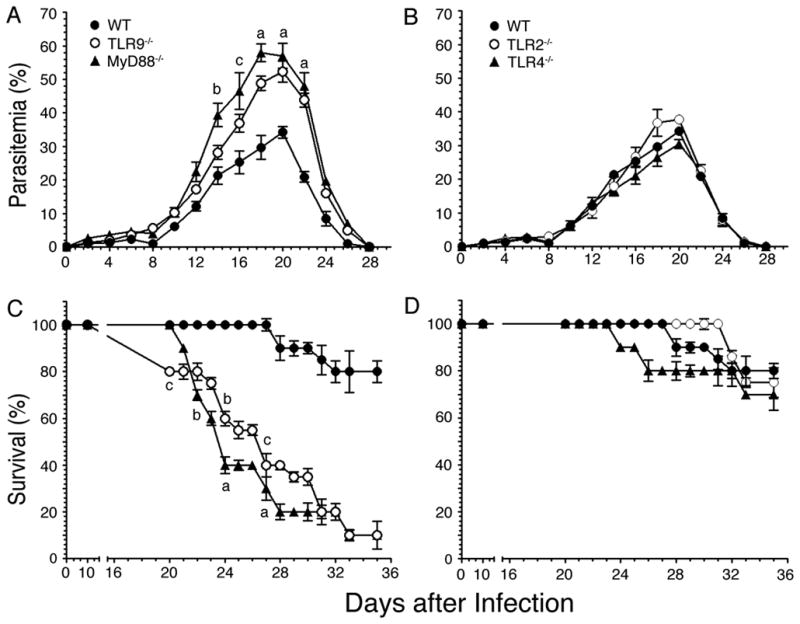
WT, TLR2−/−, TLR4−/−, TLR9−/−, and MyD88−/− mice (n = 10 in each case) were infected with P. yoelii. A and B, Blood parasitemia was measured every 2 days after infection and mean parasitemia in survived animals are plotted. C and D, The infected mice were monitored twice daily and their survival rates are plotted. Experiments were performed two times. The letters, a, b and c, denote the statistical significance of parasitemia and survival rates between TLR9−/− or MyD88−/− mice and infected WT mice. a, p <0.001; b, p <0.01; c, p <0.05.
FIGURE 1. TLR9 and MyD88 regulate pro- and anti-inflammatory cytokine production by DCs in malaria parasite-infected mice.
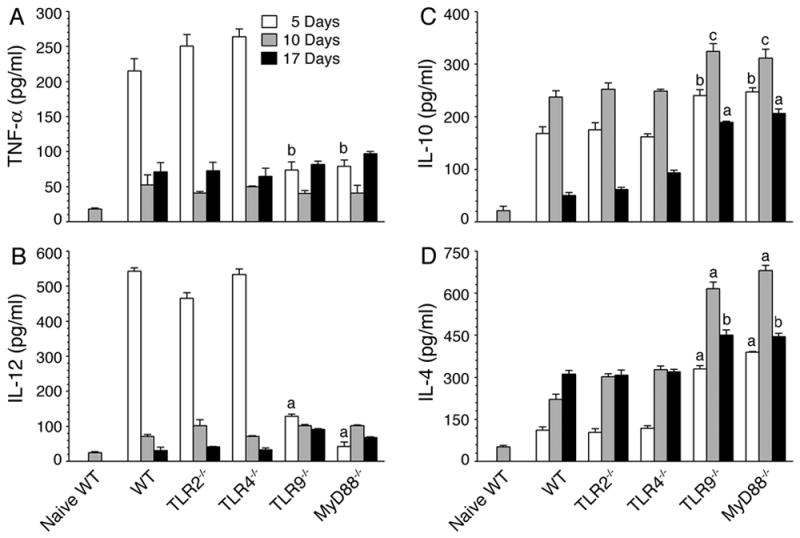
Spleen DCs, isolated from P. yoelii-infected WT, TLR2−/−, TLR4−/−, TLR9−/−, and MyD88−/− mice at 5, 10, and 17 days postinfection, were cultured in 96 well plates. After 24 h, TNF-α (A), IL-12 (B), IL-10 (C) and IL-4 (D) released into the culture media were analyzed by ELISA. Spleen DCs from naïve WT mice were analyzed as controls. Experiments were performed three times and each time ELISA was performed in duplicates. The results of a representative experiment are shown. Error bars represent mean values ± SD. The letters, a, b and c, denote the statistical significance between the levels of cytokines produced by DCs from the indicated gene knockout mice and those produced by DCs from the corresponding infected WT mice. a, p <0.001; b, p <0.01; c, p <0.05.
In contrast to infected WT, TLR2−/− and TLR4−/− mice, DCs from infected TLR9−/− and MyD88−/− mice produced low levels of TNF-α and IL-12, and higher levels of IL-10 and IL-4, at all stages of infection (Fig. 1). Notably, at early stages of infection, TNF-α and IL-12 levels produced by DCs from infected TLR9−/− or MyD88−/− mice were comparable to those produced by DCs from the infected WT, TLR2−/− and TLR4−/− mice at later stages (10 and 17 days) of infection. Collectively, these results revealed that TLR9- and MyD88-mediated signaling dominantly drives pro-inflammatory cytokine responses to malaria parasites during the early stages of infection and thus, the deficiency in TLR9 or MyD88 lead to type 2 cytokine/anti-inflammatory responses by DCs. The results are consistent with our recent report that TLR9 is the major sensor of P. falciparum that mediates pro-inflammatory responses (15).
Given that cytokine responses by DCs from infected TLR9−/− and MyD88−/− mice were markedly different from those by DCs from infected WT mice, it was of interest to determine whether DCs from these mice differ in their activation status. Therefore, we evaluated maturation of DCs in the infected animals by analyzing surface expression of costimulatory molecules. Spleen DCs from WT, TLR9−/−, and MyD88−/− mice at 3 days postinfection exhibited significantly increased levels of CD40, CD80, and CD86 than DCs from naïve WT mice (Fig. S2). These results demonstrate that, regardless of the critical requirement of TLR9 and MyD88 for the production of pro-inflammatory cytokines by DCs, malaria parasites can activate and induce maturation of DCs in TLR9- and MyD88-independent manner. These results agree with the ability of DCs from infected TLR9−/− and MyD88−/− mice to efficiently produce anti-inflammatory responses (see Fig. 1).
To further understand the role of TLR9 and MyD88 in malaria parasite-induced function of DCs, we assessed the ability of ex vivo DCs to induce cytokine responses in T cells. DCs from the spleens of mice at 5, 10 and 17 days postinfection were cocultured with OT-II T cells and treated with OVA peptide and the levels of TNF-α, IL-12, IFN-γ, IL-4 and IL-10 in the culture supernatants were analyzed by ELISA. The cytokine levels produced by the cocultures was significantly higher than that produced by the control DCs (p <0.05 to <0.001) (Fig. 2A-E). Intracellular staining with anti-cytokine antibodies and flow cytometry analysis showed that DCs from mice at both 5 and 10 days postinfection can efficiently induce the production of TNF-α and IFN-γ in OT-II T cells (Fig. 2F); cells were gated as indicated in Fig. S3A. DCs from mice at 5 days postinfection induced substantially higher levels of TNF-α and IFN-γ production by OT-II T cells than DCs from mice at 10 days postinfection. These results are consistent with a previous report, based on ELISA analysis of cytokines produced by cocultures, that the ability of DCs from malaria infected mice to induce T cells to produce pro-inflammatory responses decreases as the infection progresses (27). However, in contrast (27), DCs from mice at both 5 and 10 days postinfection unable to induce detectable levels of IL-4 and IL-10 by OT-II T cells (Fig. S3B and data not shown), even though the cocultures produced substantially higher levels of these cytokines than by control DCs. Therefore, it appears that, in vivo, besides DCs, cytokine milieu and other antigen-presenting cells such as macrophages and mast cells influence T cells to produce IL-4 and IL-10. Macrophages uptake IRBCs efficiently and produce IL-10 and IL-4 (44, and our unpublished results). Consistent with the fact that T cells produce little or no IL-12 (45), the expression of this cytokine by OT-II T cells was not evident. However, interestingly, the cocultures produced significantly higher levels of IL-12 than control DCs (Fig. 2C). Therefore, the observed increased production of IL-4, IL-10, and IL-12 by cocultures, even though OT-II T cells unable to produce these cytokines, suggested that OT-II T cells in turn influenced DCs to induce increased expression of cytokines and/or enabled DCs to survive longer, leading to increased production of cytokines. The influence of T cells in inducing DCs to produce higher levels of cytokines is also evident from enhanced production of TNF-α and IL-4 by DCs cocultured with OT-II T cells as compared to DCs cultured alone (Fig. S3C). To determine the possibility that ligation of T cells leading to increased life span of DCs in coculture, we assessed the duration of survival of cocultured DCs by measuring the levels of cell death at different time points by flow cytometry after staining with annexin V. It is known that annexin V binds phosphatidylserine expressed on the surface of apoptotic cells and the annexin V-binding assay is used for measuring the extent of cell death (46, 47). The results showed that the levels of annexin V-positive DCs were substantially higher in control DC culture than that in coculture (Fig. 2G). Together the above data clearly demonstrated that ligation of OT-II T cells lead to increased life span of DCs and hence increased production of cytokines by DCs. These results are consistent with the previous report that ligation of CD40 and CD40 ligand leads to increased production of IL-12 by DCs (48, 49).
