

LOOKING BACK
Virchow (1821 – 1902), the German pathologist and one of the 19th century's foremost leaders in medicine and pathology, observed that the artery walls of patients dying of occlusive vascular disease, such as myocardial infarction, were often thickened and irregular, and contained a yellowish fatty substance. This pathological condition was termed atheroma, the Greek word for porridge, and subsequently identified as cholesterol. Anitschkow and Chalatow in 1913 showed that feeding cholesterol to rabbits rapidly produces atheromatous disease similar to that found in man.
However, at that time, physicians were not convinced of any causal link between cholesterol and coronary heart disease (CHD) because most patients with the disease have plasma cholesterol levels not much different from that of the general population average.[1]
The Framingham study, led by Dawber, was initiated in the 1950s to prospectively study the relationship between blood cholesterol and other potential risk factors and death from coronary disease. This work established an increasingly strong correlation between high plasma cholesterol and CHD mortality,[2] which was confirmed by many other population-based studies. Moreover, the Seven Countries Study, which was initiated in the 1950s, showed that northern European countries and the United States had both high plasma cholesterol and high CHD mortality rates. By contrast, plasma cholesterol and CHD mortality were both substantially lower in southern Europe, and even more so in Japan.[3] Later investigations established that the association with CHD mortality was attributable mainly to low-density lipoprotein (LDL) cholesterol. Subsequent studies showed that LDL cholesterol comprises about 70% of total cholesterol and that high-density lipoprotein (HDL) cholesterol is inversely correlated with CHD mortality.
The above findings lead to the Lipid Hypothesis, which proposed that elevated total, or more accurately LDL, cholesterol was causally related to coronary disease and that reducing it would reduce the risk of myocardial infarction and other coronary events. This hypothesis remained controversial for many years because of the lack of clear evidence that lowering cholesterol provided any clinical benefits.
Before the discovery of statins (HMG-CoA reductase inhibitors), a number of dietary intervention studies and a few drug studies had reported a reduction in CHD events in patients with and without CHD. No individual study was compelling enough but taken together, there seemed to be a trend that lowering cholesterol reduced the risk of coronary events.[4] On the basis of those studies and the National Institutes of Health (NIH) Coronary Primary Prevention Trial (USA), an NIH Consensus Conference convened in 1984 concluded that lowering elevated LDL cholesterol with diet and drugs would reduce the risk of CHD.[5] The NIH (USA) accepted the findings of the Consensus Conference and the following year initiated a massive program to educate physicians and the public about the importance of treating hypercholesterolemia.
CHOLESTEROL SYNTHESIS
“Cholesterol is the most highly decorated small molecule in biology. Thirteen Nobel Prizes have been awarded to scientists who devoted major parts of their careers to cholesterol. Ever since it was isolated from gallstones in 1784, cholesterol has exerted an almost hypnotic fascination for scientists from the most diverse areas of science and medicine…. Cholesterol is a Janus-faced molecule. The very property that makes it useful in cell membranes, namely its absolute insolubility in water, also makes it lethal”.
Michael Brown and Joseph Goldstein Nobel Lectures (1985)
Most mammalian cells can produce cholesterol. Cholesterol biosynthesis is a complex process involving more than 30 enzymes, and the details of the biosynthetic pathway were worked out in many institutions in the 1950s and 1960s. In the hope that lowering cholesterol would reduce the risk of CHD, the simplified pathway in [Figure 1] was a natural target in the search for drugs to reduce plasma cholesterol concentrations. However, early attempts to reduce cholesterol biosynthesis were disastrous. Triparanol, which inhibits a late step in the pathway, was introduced into clinical use in the mid-1960s, but was withdrawn from the market shortly after because of the development of cataracts and various cutaneous adverse effects.[6] These side effects were attributable to tissue accumulation of desmosterol, the substrate for the inhibited enzyme.
Figure 1.
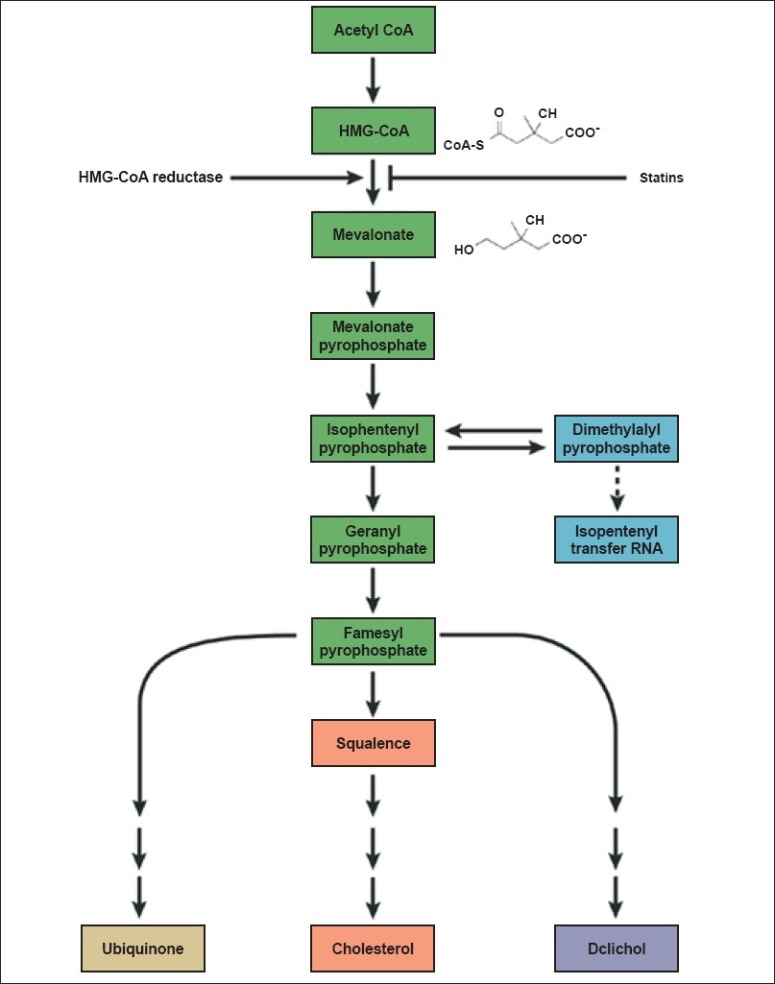
The cholesterol biosynthesis pathway. Cholesterol biosynthesis is a complex process involving more than 30 enzymes. A simplified version is shown here,which highlights the step inhibited by statins, and shown the chemical structures of the starting material (HMG-CoA)and product (mevaonate) of this step.
STATIN BREAKTHROUGHS
HMG-CoA reductase (3-hydroxy-3-methyl-glutaryl-CoA) is the rate-limiting enzyme in the cholesterol biosynthetic pathway and was an attractive target in the search for drugs to reduce plasma cholesterol concentrations.
In the 1970s, the Japanese microbiologist Akira Endo, during a search for antimicrobial agents, first discovered natural products with a powerful inhibitory effect on HMG-CoA reductase, including ML236B (compactin) in a fermentation broth of Penicillium citrinum.[4,5] Although no HMG-CoA reductase inhibitor has been shown to have useful antimicrobial activity, the possibility that an agent that inhibited the rate-limiting step in the cholesterol biosynthesis pathway could have useful lipid-lowering properties was quickly appreciated by Endo and others.
In the rabbit, monkey, and dog, compactin was shown to lower plasma cholesterol.[6] The prototype compound compactin was developed by Sankyo, and was shown to be highly effective in reducing concentrations of total and LDL cholesterol in the plasma of patients with heterozygous familial hypercholesterolaemia.[7,8] In 1978, Alberts, Chen and others at Merck Research Laboratories found a potent inhibitor of HMG-CoA reductase in a fermentation broth of Aspergillus terreus.[9] They named their discovery mevinolin and later named officially as lovastatin.