FIGURE 2. DCs from malaria infected mice induced cytokine expression by OT-II T cells.
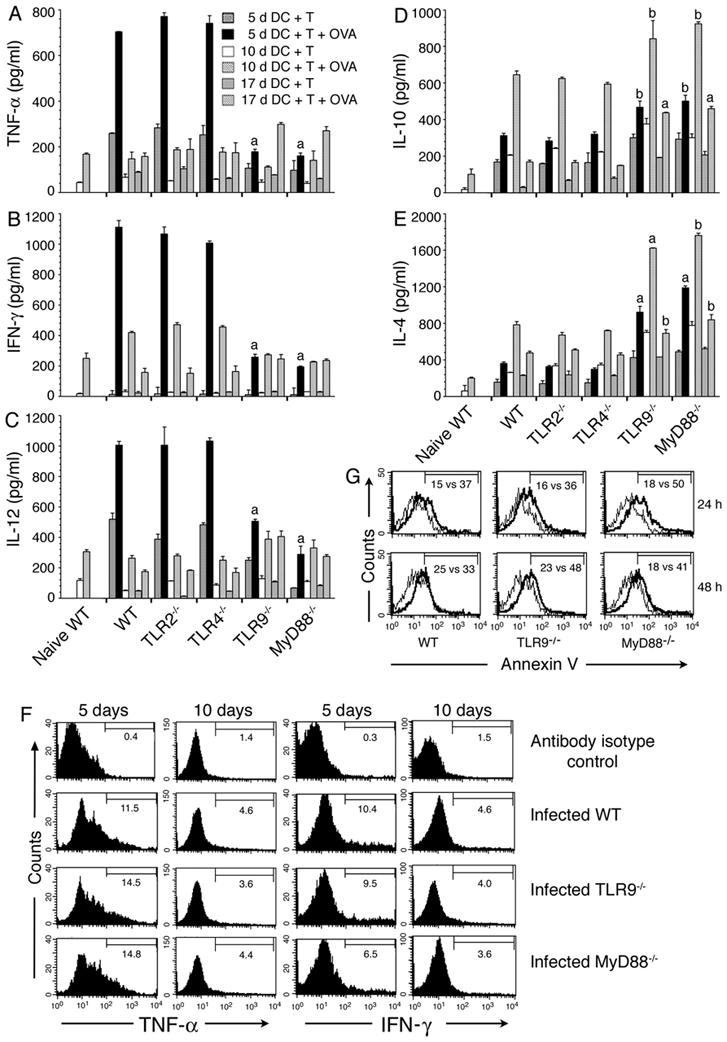
Spleen T cells from naïve OT-II transgenic mice were cocultured with spleen DCs from P. yoelii-infected WT, TLR2−/−, TLR4−/−, TLR9−/− and MyD88−/− mice at the indicated days postinfection in the presence of OVA323–339 peptide. After 72 h, the culture supernatants were collected and assayed for TNF-α (A), IFN-γ (B), IL-12 (C), IL-10 (D) and IL-4 (E) by ELISA. Cocultures not treated with OVA peptide were used as controls. Data are a representative of three independent experiments, each performed in duplicates. Note: 5 d DC plus T, 10 d DC plus T, and 17 d DC plus T refers to cocultures of spleen DCs from mice at 5, 10, and 17 days postinfection and OT-II T cells from naïve mice. The letters, a and b, indicate the statistical significance between the levels of cytokines produced by OT-II T cells activated with DCs from the indicated gene knockout mice and those produced by OT-II T cells activated with DCs from the corresponding infected WT mice. a, p <0.001; b, p <0.01. (F) OT-II T cells and DCs from mice at 5 and 10 days postinfection were cocultured for 6–12 h in the presence of OVA peptide and then added GolgiPlug. Production of TNF-α and IFN-γ by T cells were analyzed by flow cytometry after intracellular staining with anti-cytokine antibodies. Histograms show the percent positive OT-II T cells for each cytokine. (G) Spleen DCs from mice at 5 days postinfection were cultured either alone or with OT-II T cells in presence of OVA peptide. After 24 or 48 h, DCs were surface stained with annexin V and analyzed by flow cytometry. Histograms indicate the percentage of annexin V positive cells in gated DCs in coculture vs. control DC culture.
Furthermore, in three independent experiments, we consistently found that OT-II T cells cocultuted with spleen DCs from infected TLR9−/− and MyD88−/− mice produced noticeably lower levels of IFN-γ than OT-II cells cocultured with DCs from infected WT mice, although there was no difference in the level of TNF-α production (Fig. 2F). However, in all three groups, OT-II T cells cocultured with DCs from mice at 5 days postinfection produced markedly higher levels of TNF-α and IFN-γ than OT-II T cells cocultured with DCs from mice at 10 days postinfection. Thus, these results demonstrated that DCs are programmed to induce a strong Th1 development at early stages of infection in a TLR9- and MyD88-dependent manner, and that the ability of DCs to induce Th1 polarization decreases as the infection progresses.
TLR9 and MyD88 differentially regulate cytokine production in response to malaria infection
In addition to DCs, various other cell types, including T and B cells produce cytokines, contributing to serum cytokine profiles of malaria-infected mice. This is especially the case at later stages of infection. To determine the role of TLR9 and MyD88 on cytokine production in vivo in response to malaria infection, we analyzed cytokine responses in P. yoelii-infected WT, TLR2−/−, TLR4−/−, TLR9−/−, and MyD88−/− mice. The serum TNF-α, IL-12, IL-10 and IL-4 profile in WT, TLR2−/−, TLR4−/−, and TLR9−/− mice at the early stages of infection were similar to those produced by DC ex vivo (Fig. 3). That is, higher levels of pro-inflammatory cytokines, including IFN-γ, and lower Th2 type/anti-inflammatory cytokines by WT, TLR2−/−, and TLR4−/− mice, and vice versa by TLR9−/− mice. However, at the later stages of infection, while the profiles of TNF-α and IL-10 in the sera of infected WT, TLR2−/−, and TLR4−/− mice were comparable to those produced by ex vivo DCs, the serum levels of IL-12 and IL-4 was substantially increased in all these mice.
FIGURE 3. Malaria infected TLR9−/− and MyD88−/− mice differentially produce cytokines.
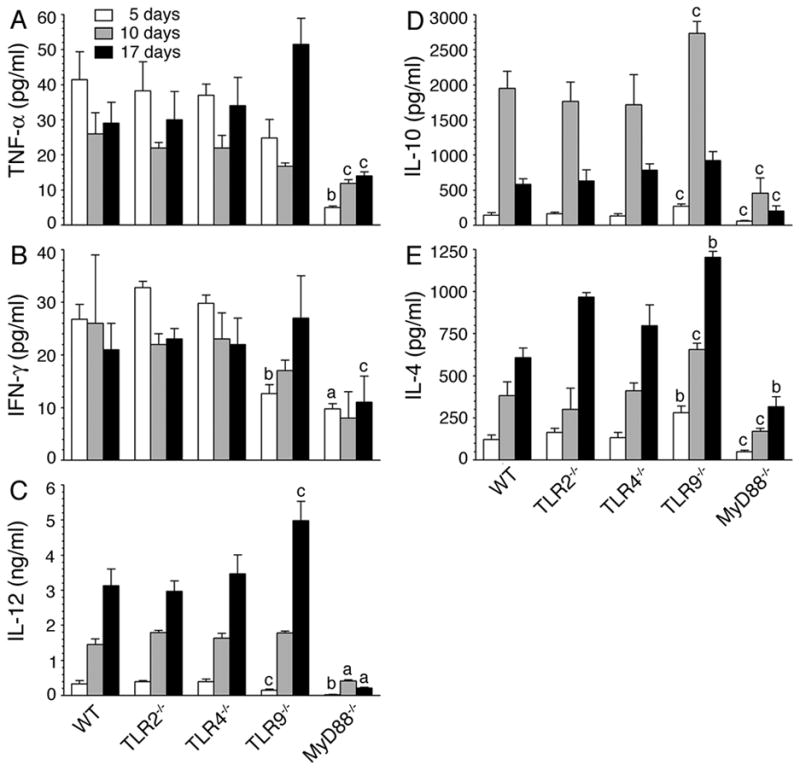
WT, TLR2−/−, TLR4−/−, TLR9−/− and MyD88−/− mice (n = 5 in each group) were infected with P. yoelii. Blood from the infected mice at 5, 10, and 17 days postinfection was collected, and sera prepared. TNF-α (A), IFN-γ (B), IL-12 (C), IL-10 (D) and IL-4 (E) present in the pooled sera were measured by ELISA. Experiments were performed two times and each time ELISA was performed in duplicates. The letters, a, b and c, represent the statistical significance between the levels of cytokines produced by the indicated gene knockout mice and those produced by the corresponding infected WT mice. a, p <0.001; b, p <0.01; c, p <0.05.