LOVASTATIN – THE FIRST STATIN
Merck began clinical trials of lovastatin in healthy volunteers in 1980. Lovastatin was shown to be dramatically effective for lowering LDL cholesterol in healthy volunteers, with no obvious adverse effects.[10,11] However, clinical trials of compactin were stopped because of concerns with serious animal toxicity. Compactin and lovastatin have close structural similarity; hence Merck suspended clinical trials with lovastatin and initiated additional animal safety studies. The future of lovastatin was uncertain. However in 1982, small-scale clinical investigations asked Merck for lovastatin to test its effect in selected small groups of patients with severe heterozygous familial hypercholesterolaemia (FH) refractory to existing therapy. The investigators found dramatic reductions in LDL cholesterol.[12,13] with very few adverse effects. Later, Thompson in London found that lovastatin considerably enhanced the hypolipidemic effect of apheresis in patients with heterozygous FH.[14]
In 1984, in randomized, double-blind Phase IIb placebo-controlled studies, lovastatin was found to be as effective in patients with heterozygous FH[15] and patients with CHD and non-familial hypercholesterolaemia[16] as it had been in healthy volunteers.[11] These effects were confirmed in larger Phase III studies, in which lovastatin produced much greater reductions in LDL cholesterol than the control agents cholestyramine[17] and probucol,[18] with very few adverse effects.
Lovastatin produced a profound reduction of apolipoprotein-B-containing lipoproteins, especially LDL cholesterol and, to a lesser extent, plasma triglycerides, and a small increase in HDL cholesterol. Observed tolerability continued to be excellent, with very few patients withdrawing from treatment due to adverse effects. In November 1986, Merck applied for regulatory approval of lovastatin, and the US FDA advisory panel voted unanimously for the approval of the drug, with approval given in 1987.
When lovastatin became available for prescription use, physicians were able to obtain large reductions in plasma cholesterol – for the first time. Lovastatin at its maximal recommended dose of 80 mg daily produced a mean reduction in LDL cholesterol of 40%,[15–18] a far greater reduction than could be obtained with any of the treatments available at the time. The drug produced very few adverse effects, and with once- or twice-daily dosing, was easy for patients to take. Lovastatin, went on to revolutionize the treatment of hypercholesterolaemia, achieving peak annual sales of more than US $1 billion initially. Statins generated $35.3 billion worldwide in 2009. So, lovastatin became a success story despite initial skepticism from the medical community.
STATINS AND CLINICAL USE
Before the commercial availability of lovastatin in 1987, lipid lowering was limited to dietary changes, bile acid sequestrants such as cholestyramine and colestipol, nicotinic acid, the fibrates, and probucol. However, these treatments have limited efficacy or tolerability, or both. Dietary changes acceptable to patients produced only small changes in total and LDL cholesterol.[19]
On the other hand, lovastatin, the first statin developed, produced a significant mean reduction in LDL cholesterol, had few adverse effects, and was easy for patients to take, requiring only once- or twice-daily dosing. For these reasons, lovastatin was rapidly accepted by patients.
Simvastatin was the second statin used clinically. Simvastatin which differs from lovastatin in an additional side chain methyl group was approved for marketing in Sweden in 1988, and then worldwide. Pravastatin followed in 1991, fluvastatin in 1994, atorvastatin in 1997, cerivastatin in 1998 (of which more below), and rosuvastatin in 2003.
Lovastatin is a fermentation product. Simvastatin is a semisynthetic derivative of lovastatin, and pravastatin is derived from the natural product compactin by biotransformation. All other HMG-CoA reductase inhibitors are totally synthetic products. The mean reduction in LDL cholesterol attainable with the maximal recommended dose of different statins ranges from 35 to 55%.
STATIN ADVERSE EFFECTS
In animals, statins produce significant toxicity at high doses: increases in hepatic transaminases, atypical focal hyperplasia of the liver, squamous epithelial hyperplasia of the rat fore stomach (an organ not present in man), cataracts, vascular lesions in the central nervous system (CNS), skeletal muscle toxicity, testicular degeneration and, although the statins are clearly not genotoxic, tumours of the liver and other sites (details can be found in the product circulars of the individual statins). Fortunately, except for rare cases of myopathy and marked but asymptomatic increases in hepatic transaminases, none of the adverse effects found in animals occur at human therapeutic doses.