Notably, in contrast to cytokine profiles observed by DC ex vivo (see Fig. 1), the infected TLR9−/− mice presented a distinct cytokine profile at the later stages of infection; that is, significantly higher levels of IL-12 and IL-4 compared to WT, TLR2−/−, and TLR4−/− mice (Fig. 3). Furthermore, interestingly, in the case of infected MyD88−/− mice, the levels of both pro- and anti-inflammatory cytokines were markedly low at all stages of infection, despite the fact that ex vivo DCs produced significantly higher levels of IL-10 and IL-4 than DCs from the infected WT, TLR2−/− and TLR4−/− mice (compare Figs. 1 and 3). Since cytokine profiles of DCs from WT, TLR2−/− and TLR4−/− mice were more or less comparable to those observed in vivo in these mice at early stages of infection (compare Figs.1 and 3), it appears that, during early stages of infection, DCs are the major contributors to the cytokine profiles observed in infected mice. However, at later stages of infection, it appears that other cell types such as T and B cells contribute to the production of cytokines in a TLR9-independent and MyD88-dependent manner. Thus, the results demonstrate that MyD88−/− mice but not TLR9−/− mice have intrinsic defect in producing pro- and anti-inflammatory cytokines in response to malaria infection (Fig. 3), although DCs deficient in TLR9 and MyD88 produce similar cytokine profiles (see Fig. 1).
TLR9 and MyD88 contribute to cytokine production by T and B cells in response to malaria infection
To test the prediction that, at late infection stages, T and B cells significantly contribute to the observed serum cytokine profiles (see above), we first analyzed cytokines produced by T cells. Intracellular staining showed that T cells from WT, TLR9−/−, and MyD88−/− mice at 10 and 17 days postinfection produced substantial levels of IFN-γ and IL-4 (Fig. 4A, 4B). The pro-inflammatory cytokine expression by T cells from the infected TLR9−/− and MyD88−/− mice was appreciably lower than that by T cells from WT mice. We next analyzed cytokine responses by T cells isolated from mice at 10 days postinfection after restimulation with P. yoelii IRBC antigens presented by in vitro generated DCs. Previously, we showed that IRBCs can induce efficient production of cytokines by FL-DCs and spleen DCs, and that the cytokine profiles produced by these cells are similar (15). Here, we used FL-DCs from WT mice as antigen-presenting cells for the restimulation of ex vivo T cells. Cytokine profiles produced by T cells restimulated with IRBC-treated FL-DCs were similar to those produced by unstimulated ex vivo T cells (Fig. S4A, and compare Fig. S4A with Fig. 4B). Together the above results indicate that cytokine production by T cells in response to malaria infection is to certain extent TLR9- and MyD88-dependent and that the cytokines produced by T cells contribute significantly to the serum cytokine profiles of parasite-infected WT, TLR9−/−, and MyD88−/− mice.
FIGURE 4. TLR9 and MyD88 are critical for production of cytokines by T and B cells.

Total spleen cells from WT, TLR9−/− and MyD88−/− mice at 10 and 17 days post infection were cultured for 4 h in the presence of GolgiPlug, surface stained with anti-mouse CD3ε antibody followed by intracellular staining using anti-cytokine antibodies, and analyzed by flow cytometry. (A) Cells were selected by side scattering (SSC) and forward scattering (FSC) and were gated for total T cells (CD3ε+; B) and B cells (CD19+; G). (B) Percent positive cells for each cytokine in gated T cells are shown in the histograms. Cells from naïve WT mice were analyzed as controls. Isolated spleen T cells from WT, TLR9−/− and MyD88−/− mice at 10 days (C and D) and 17 days (E and F) after infection were cocultured with WT FL-DCs in the presence or absence of P. yoelii IRBCs (C–F; D and F are controls for C and E, respectively). In parallel, WT FL-DCs alone was also stimulated with IRBCs as controls for cocultures. After 72 h, cytokines secreted into the culture medium were analyzed by ELISA. The letters, a, b and c represent statistical significance between the levels of cytokines in indicated groups of infected mice. a, p <0.001; b, p <0.01; c, p <0.05. (G) The cultured total spleen cells were surface stained for B cells using anti-mouse CD19 antibody followed by intracellular staining using antibodies against mouse IL-12 and IL-4. Histograms show the percent B cells positive for each cytokine. B cells from naïve WT mice were similarly stained and analyzed as controls.
We next measured cytokine levels in culture supernatants of spleen T cells from WT, TLR9−/− and MyD88−/− mice at 10 and 17 days postinfection restimulated with P. yoelii IRBCs presented by FL-DCs. The profiles of TNF-α, IFN-γ, IL-12, IL-4, and IL-10 produced were nearly comparable to those of serum cytokines in the respective infected mice (Fig. 4C and E, and see Fig. 3). Further, the levels of cytokines produced by restimulated T cells were significantly higher than those secreted by the IRBC-stimulated control DCs (Fig. 4C and 4E). Furthermore, in the case of T cells from mice at 10 day postinfection, the restimulated T cells from WT mice produced significantly higher levels of TNF-α, IFN-γ, and IL-12 than the TLR9−/−T cells, whereas the restimulated TLR9−/− T cells produced significantly higher levels of IL-4 and IL-10 (Fig. 4C and 4D). However, in the case of T cells from mice at 17 days postinfection, the restimulated T cells from TLR9−/− mice produced significantly higher levels of TNF-α, IL-12, and IL-4 than the T cells from WT mice (Fig. 4E and 4F). Compared to these results, the T cells from MyD88−/− mice at 10 and 17 days postinfection produced substantially lower levels of both pro- and anti-inflammatory cytokines. FL-DCs also produced appreciable levels of cytokines in the presence of T cells from infected mice (Fig. S4B). Together the above results confirm that DC and T cell interactions lead to cytokine production by both cell types in response to malaria parasites in a TLR9- and MyD88-dependent manner.
We also assessed cytokine responses by B cells at 10 and 17 days postinfection by intracellular staining and flow cytometry. Although B cells are known to produce a wide range of cytokines, including TNF-α, IFN-γ, IL-12, IL-10, and IL-4, we analyzed IL-12 and IL-4 production as representatives of pro-inflammatory and type 2 cytokine responses, respectively. B cells from infected WT, TLR9−/−, and MyD88−/− mice produced appreciable levels of both IL-12 and IL-4. However, the levels of cytokines produced by B cells from MyD88−/− mice were lower than those produced WT and TLR9−/− mice (Fig. 4G). Although, detailed analysis of cytokine responses by B cells from WT and TLR knockout mice has not been done in the present study, the above results suggests that B cells produce substantial levels of cytokines in response to malaria infection, contributing to the serum cytokine profiles of parasite-infected mice.
Overall the results of the above analyses demonstrate that the cytokines produced by T and B cells from infected WT, TLR9−/−, and MyD88−/− mice contribute substantially to the serum cytokine profiles of the respective mice. The results further demonstrate that TLR9 and MyD88 distinctively regulate cytokine responses by T and B cells in response to malaria infection.
TLR9-independent/MyD88-dependent and/or IL-1R/IL-18R-mediated signaling also contribute to cytokine responses to malaria parasites
While the malaria parasite-induced function of DCs was dependent on both TLR9 and MyD88 (see Fig. 1, and also see Ref. 15), the serum cytokine responses to malaria infection were significantly independent of TLR9 but largely dependent on MyD88 (see Fig. 3). To gain insight into how TLR9 and MyD88 differentially regulate immune responses to malaria, we performed the following studies. The malaria parasite hemozoin has been reported to activate inflammasome-mediated signaling to induce the production of IL-1β (22, 23). To determine the contribution of inflammasome-mediated signaling, we measured the levels of IL-1β and IL-18 in the sera of P. yoelii-infected mice. Both IL-1β and IL-18 were produced throughout the course of infection (Fig. 5). The production of IL-1β decreased as the infection progressed, whereas that of IL-18 increased with increasing parasitemia. Further, although the production of both cytokines was dependent, to some extent, on TLR9 at early stages of infection, it was mostly independent of TLR9 at later stages of infection (Fig. 5). These results suggested that inflammasome also plays an important role in immune responses to malaria infection. Furthermore, since infected MyD88−/− but not TLR9−/− mice showed markedly low levels of IL-1β and IL-18, collectively, the above results suggested that TLR9-independent/MyD88-dependent- and/or IL-1R/IL-18R-mediated signaling is also involved in the inflammatory cytokine responses to malaria parasites. Since several cell types including monocytes, NK, T and B cells have IL-1/IL-18 receptor (50), which trigger signaling through MyD88 (51), it is likely that high levels of IL-18 produced during later stages of infection induce IL-12 and IL-4 production by these cells. This explains the increased production of IL-12 and IL-4 by infected TLR9−/− mice. Furthermore, since production of IL-1β and IL-18 by inflammasome-mediated signaling requires the pathogen recognition receptor-mediated expression of pro-IL-1β and pro-IL-18, it appears that TLRs other than TLR9 also contribute to certain extent in these cytokine responses. Since serum cytokine profiles of infected TLR2−/− and TLR4−/− mice were similar to those of the infected WT mice (see Fig. 3), despite parasites having TLR2 and TLR4 as well as TLR11 ligands (10–17), it appears that collective signaling strength of TLR2, TLR4 and TLR11 is responsible for the MyD88-dependent cytokine-inducing activity seen in TLR9 deficient mice.
FIGURE 5. Production of IL-1β and IL-18 during malaria infection is TLR9-independent but MyD88-dependent.
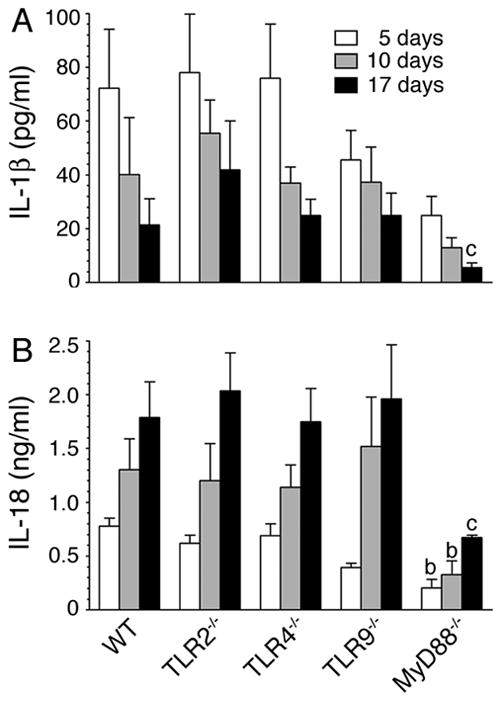
Sera from P. yoelii-infected mice at 5, 10, and 17 days postinfection were prepared as outlined in legends to Fig. 3. The levels of IL-1β (A) and IL-18 (B) in pooled sera were measured by ELISA. The letters, b and c, denote the statistical significance between the levels of IL-1β and IL-18 produced by the indicated mice and those produced by the corresponding infected WT mice. b, p <0.01; c, p <0.05.
TLR9 and MyD88 are essential for the development of cell-mediated immunity to malaria
As shown in Fig. 1, at the early stages of infections, pro-inflammatory cytokine responses to malaria parasites by DCs is essentially dependent on TLR9 and MyD88, whereas as the infection progresses TLR9 and MyD88 differentially regulate these cytokine responses in vivo. To determine whether TLR9 and MyD88 similarly or differentially regulate cell-mediated immunity to malaria parasites, we measured cytotoxic activity of NK and CD8+ T cells from infected WT, TLR9−/− and MyD88−/− mice at different stages of infection. It has been shown that GM-DCs efficiently internalize IRBCs by the scavenger receptor (CD36)-mediated and nonspecific phagocytosis and present antigens to T cells (52–54). Therefore, we used IRBC-pulsed GM-DCs as target cells for measuring T cell cytotoxic activity and the cytotoxic activity of NK cells from infected mice was measured using YAC-1 cells as a target. Both NK and CD8+ T cells from infected WT mice exhibited noticeable levels of cytotoxic activity to target cells even at 5 days postinfection and the activity was substantially increased at later stages of infection (Fig. 6). In contrast, NK and CD8+ T cells from infected MyD88−/− mice exhibited little or no cytotoxic activity at all stages of infection, and cells from infected TLR9−/− mice exhibited low levels of cytotoxic activity. To further confirm the cytotoxic activity of CD8+ T cells we also measured their ability to lyse anti-CD3ε-redirected EL-4 target cells. As in the case of IRBC-internalized DCs, EL-4 cells were efficiently lysed by WT CD8+ T cells but not by CD8+ T cells from MyD88−/− mice; cells from TLR9−/− mice showed low levels of cytolytic activity (data not shown). These results demonstrate that both TLR9 and MyD88 play important roles in NK and T cells acquiring cytotoxic effector activity. These results together with the data that pro-inflammatory cytokine responses by DCs at early stages of infection are critically dependent on both TLR9 and MyD88 (see Fig. 1) demonstrate that DCs contribute predominantly to the development of cytotoxic activity of NK and CD8+ T cells to malaria. Furthermore, the observed differences, albeit low, in the cytotoxic activity of TLR9 and MyD88 deficient cells suggest that TLR9-independent/MyD88-dependent- and/or IL-1R/IL-18R-mediated signaling also plays a role to certain extent in the development of cell-mediated immunity.
FIGURE 6. TLR9 and MyD88 are essential for the development of NK and T cell cytotoxic activity.
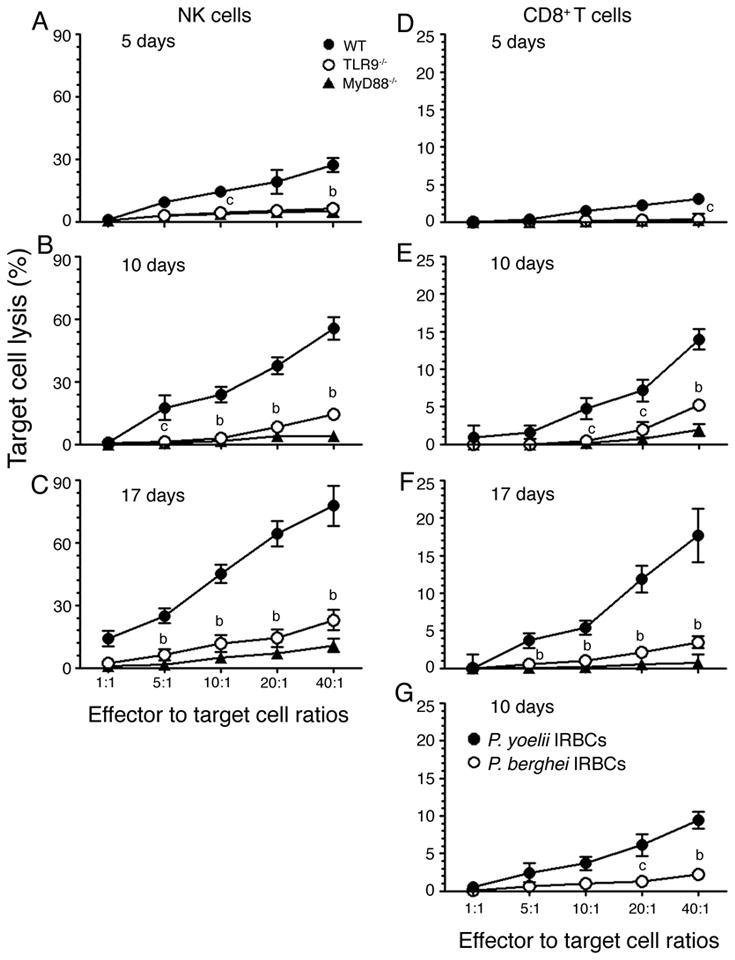
Liver NK cells (A, B and C) and spleen CD8+ T cells (D, E and F) from P. yoelii-infected WT, TLR9−/− and MyD88−/− mice at 5, 10, and 17 days postinfection were cocultured, respectively, with YAC-1 and P. yoelii IRBC-pulsed GM-DCs as target cells as described in the “Materials and Methods”. The percentages of target cell lysed were plotted. Data shown are a representative of two independent experiments. (G) Plots of percent cell lysis of GM-DCs pulsed with either P. yoelii (closed circle) or P. berghei (open circle) IRBCs by spleen CD8+ T cells from WT mice at 10 days postinfection. The letters, b and c, represent the statistical significance between the cytotoxic activity of NK and T cells from the infected TLR9−/− mice and that of NK and T cells from the infected WT mice. b, p <0.01; c, p <0.05.
To determine whether the observed T cell cytotoxic activity was antigen specific, we analyzed the ability of CD8+ T cells from P. yoelii-infected WT mice to lyse GM-DCs pulsed with P. berghei IRBCs; GM-DCs pulsed with P. yoelii IRBCs were used as a control. The cytotoxic activity against P. berghei antigens was about ~20% of that against P. yoelii antigens. These results agree with the fact that many proteins of P. yoelii and P. berghei are homologous (55), and strongly suggest that the P. yoelii-induced cytotoxic activity of CD8+ T cells is largely, if not completely, antigen specific.
TLR9 and MyD88 modulate antibody responses to malaria infection
To determine whether TLR9 and MyD88 also contribute to the humoral responses to malaria infection, we analyzed total antibody responses and IgG subclasses in sera of parasite-infected mice. As shown in Fig. 7, TLR9−/− and MyD88−/− mice produced similar levels of antibody titer and similar antibody subclass profiles. Further, interestingly, WT mice showed significantly lower levels of total IgG than TLR9−/− and MyD88−/− mice (Fig. 7A); this appears to be in response to the significantly lower levels of parasitemia in WT mice than in TLR9−/− and MyD88−/− mice. Immunoglobulin subclass analysis showed significantly higher levels of IgG1 in the sera of parasite-infected TLR9−/− and MyD88−/− mice than in the sera of WT mice (Fig. 7B). On the other hand, levels of IgG2a and IgG2b were significantly higher in infected WT mice than in TLR9−/− and MyD88−/− mice. The observed high levels of Th1 type antibodies in WT mice and higher level of Th2 type antibodies in TLR9−/− and MyD88−/− mice are consistent with robust Th1 responses driven by WT DCs and significantly higher type 2 cytokine responses by TLR9−/− and MyD88−/− DCs. These results are consistent with the previous report that in the case of P. chabaudi chabaudi AS infection, MyD88−/− mice produced higher levels of IgG1 than WT and TLR2−/− mice (18). Overall, the above results demonstrate that TLR9 and MyD88 significantly regulate humoral responses to malaria parasites that are likely driven by DCs at early stages of infection.
FIGURE 7. TLR9 and MyD88 play roles in antibody responses to malaria parasites.