Establishment of the safety of statins required considerable effort in large long-term clinical trials and shorter specialized studies. Five-year trials have been published with simvastatin,[20–22] pravastatin[23–25] and lovastatin.[26] A smaller four-year trial with fluvastatin has also been reported,[27] as has a recent large trial with atorvastatin with 3.3 years follow-up.[28]
Of particular concern was the observation of cataracts in animal toxicology studies. When lovastatin and simvastatin were first marketed, slit-lamp examination was required to detect possible lens opacities at an early stage. Several studies using either specialized techniques in relatively small patient populations,[29–31] or routine slit-lamp examination in large populations,[21,32] showed that lens opacities occurred with similar frequencies in the active and placebo groups. However, in 2010, the British Medical Journal published a study that statins may raise cataract and kidney risk. Researchers estimated that for every 10,000 people taking a statin, there were about 271 fewer cases of heart disease, 8 fewer cases of esophageal cancer, 307 extra patients with cataracts, 23 additional patients with acute kidney failure, and 74 extra patients with liver dysfunction.[33]
There has been no good evidence of any increase in the risk of cancer at any particular site.[22,34] In the Scandinavian Simvastatin Survival Study (4S), there was a trend towards fewer cancers among the patients originally randomized to simvastatin[35] after seven years (an additional two years after the end of the study). Researchers have been unable to find a link between statins and cancer – neither an increase nor reduction in cancer in risk.
On March 3, 2012, the FDA announced changes to the safety information on the labels of statins: there is a small increased risk of higher blood sugar levels and of being diagnosed with type 2 diabetes. In addition, the statin labels will also now reflect reports of certain cognitive effects such as memory loss and confusion experienced by some patients taking the drugs. The announcement said the risk was “small” and should not materially affect the use of these medications.
It seems that the risk of diabetes is real only with the more potent statins such as simvastatin (Zocor), atorvastatin (Lipitor), and rosuvastatin (crestor), and particularly at higher doses.
The latest FDA change to the safety information is that patients taking statins will no longer need routine periodic monitoring of liver enzymes. FDA has concluded that serious liver injury with statins is rare and unpredictable in individual patients, and that routine periodic monitoring of liver enzymes does not appear to be effective in detecting or preventing this rare side effect.
STATIN-INDUCED MYOPATHY
Muscle complaints are common in statin users, occurring in more than 10% of this population.[36]
Myalgia or muscle pain occurs in about 7% of statin users. Interestingly, myopathy was not among the numerous abnormalities detected in the original animal safety studies with statins.
It was first reported in a cardiac transplant patient receiving the immunosuppressive cyclosporine, in addition to gemfibrozil.[37] Both of these drugs were later found to substantially increase the risk of myopathy with statins.[38,39] The mechanism of statin-associated myotoxicity has not been satisfactorily defined and is likely due to multiple factors, including membrane instability, mitochondrial dysfunction, and defects in myocyte duplication.
Cerivastatin was introduced in 1998 but was withdrawn in August 2001 by the manufacturer because of a large number of reports of rhabdomyolysis, of which more than 50 cases were fatal.[40,41] The risk of rhabdomyolysis was much higher with cerivastatin than with the other statins.[40–42]
Some of the reported cases of rhabdomyolysis occurred during concomitant use of gemfibrozil and cerivastatin.[42] Gemfibrozil increases the risk of myopathy with all statins; the mechanism is probably partly pharmacodynamic, as gemfibrozil can cause myopathy alone,[43] and partly pharmacokinetic. In the case of cerivastatin, the pharmacokinetic interaction is particularly marked; gemfibrozil was recently reported to increase the plasma concentration of cerivastatin approximately fivefold.[44]
It is not known why cerivastatin is more myotoxic than other statins. Its withdrawal shook the confidence of some physicians in the safety of statins in general.[40] Prescription growth rates for the class, which had approached 20% annually in many countries, fell dramatically.
HOW STATINS LOWER CHOLESTEROL
The Nobel Prize in Physiology or Medicine 1985 was awarded jointly to Michael S. Brown and Joseph L. Goldstein “for their discoveries concerning the regulation of cholesterol metabolism.”
They found that cells on their surfaces have receptors which mediate the uptake of the cholesterol-containing particles called low-density lipoprotein (LDL) that circulate in the blood stream. They discovered that the underlying mechanism to the severe hereditary familial hypercholesterolemia is a complete, or partial, lack of functional LDL-receptors. In normal individuals the uptake of dietary cholesterol inhibits the cells own synthesis of cholesterol. As a consequence the number of LDL-receptors on the cell surface is reduced. This leads to increased levels of cholesterol in the blood which subsequently may accumulate in the wall of arteries, causing atherosclerosis and eventually a heart attack or a stroke.
Statins lower human plasma cholesterol by increasing the uptake of LDL via the LDL receptor. Although LDL receptor upregulation is clearly the primary mechanism of action, these drugs also decrease the production of apolipoprotein-B-containing lipoproteins by the liver.[44] Consistent with this mechanism is the fact that high doses of atorvastatin[43] and simvastatin[45] produce moderate reductions of LDL cholesterol in patients with homozygous FH, who lack the LDL receptor.