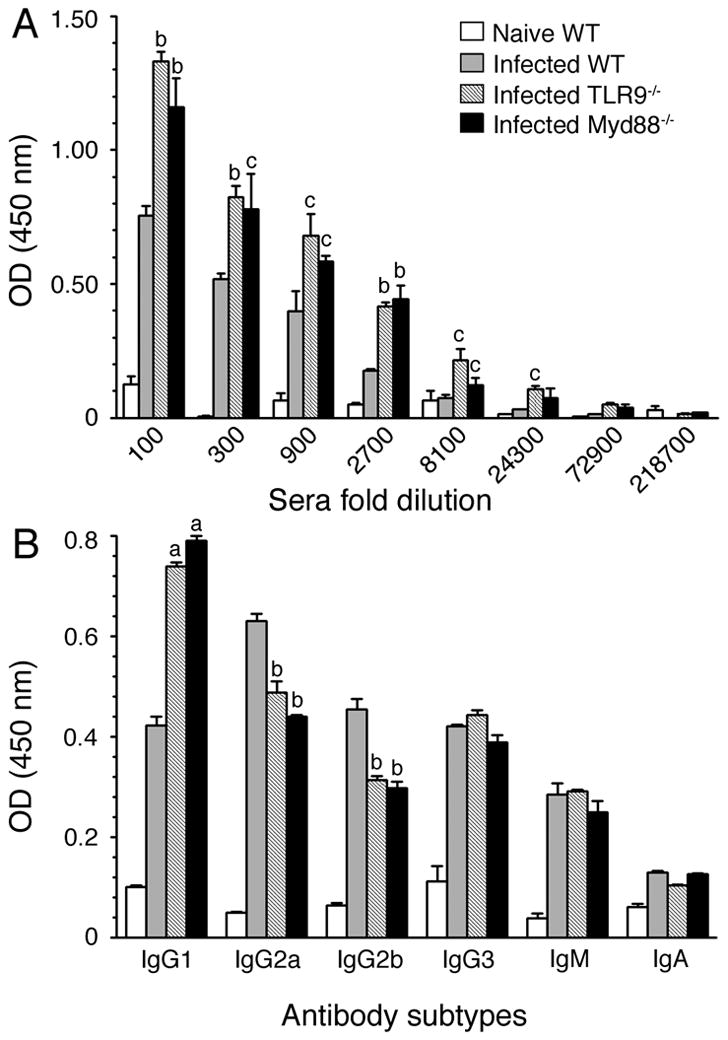
Total IgG levels (A) and antibody isotypes (B) in the pooled sera of P. yoelii-infected WT, TLR9−/− and MyD88−/− mice at 40 days postinfection were analyzed by ELISA. Sera were 1:3 serially diluted for total IgG analysis and antibody isotype analysis was performed using 1:8100 diluted sera. Serum from naïve WT mice was used as a control. The letters, a, b and c represent statistical significance between the levels of total IgG or antibody isotypes in sera of infected TLR9−/− or MyD88−/− mice and those in sera of infected WT mice. a, p <0.001; b, p <0.01; c, p <0.05. Data shown are a representative of two independent experiments.
TLR9 and MyD88 significantly contribute to malaria protective immunity
Finally, we assessed the contributions of TLRs and MyD88 to the development of overall protective immunity to malaria by measuring the progression of infection in WT, TLR2−/−, TLR4−/−, TLR9−/−, and MyD88−/− mice and by assessing the survival of infected animals. In all mouse types studied, parasitemia remained low during the first week of infection and thereafter parasites grew exponentially, reaching peak parasitemia between 18 and 20 days postinfection (Fig. 8A and 8B). After three weeks, parasitemia rapidly decreased in survived mice, and infection was completely cleared at the end of fourth week. During the exponential growth, parasitemia was 2-fold higher in MyD88−/− than in WT, TLR2−/− and TLR4−/− mice. TLR9−/− mice showed noticeably lower levels of parasitemia than MyD88−/− mice, but exhibited considerably higher parasitemia than WT, TLR2−/− and TLR4−/− mice. Compared to WT mice, both MyD88−/− and TLR9−/− mice were more susceptible to infection and 80–90% of mice eventually succumbed to death (Fig. 8C and 8D). These results are consistent with the higher cytotoxic activity and Th1 type antibody responses by WT mice than TLR9−/− and MyD88−/− mice. Thus, collectively, our results demonstrate that TLR9 and MyD88 regulate innate immune responses as well as cellular and humoral immunity to malaria infection, contributing to the development of protective immunity.
Discussion
The results presented here allow us to make two important conclusions that have broader implications in understanding the molecular mechanisms involved in the regulation of innate and adaptive immune responses to malaria. For one, TLR9 and MyD88 play central roles in the regulation of pro- and anti-inflammatory responses, Th1 and Th2 responses, and in the development of cytotoxic effector function and antibody responses to malaria infection. This conclusion is supported by the following findings: The deficiency in either TLR9 or MyD88 but not that in TLR2 or TLR4 leads to (i) markedly decreased production of pro-inflammatory cytokines with concomitant increase in the production of type 2/anti-inflammatory cytokines by DCs; (ii) increased commitment to Th2 development; (iii) impaired NK and T cell cytolytic activity; (iv) significantly lower levels of Th1 type antibody responses and increased Th2 type antibodies; (v) significantly higher levels of parasitemia and increased susceptible to malaria. Secondly, the inflammasome as well as TLR9-independent/MyD88-dependent- and/or IL-1R/IL-18R-mediated signaling also contribute substantially to the development of protective immunity to malaria. This conclusion is evident from our findings that infected TLR9−/− mice produced high levels of IL-1β and IL-18 than infected MyD88−/− mice. Further, NK and CD8+ T cells from infected TLR9−/− mice exhibit cytotoxic activity, albeit at marginal levels, whereas the corresponding cells from the infected MyD88−/− mice showed little or no cytotoxic activity (see Fig. 6). Thus, our study provides valuable insights into role of TLRs in the regulation of innate and adaptive immune responses to malaria infection.
Our data also clearly demonstrate that TLR9 and MyD88 play critical roles in the development of protective immunity to malaria infection. Studies have shown that protective immunity to malaria involves strong early pro-inflammatory cytokine responses and Th1 polarization leading to effective cell-mediated and humoral responses (2–4). On the other hand, higher anti-inflammatory and type 2 cytokine responses at the early stages of infection result in inability to control infection, leading to pathogenesis (2–4). In agreement with this notion, in the present study parasite-infected WT mice that were protected from infection but not susceptible TLR9−/− and MyD88−/− mice produced high levels of pro-inflammatory cytokines and low levels of type 2/anti-inflammatory cytokines, and were strongly Th1 polarized, elicited efficient NK and T cell cytotoxic activity and produced higher levels of protective Th1 type (56), opsonizing IgG2a and IgG2b, which are likely to aid effective parasite clearance. In contrast, the deficiency in TLR9 resulted in substantially lower levels of pro-inflammatory cytokines and higher levels of type 2/anti-inflammatory cytokines at early stages of infection, increased production of both pro-and anti-inflammatory responses at later stages of infection, and significantly impaired NK and CD8+ T cell cytotoxic activity. The MyD88−/− deficiency on the other hand caused low levels of both pro-inflammatory and anti-inflammatory cytokine responses at all stages of infection and completely impaired NK and CD8+ T cell cytotoxic activity. Furthermore, TLR9 or MyD88 deficiency significantly limited the production of Th1 type IgGs, which are known to be protective (56), while increasing the levels of Th2 type IgGs. The pro-inflammatory cytokines and strong Th1 polarization is known to increase macrophage phagocytic activity, proliferation of cytotoxic CD8+ T cells, and promote the production of opsonizing antibodies (57, 58). All of these can contribute to efficient parasite clearance, thereby providing protection against infection in WT mice. Effective clearance of IRBC-internalized macrophages, DCs and other phagocytic cells is also likely to be important for allowing the immune system to function efficiently against malaria infection; this function is severely compromised in TLR9−/− and MyD88−/− mice. Thus, our conclusion that TLR9 and MyD88 are crucial for the development of protective immunity to malaria is supported by the observed markedly lower ability of TLR9−/− and MyD88−/− mice to control parasitemia and substantially higher susceptibility to death compared to WT mice. Our conclusion also agrees with the results of recent studies that TLR9 provides protection against cerebral malaria in P. berghei ANKA-infected mice (59), MyD88 protects P. chabaudi chabaudi infected mice from malaria illness (18), and MyD88 is also involved in the control of early parasitemia in nonlethal P. yoelii infection (60).
The results of this study further demonstrate that TLR9-independent/MyD88-dependent and/or IL-1R/IL-18R-mediated signaling also significantly involved in the regulation of immune responses to malaria infection. This is evident from our observation that P. yoelii-infected MyD88−/− mice produced markedly low levels of both pro- and anti-inflammatory cytokines, whereas the infected TLR9−/− mice produced substantial levels of both types of cytokines (see Fig. 3). Further, in a preliminary study using FL-DCs, we noted that DCs deficient in TLR9 produced appreciable levels of TNF-α and IL-12 upon stimulation with P. yoelii-infected erythrocytes, whereas DCs deficient in MyD88 unable to produce these cytokines (N. M. Gowda, X. Wu and D. C. Gowda, unpublished results). Malarial GPIs, microparticles from IRBCs, and heme released from the parasite-infected erythrocytes have been shown to activate macrophages through TLR2 or TLR4 (10–12). However, the effects of TLR2 or TLR4 were not evident either from the serum cytokine profiles of the infected mice or from the cytokine pattern produced by DCs ex vivo (see Figs. 1–3). Compared to other protozoan parasites such as Trypanosoma and Leishmania, malaria parasites express relatively low levels of GPIs (61, 62), and also it is possible that levels of microparticles released by parasite-infected erythrocytes are low. Furthermore, although malaria parasite’s profilin has been reported to activate DCs via TLR11 to produce IL-12 (16, 17), its activity has been reported to be low (63). Therefore, it appears that individually these receptors are unable to activate cells to a significant extent to induce strong immune responses, but collective signaling by TLR2, TLR4 and TLR11 significantly activate the innate immune system in TLR9−/− mice in vivo to induce cytokine responses by DCs and other cells. Moreover, the production of IL-1β and IL-18 at high levels by P. yoelii-infected TLR9−/− mice but not by MyD88−/− mice (see Fig. 5), suggests that MyD88-dependent, inflammasome- and IL-1R/IL-18R-mediated signaling also contributes substantially to the activation of cells of the immune system, especially during later stages of infection when IL-18 is produced at higher levels. Therefore, cytokines produced in response to MyD88-mediated signaling, involving TLR2, TLR4 and TLR11 and/or IL-1R/IL-18R, collectively account for the observed high levels of serum cytokines in P. yoelii-infected TLR9−/− mice. This explains why parasite infected MyD88−/− mice produce only marginal levels of cytokines.
The results presented here additionally demonstrate that IL-18, which has been shown to play an important role in malaria infection (60, 64–66), is produced in a TLR9-independent and MyD88-dependent manner. IL-18 is known to activate NK and T cells through its receptor-mediated signaling to produce IFN-γ, contributing to Th1 cell development (50, 67). Additionally, IL-18 enhances NK cell cytotoxicity, Fas ligand-mediated Th1 cell cytotoxicity, and proliferation of activated T cells (67, 68). In the parasite-infected mice, parallel to IL-18 induction, IL-12 is produced at increased levels as infection progresses (see Figs. 3C and 5B). IL-12 and IL-18 have shared functions and thus, they synergistically and independently activate NK, T, and B cells through their receptors to produce cytokines (50, 69, 70). IL-18 is also known to up-regulate IL-12 receptor and IL-12 up-regulates IL-18 receptor (70). Thus, during malaria infection, IL-18 appears to collaborate with IL-12 in the development of Th1 cells and enhancement of NK and T cell cytotoxic activity by promoting cell proliferation. This is apparent from the observed high levels of pro-inflammatory cytokines in mice at later stages of infection and increased cytotoxic activity of NK and T cells as infection progresses. Previous studies have shown that IL-18 and IL-12 are protective to P. yoelii and P. chabaudi malaria (71–73). IL-18 has also been shown to be protective against severe malaria in human (64–66). Based on these observations, it is expected that TLR9−/− mice should be protected from malaria infection as they produce high levels of IL-18 and IL-12. However, IL-18 is also known to induce anti-inflammatory cytokines and Th2 responses depending on the cytokine milieu of the system (74). Therefore, unusually high levels of IL-4 seen in the infected TLR9−/− mice in parallel with increased production of IL-18 is most likely due to the IL-18-mediated Th2 responses. Since, the deficiency in TLR9 caused Th2-biased responses, IL-18 can efficiently augment Th2 responses in TLR9−/− mice. Therefore, it appears that IL-18 by efficiently driving both Th1 and Th2 responses neutralizes the protective effect of Th1 responses by Th2 responses in TLR9−/− mice. Thus, this explains why TLR9−/− mice, similar to MyD88−/− mice, have impaired cell-mediated and protective antibody responses and thus are more susceptible to malaria infection than WT mice.
Overall results of the present study demonstrate that initial robust pro-inflammatory cytokines are critical for the development of protective immunity to malaria. DCs are the major cells of the innate immune system, that play crucial role in the initiation of innate immune responses and development of adaptive immunity by connecting the innate immune arm to that of adaptive immune system. This conclusion is evident from our observation that, DCs from the infected WT mice produced substantially high levels of pro-inflammatory cytokines at early stages of infection that correlated with high levels of NK and CD8+ T cells cytotoxicity and protective Th1 antibodies production. In contrast, DCs from TLR9−/− and MyD88−/− mice produced low levels of pro-inflammatory cytokines and that resulted in complete absence or marginal levels of cytotoxic activity and decreased Th1 antibody production.
Finally, our observations have broader implication in understanding the roles of TLRs in the regulation of innate and adaptive immunity in other pathogenic infections as well. Although TLRs have been recognized to be crucial for producing pro-inflammatory responses during infections by diverse group of pathogens such as bacteria, fungi and parasites, the roles of these receptors in the regulation of immune responses remain poorly understood. The results presented here implicate that TLRs play central roles in the regulation of innate and adaptive immunity to various pathogenic microorganisms. Detailed understanding of the TLR-dependent immune regulations is likely to provide strategies for the development of therapeutics or vaccines against pathogenic infections.
Supplementary Material
Acknowledgments
This work was supported by grants AI 41139 from NIAID, from the National Institutes of Health and a grant from The Pennsylvania Department of Health.
We thank Drs. Shizuo Akira and Tatoshi Uematsu, Research Institute for Microbial Diseases, Osaka University, Osaka, Japan, for providing TLR and MyD88 knockout mice; Dr. Glenn Dranoff, Dana-Farber Cancer Institute, Harvard University Medical School for giving FLT3-expressing B16 cells; Drs. Christopher Norbury and Todd Schell, Department of Microbiology and Immunology, Hershey Medical Center, Hershey, respectively for giving OT-II transgenic mice and GM-CSF producing X63 melanoma cells, and EL-4 cells; Dr. Nikki Keasey, Department of Hematology/Oncology, Hershey Medical Center, Hershey, PA, for providing YAC-1 cell line.
References
- 1.Snow RW, Guerra CA, Noor AM, Myint HY, Hay SI. The global distribution of clinical episodes of Plasmodium falciparum malaria. Nature. 2005;434:214–217. doi: 10.1038/nature03342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stevenson MM, Riley EM. Innate Immunity to malaria. Nat Rev Immunol. 2004;4:169–180. doi: 10.1038/nri1311. [DOI] [PubMed] [Google Scholar]
- 3.Schofield L, Grau GE. Immunological processes in malaria pathogenesis. Nat Rev Immunol. 2005;5:722–735. doi: 10.1038/nri1686. [DOI] [PubMed] [Google Scholar]
- 4.Riley EM, Wahl S, Perkins DJ, Schofield L. Regulating immunity to malaria. Parasite Immunol. 2006;28:35–49. doi: 10.1111/j.1365-3024.2006.00775.x. [DOI] [PubMed] [Google Scholar]
- 5.Hunt NH, Grau GE. Cytokines: accelerators and brakes in the pathogenesis of cerebral malaria. Trends Immunol. 2003;24:491–499. doi: 10.1016/s1471-4906(03)00229-1. [DOI] [PubMed] [Google Scholar]
- 6.Trinchieri G, Sher A. Cooperation of Toll-like receptor signals in innate immune defense. Nat Rev Immunol. 2007;7:179–190. doi: 10.1038/nri2038. [DOI] [PubMed] [Google Scholar]
- 7.Akira S. Innate immunity to pathogens: diversity in receptors for microbial recognition. Immunol Rev. 2009;227:5–8. doi: 10.1111/j.1600-065X.2008.00739.x. [DOI] [PubMed] [Google Scholar]
- 8.Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637–650. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 9.Blasius AL, Beutler B. Intracellular toll-like receptors. Immunity. 2010;32:305–315. doi: 10.1016/j.immuni.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 10.Krishnegowda G, Hajjar AM, Zhu J, Douglass EJ, Uematsu S, Akira S, Woods AS, Gowda DC. Induction of proinflammatory responses in macrophages by the glycosylphosphatidylinositols of Plasmodium falciparum: cell signaling receptors, glycosylphosphatidylinositol (GPI) structural requirement, and regulation of GPI activity. J Biol Chem. 2005;280:8606–8616. doi: 10.1074/jbc.M413541200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Figueiredo RT, Fernandez PL, Mourao-Sa DS, Porto BN, Dutra FF, Alves LS, Oliveira MF, Oliveira PL, Graca-Souza AV, Bozza MT. Characterization of heme as activator of Toll-like receptor 4. J Biol Chem. 2007;282:20221–20229. doi: 10.1074/jbc.M610737200. [DOI] [PubMed] [Google Scholar]
- 12.Couper KN, Barnes T, Hafalla JC, Combes V, Ryffel B, Secher T, Grau GE, Riley EM, de Souza JB. Parasite-derived plasma microparticles contribute significantly to malaria infection-induced inflammation through potent macrophage stimulation. PLoS Pathog. 2010;6 doi: 10.1371/journal.ppat.1000744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pichyangkul S, Yongvanitchit K, Kum-arb U, Hemmi H, Akira S, Krieg AM, Heppner DG, Stewart VA, Hasegawa H, Looareesuwan S, Shanks GD, Miller RS. Malaria blood stage parasites activate human plasmacytoid dendritic cells and murine dendritic cells through a Toll-like receptor 9-dependent pathway. J Immunol. 2004;172:4926–4933. doi: 10.4049/jimmunol.172.8.4926. [DOI] [PubMed] [Google Scholar]