THE CHOLESTEROL CONTROVERSY
For many years, the role of cholesterol in the pathogenesis of atherosclerosis was controversial.[46] There was skepticism in the medical community that elevated cholesterol is an important cause of atherosclerosis and coronary heart disease even though there was growing body of supportive evidence from experimental studies in animals, striking findings in humans with familial hypercholesterolemia, consistent epidemiologic correlations, and several small but impressive clinical trials.[47] Many leaders in the field of lipoprotein and atherosclerosis research however, were convinced that the accumulated evidence justified intervention to lower blood cholesterol levels.[47]
The American Heart Association began recommending dietary changes to control blood cholesterol levels as early as 1960. Diet alone, however, was insufficient, and the drugs available at that time had limited effectiveness. Cholestyramine, an inhibitor of bile acid reabsorption, was among the more effective drugs in use, but it was unpleasant to take, and compliance was poor.
The results of the Coronary Primary Prevention Trial,[48] a 7-year, double-blind, randomized clinical trial of cholestyramine among 3800 men with hypercholesterolemia was a turning point in medical thinking. That study showed a significant 20% decrease in the rate of fatal coronary heart disease or nonfatal myocardial infarction. The American National Institute of Health, after a panel review of the findings, declared in 1985 that lowering blood cholesterol should be a major national health goal.
The publication in 1994 of the results of the Scandinavian Simvastatin Survival Study (4S)[20] further boosted the status of statins in preventive cardiology. A total of 4,444 patients with CHD and total plasma cholesterol 5.5–8.0 mmol l-1 on a lipid-lowering diet were randomly allocated on a double-blind basis to simvastatin 20–40 mg once daily or placebo for five years. There was an unequivocal 30% reduction in all-cause mortality (P = 0.0003), due to a 42% reduction in coronary deaths. These effects on mortality were accompanied by a 34% reduction in major coronary events (non-fatal myocardial infarction plus CHD death) and a 37% reduction in revascularization procedures. There was no indication of any increase in non-cardiovascular mortality, and in particular, no increase in violent deaths or the incidence of cancer.[20,21] Only one patient in the simvastatin group developed myopathy. These results reassured those who argued that lowering cholesterol might reduce CHD events but not total mortality.
The magnitude of the reduction in LDL cholesterol obtainable with statins is much greater than with the earlier treatments used in previous clinical trials. Subsequent clinical trials with simvastatin, pravastatin, lovastatin, fluvastatin, and atorvastatin established that these staatins not only substantially reduced the risk of caardiovasccular events but did so without any increase in non-cardiovascular mortality such as that of cancer. The risk of CHD events was reduced both in patients who already had CHD (secondary prevention),[20,22,27,28] and in those who did not (primary prevention).[22,23,26]
The Heart Protection Study (HPS)[22] is the largest of the placebo-controlled five-year statin trials involving more than 20,000 patients in the United Kingdom with CHD, or at high risk of CHD due to cerebrovascular or peripheral vessel disease, or diabetes. Participants were randomized to simvastatin 40 mg or placebo for five years. HPS confirmed and expanded previous evidence, including firmly establishing the benefit of simvastatin in women, and its effectiveness for reduction of the risk not only of CHD events such as myocardial infarction, but also of strokes. It also provided new and compelling evidence for the beneficial effects of simvastatin on clinical outcomes in various large patient groups that had scarcely been studied. Most importantly, significant reductions in the risk of major vascular events were observed in patients with diabetes but no CHD, patients with cerebrovascular or peripheral vessel disease but no CHD, patients aged 70 or older, and patients with LDL cholesterol well below average <100 mg dl-1 (2.6 mmol l-1) at entry. These effects had not been previously reported for any statin. The tolerability and safety of simvastatin was confirmed yet again; the incidence of myopathy, including rhabdomyolysis, was <0.1%.
CONCLUSIONS
Coronary artery disease is still the leading cause of death in the industrialized and developing world. Studies have shown that elevated blood cholesterol is an important cause for atherosclerosis and large-scale trials have shown that lowering blood cholesterol significantly reduces major coronary events and coronary heart disease deaths.