- 14.Parroche P, Lauw FN, Goutagny N, Latz E, Monks BG, Visintin A, Halmen KA, Lamphier M, Olivier M, Bartholomeu DC, Gazzinelli RT, Golenbock DT. Malaria hemozoin is immunologically inert but radically enhances innate responses by presenting malaria DNA to toll-like receptor 9. Proc Natl Acad Sci USA. 2007;104:1919–1924. doi: 10.1073/pnas.0608745104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu X, Gowda NM, Kumar S, Gowda DC. Protein-DNA complex is the exclusive malaria parasite component that activates dendritic cells and triggers innate immune responses. J Immunol. 2010;184:4338–4348. doi: 10.4049/jimmunol.0903824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yarovinsky F, Zhang D, Andersen JF, Bannenberg GL, Serhan CN, Hayden MS, Hieny S, Sutterwala FS, Flavell RA, Ghosh S, Sher A. TLR11 activation of dendritic cells by a protozoan profilin-like protein. Science. 2005;308:1626–1629. doi: 10.1126/science.1109893. [DOI] [PubMed] [Google Scholar]
- 17.Kursula I, Kursula P, Ganter M, Panjikar S, Matuschewski K, Schüler H. Structural basis for parasite-specific functions of the divergent profilin of Plasmodium falciparum. Structure. 2008;16:1638–1648. doi: 10.1016/j.str.2008.09.008. [DOI] [PubMed] [Google Scholar]
- 18.Franklin BS, Rodrigues SO, Antonelli LR, Oliveira RV, Goncalves AM, Sales-Junior PA, Valente EP, Alvarez-Leite JI, Ropert C, Golenbock DT, Gazzinelli RT. MyD88-dependent activation of dendritic cells and CD4(+) T lymphocytes mediates symptoms, but is not required for the immunological control of parasites during rodent malaria. Microbes Infect. 2007;9:881–890. doi: 10.1016/j.micinf.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 19.Coban C, Ishii KJ, Uematsu S, Arisue N, Sato S, Yamamoto M, Kawai T, Takeuchi O, Hisaeda H, Horii T, Akira S. Pathological role of Toll-like receptor signaling in cerebral malaria. Int Immunol. 2007;19:67–79. doi: 10.1093/intimm/dxl123. [DOI] [PubMed] [Google Scholar]
- 20.Seixas E, Moura Nunes JF, Matos I, Coutinho A. The interaction between DC and Plasmodium berghei/chabaudi-infected erythrocytes in mice involves direct cell-to-cell contact, internalization and TLR. Eur J Immunol. 2009;39:1850–1863. doi: 10.1002/eji.200838403. [DOI] [PubMed] [Google Scholar]
- 21.Fritz JH, Ferrero RL, Philpott DJ, Girardin SE. Nod-like proteins in immunity, inflammation and disease. Nat Immunol. 2006;7:1250–1257. doi: 10.1038/ni1412. [DOI] [PubMed] [Google Scholar]
- 22.Griffith JW, Sun T, McIntosh MT, Bucala R. Pure hemozoin is inflammatory in vivo and activates the NALP3 inflammasome via release of uric acid. J Immunol. 2009;183:5208–5220. doi: 10.4049/jimmunol.0713552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shio MT, Eisenbarth SC, Savaria M, Vinet AF, Bellemare MJ, Harder KW, Sutterwala FS, Bohle DS, Descoteaux A, Flavell RA, Olivier M. Malarial hemozoin activates the NLRP3 inflammasome through Lyn and Syk kinases. PLoS Pathog. 2009;5:e1000559. doi: 10.1371/journal.ppat.1000559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wilson NS, Villadangos JA. Regulation of antigen presentation and cross-presentation in the dendritic cell network: facts, hypothesis, and immunological implications. Adv Immunol. 2005;86:241–305. doi: 10.1016/S0065-2776(04)86007-3. [DOI] [PubMed] [Google Scholar]
- 25.Steinman RM, Hemmi H. Dendritic cells: translating innate to adaptive immunity. Curr Top Microbiol Immunol. 2006;311:17–58. doi: 10.1007/3-540-32636-7_2. [DOI] [PubMed] [Google Scholar]
- 26.Lopez-Bravo M, Ardavin C. In vivo induction of immune responses to pathogens by conventional dendritic cells. Immunity. 2008;29:343–351. doi: 10.1016/j.immuni.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 27.Perry JA, Olver CS, Burnett RC, Avery AC. Cutting edge: the acquisition of TLR tolerance during malaria infection impacts T cell activation. J Immunol. 2005;174:5921–5925. doi: 10.4049/jimmunol.174.10.5921. [DOI] [PubMed] [Google Scholar]
- 28.Villadangos JA, Schnorrer P. Intrinsic and cooperative antigen-presenting functions of dendritic-cell subsets in vivo. Nat Rev Immunol. 2007;7:543–555. doi: 10.1038/nri2103. [DOI] [PubMed] [Google Scholar]
- 29.Pulendran B, Tang H, Manicassamy S. Programming dendritic cells to induce T(H)2 and tolerogenic responses. Nat Immunol. 2010;11:647–655. doi: 10.1038/ni.1894. [DOI] [PubMed] [Google Scholar]
- 30.Babu S, Blauvelt CP, Kumaraswami V, Nutman TB. Cutting edge: diminished T cell TLR expression and function modulates the immune response in human filarial infection. J Immunol. 2006;176:3885–3889. doi: 10.4049/jimmunol.176.7.3885. [DOI] [PubMed] [Google Scholar]
- 31.Dasari P, I, Nicholson C, Hodge G, Dandie GW, Zola H. Expression of toll-like receptors on B lymphocytes. Cell Immunol. 2005;236:140–145. doi: 10.1016/j.cellimm.2005.08.020. [DOI] [PubMed] [Google Scholar]
- 32.Hou B, Saudan P, Ott G, Wheeler ML, Ji M, Kuzmich L, Lee LM, Coffman RL, Bachmann MF, DeFranco AL. Selective utilization of Toll-like Receptor and MyD88 signaling in B cells for enhancement of the antiviral germinal center response. Immunity. 2011;34:375–384. doi: 10.1016/j.immuni.2011.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.LaRosa DF, Stumhofer JS, Gelman AE, Rahman AH, Taylor DK, Hunter CA, Turka LA. T cell expression of MyD88 is required for resistance to Toxoplasma gondii. Proc Natl Acad Sci USA. 2008;105:3855–3860. doi: 10.1073/pnas.0706663105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Quigley M, Martinez J, Huang X, Yang Y. A critical role for direct TLR2-MyD88 signaling in CD8 T-cell clonal expansion and memory formation following vaccinia viral infection. Blood. 2009;113:2256–2264. doi: 10.1182/blood-2008-03-148809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rahman AH, Zhang R, Blosser CD, Hou B, Defranco AL, Maltzman JS, Wherry EJ, Turka LA. Antiviral memory CD8 T-cell differentiation, maintenance, and secondary expansion occur independently of MyD88. Blood. 2011;117:3123–3130. doi: 10.1182/blood-2010-11-318485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou S, Kurt-Jones EA, Cerny AM, Chan M, Bronson RT, Finberg RW. MyD88 intrinsically regulates CD4 T-cell responses. J Virol. 2009;83:1625–1634. doi: 10.1128/JVI.01770-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barr TA, Brown S, Mastroeni P, Gray D. B cell intrinsic MyD88 signals drive IFN-γ production from T cells and control switching to IgG2c. J Immunol. 2009;183:1005–1012. doi: 10.4049/jimmunol.0803706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kang SM, Yoo DG, Kim MC, Song JM, Park MK, Quan EOFS, Akira S, Compans RW. MyD88 plays an essential role in inducing B cells capable of differentiating into antibody-secreting cells after vaccination. J Virol. 2011;85:11391–11400. doi: 10.1128/JVI.00080-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gowda NM, Wu X, Gowda DC. The nucleosome (histone-DNA complex) is the TLR9-specific immunostimulatory component of Plasmodium falciparum that activates DCs. PLoS One. 2011;6:e20398. doi: 10.1371/journal.pone.0020398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brasel K, De Smedt T, Smith JL, Maliszewski CR. Generation of murine dendritic cells from flt3-ligand-supplemented bone marrow cultures. Blood. 2000;96:3029–3039. [PubMed] [Google Scholar]
- 41.Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, Muramatsu S, Steinman RM. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. 1992;176:1693–1702. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baran K, Ciccone A, Peters C, Yagita H, Bird PI, Villadangos JA, Trapani JA. Cytotoxic T Lymphocytes from cathepsin B-deficient mice survive normally in vitro and in vivo after encountering and killing target cells. J Biol Chem. 2006;281:30485–30491. doi: 10.1074/jbc.M602007200. [DOI] [PubMed] [Google Scholar]
- 43.Piontek GE, Taniguchi K, Ljunggren HG, Grönberg A, Kiessling R, Klein G, Kärre K. YAC-1 MHC class I variants reveal an association between decreased NK sensitivity and increased H-2 expression after interferon treatment or in vivo passage. J Immunol. 1985;135:4281–4288. [PubMed] [Google Scholar]
- 44.Sinniah R, Rui-Mei L, Kara AU. Up-regulation of cytokines in glomerulonephritis associated with murine malaria infection. Int J Exp Pathol. 1999;80:87–95. doi: 10.1046/j.1365-2613.1999.00101.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Watford WT, Moriguchi M, Morinobu A, O’Shea JJ. The biology of IL-12: coordinating innate and adaptive immune responses. Cytokine Growth Factor Rev. 2003;14:361–368. doi: 10.1016/s1359-6101(03)00043-1. [DOI] [PubMed] [Google Scholar]
- 46.Cederholm A, Frostegård J. Annexin A5 as a novel player in prevention of atherothrombosis in SLE and in the general population. Ann N Y Acad Sci. 2007;1108:96–103. doi: 10.1196/annals.1422.011. [DOI] [PubMed] [Google Scholar]
- 47.Schlaepfer DD, Jones J, Haigler HT. Inhibition of protein kinase C by annexin V. Biochemistry. 1992;31:1886–1891. doi: 10.1021/bi00121a043. [DOI] [PubMed] [Google Scholar]
- 48.Cella M, Scheidegger D, Palmer-Lehmann K, Lane P, Lanzavecchia A, Alber G. Ligation of CD40 on dendritic cells triggers production of high levels of interleukin-12 and enhances T cell stimulatory capacity: T-T help via APC activation. J Exp Med. 1996;184:747–752. doi: 10.1084/jem.184.2.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Snijders A, Kalinski P, Hilkens CM, Kapsenberg ML. High-level IL-12 production by human dendritic cells requires two signals. Int Immunol. 1998;10:1593–1598. doi: 10.1093/intimm/10.11.1593. [DOI] [PubMed] [Google Scholar]
- 50.Caliguiri G, Kaveri S, Nicoletti A. When interleukin-18 conducts, the Preludio sounds the same no matter who plays. Arterioscler Thromb Vasc Biol. 2005;25:655–657. doi: 10.1161/01.ATV.0000154921.49792.ef. [DOI] [PubMed] [Google Scholar]
- 51.O’Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signaling. Nat Rev Immunol. 2007;7:353–364. doi: 10.1038/nri2079. [DOI] [PubMed] [Google Scholar]
- 52.Urban BC, Ferguson DJ, Pain A, Willcox N, Plebanski M, Austyn JM, Roberts DJ. Plasmodium falciparum-infected erythrocytes modulates the maturation of dendritic cells. Nature. 1999;400:73–77. doi: 10.1038/21900. [DOI] [PubMed] [Google Scholar]
- 53.Urban BC, Willcox N, Roberts DJ. A role for CD36 in the regulation of dendritic cell function. Proc Natl Acad Sci USA. 2001;98:8750–8755. doi: 10.1073/pnas.151028698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Elliott SR, Spurck TP, Dodin JM, Maier AG, Voss TS, Yosaatmadja F, Payne PD, McFadden GI, Cowman AF, Rogerson SJ, Schofield L, Brown GV. Inhibition of dendritic cell maturation by malaria is dose dependent and does not require Plasmodium falciparum erythrocyte membrane protein 1. Infect Immun. 2007;75:3621–3632. doi: 10.1128/IAI.00095-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang T, Fujioka H, Drazba JA, Sam-Yellowe TY. Rhop-3 protein conservation among Plasmodium species and induced protection against lethal P. yoelii and P. berghei challenge. Parasitol Res. 2006;99:238–252. doi: 10.1007/s00436-006-0136-9. [DOI] [PubMed] [Google Scholar]
- 56.Tangteerawatana P, Krudsood S, Chalermrut K, Looareesuwan S, Khusmith S. Natural human IgG subclass antibodies to Plasmodium falciparum blood stage antigens and their relation to malaria resistance in an endemic area of Thailand. Southeast Asian J Trop Med Public Health. 2001;32:247–254. [PubMed] [Google Scholar]
- 57.Mosser DM. The many faces of macrophage activation. J Leukoc Biol. 2003;73:209–212. doi: 10.1189/jlb.0602325. [DOI] [PubMed] [Google Scholar]
- 58.Lefeber DJ, Benaissa-Trouw B, Vliegenthart JF, Kamerling JP, Jansen WT, Kraaijeveld K, Snippe H. Th1-directing adjuvants increase the immunogenicity of oligosaccharide-protein conjugate vaccines related to Streptococcus pneumoniae type 3. Infect Immun. 2003;71:6915–6920. doi: 10.1128/IAI.71.12.6915-6920.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Franklin BS, Ishizaka ST, Lamphier M, Gusovsky F, Hansen H, Rose J, Zheng W, Ataíde MA, de Oliveira RB, Golenbock DT, Gazzinelli RT. Therapeutical targeting of nucleic acid-sensing Toll-like receptors prevents experimental cerebral malaria. Proc Natl Acad Sci USA. 2011;108:3689–3694. doi: 10.1073/pnas.1015406108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cramer JP, Lepenies B, Kamena F, Hölscher C, Freudenberg MA, Burchard GD, Wagner H, Kirschning CJ, Liu X, Seeberger PH, Jacobs T. MyD88/IL-18-dependent pathways rather than TLRs control early parasitemia in non-lethal Plasmodium yoelii infection. Microbes Infect. 2008;10:1259–1265. doi: 10.1016/j.micinf.2008.07.024. [DOI] [PubMed] [Google Scholar]
- 61.Ferguson MA. The structure, biosynthesis and functions of glycosylphosphatidylinositol anchors, and the contributions of trypanosome research. J Cell Sci. 1999;112:2799–2809. doi: 10.1242/jcs.112.17.2799. [DOI] [PubMed] [Google Scholar]
- 62.Naik RS, Branch OH, Woods AS, Vijaykumar M, Perkins DJ, Nahlen BL, Lal AA, Cotter RJ, Costello CE, Ockenhouse CF, Davidson EA, Gowda DC. Glycosylphosphatidylinositol anchors of Plasmodium falciparum: molecular characterization and naturally elicited antibody response that may provide immunity to malaria pathogenesis. J Exp Med. 2000;192:1563–1576. doi: 10.1084/jem.192.11.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Plattner F, Yarovinsky F, Romero S, Didry D, Carlier MF, Sher A, Soldati-Favre D. Toxoplasma profilin is essential for host cell invasion and TLR11-dependent induction of an interleukin-12 response. Cell Host Microbe. 2008;3:77–87. doi: 10.1016/j.chom.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 64.Chaisavaneeyakorn S, Othoro C, Shi YP, Otieno J, Chaiyaroj SC, Lal AA, Udhayakumar V. Relationship between plasma Interleukin-12 (IL-12) and IL-18 levels and severe malarial anemia in an area of holoendemicity in western Kenya. Clin Diagn Lab Immunol. 2003;10:362–366. doi: 10.1128/CDLI.10.3.362-366.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kojima S, Nagamine Y, Hayano M, Looareesuwan S, Nakanishi K. A potential role of interleukin 18 in severe falciparum malaria. Acta Trop. 2004;89:279–284. doi: 10.1016/j.actatropica.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 66.Torre D. Early production of gamma-interferon in clinical malaria: role of interleukin-18 and interleukin-12. Clin Infect Dis. 2009;48:1481–1482. doi: 10.1086/598508. [DOI] [PubMed] [Google Scholar]
- 67.Takeda K, Tsutsui H, Yoshimoto T, Adachi O, Yoshida N, Kishimoto T, Okamura H, Nakanishi K, Akira S. Defective NK cell activity and Th1 response in IL-18-deficient mice. Immunity. 1998;8:383–390. doi: 10.1016/s1074-7613(00)80543-9. [DOI] [PubMed] [Google Scholar]
- 68.Heidemann SC, Chavez V, Landers CJ, Kucharzik T, Prehn JL, Targan SR. TL1A selectively enhances IL-12/IL-18-induced NK cell cytotoxicity against NK-resistant tumor targets. J Clin Immunol. 2010;30:531–538. doi: 10.1007/s10875-010-9382-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xu D, Chan WL, Leung BP, Hunter D, Schulz K, Carter RW, McInnes IB, Robinson JH, Liew FY. Selective expression and functions of interleukin 18 receptor on T helper (Th) type 1 but not Th2 cells. J Exp Med. 1998;188:1485–1492. doi: 10.1084/jem.188.8.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yoshimoto T, Takeda K, Tanaka T, Ohkusu K, Kashiwamura S, Okamura H, Akira S, Nakanishi K. IL-12 up-regulates IL-18 receptor expression on T cells, Th1 cells, and B cells: synergism with IL-18 for IFN-γ production. J Immunol. 1998;161:3400–3407. [PubMed] [Google Scholar]
- 71.Hoffman SL, Crutcher JM, Puri SK, Ansari AA, Villinger F, Franke ED, Singh PP, Finkelman F, Gately MK, Dutta GP, Sedegah M. Sterile protection of monkeys against malaria after administration of interleukin-12. Nat Med. 1997;3:80–83. doi: 10.1038/nm0197-80. [DOI] [PubMed] [Google Scholar]
- 72.Su Z, Stevenson MM. IL-12 is required for antibody-mediated protective immunity against blood-stage Plasmodium chabaudi AS malaria infection in mice. J Immunol. 2002;168:1348–1355. doi: 10.4049/jimmunol.168.3.1348. [DOI] [PubMed] [Google Scholar]
- 73.Singh RP, Kashiwamura S, Rao P, Okamura H, Mukherjee A, Chauhan VS. The role of IL-18 in blood-stage immunity against murine malaria Plasmodium yoelii 265 and Plasmodium berghei ANKA. J Immunol. 2002;168:4674–4681. doi: 10.4049/jimmunol.168.9.4674. [DOI] [PubMed] [Google Scholar]
- 74.Nakanishi K, Yoshimoto T, Tsutsui H, Okamura H. Interleukin-18 is a unique cytokine that stimulates both Th1 and Th2 responses depending on its cytokine milieu. Cytokine Growth Factor Rev. 2001;12:53–72. doi: 10.1016/s1359-6101(00)00015-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.