Statin drugs lower blood cholesterol levels much more effectively than any diet or drug regimens that were available before the discovery of stains by Akira Endo in 1976. Endo's discovery opened the door to a new era in preventive cardiology. The statins are remarkably free of serious side effects. Nevertheless, a significant number of patients still have adverse cardiovascular events despite statin treatment. Obviously, more need to be done. Statins can reduce blood cholesterol levels by about 30% but this can be reduced by more than 50% if statins are combined with other drugs. The statins are not recommended to use with other drugs such as cholestyramine and gemfibrozil due to unwanted drug interactions. More aggressive targeting of treatable risks for coronary artery disease in addition to lowering LDL cholesterol levels can further lower morbidity and mortality associated with atherosclerosis.
The recent FDA warning that statins may cause diabetes is worrisome and presents a dilemma for those taking statins for primary prevention. Unlike those with coronary heart disease, the benefit of taking statins for primary prevention is only 2 per 100. It is estimated that about one in 200 patients treated with any of the three most potent statins will get the side effect of diabetes. At this point in time, in the primary prevention group, we do not know who will benefit from treatment and who will get diabetes. Diabetes is an important and serious side effect and dose reduction or use of a less potent statin should be considered in the primary prevention group. For those who already have coronary heart disease, the benefit of statins is quite clear and its use strongly recommended.
Why do statins cause diabetes? It is not known at present. Perhaps, genetic studies might help us determine who is at risk to develop statin induced diabetes. It is a problem in need of a solution.
REFERENCES
- 1.Steinberg D, Gotto AM., Jr Preventing coronary artery disease by lowering cholesterol levels: fifty years from bench to bedside. JAMA. 1999;282:2043–50. doi: 10.1001/jama.282.21.2043. [DOI] [PubMed] [Google Scholar]
- 2.Kannel WB. Clinical misconceptions dispelled by epidemiological research. Circulation. 1995;92:3350–60. doi: 10.1161/01.cir.92.11.3350. [DOI] [PubMed] [Google Scholar]
- 3.Keys A, Menotti A, Aravanis C, Blackburn H, Djordevic BS, Buzina R, et al. The seven countries study: 2,289 deaths in 15 years. Prev Med. 1984;13:141–54. doi: 10.1016/0091-7435(84)90047-1. [DOI] [PubMed] [Google Scholar]
- 4.Endo A, Kuroda M, Tsujita Y. ML-236A, ML-236B, and ML-236C, new inhibitors of cholesterogenesis produced by Penicillium citrinium. J Antibiot. 1976;29:1346–8. doi: 10.7164/antibiotics.29.1346. [DOI] [PubMed] [Google Scholar]
- 5.Endo A, Tsujita Y, Kuroda M, Tanzawa K. Inhibition of cholesterol synthesis in vitro and in vivo by ML-236A and ML-236B, competitive inhibitors of 3-hydroxy-3-methylglutaryl-coenzyme A reductase. Eur J Biochem. 1977;77:31–6. doi: 10.1111/j.1432-1033.1977.tb11637.x. [DOI] [PubMed] [Google Scholar]
- 6.Tsujita Y, Kuroda M, Tanzawa K, Kitano N, Endo A. Hypolipidemic effects in dogs of ML-236B, a competitive inhibitor of 3-hydroxy-3-methylglutaryl coenzyme A reductase. Atherosclerosis. 1979;32:307–13. doi: 10.1016/0021-9150(79)90174-6. [DOI] [PubMed] [Google Scholar]
- 7.Mabuchi H, Haba T, Tatami R, Miyamoto S, Sakai Y, Wakasugi T, et al. Effect of an inhibitor of 3-hydroxy-3-methyglutaryl coenzyme A reductase on serum lipoproteins and ubiquinone-10-levels in patients with familial hypercholesterolemia. N Engl J Med. 1981;305:478–82. doi: 10.1056/NEJM198108273050902. [DOI] [PubMed] [Google Scholar]
- 8.Mabuchi H, Sakai T, Sakai Y, Yoshimura A, Watanabe A, Wakasugi T, et al. Reduction of serum cholesterol in heterozygous patients with familial hypercholesterolemia. Additive effects of compactin and cholestyramine. N Engl J Med. 1983;308:609–13. doi: 10.1056/NEJM198303173081101. [DOI] [PubMed] [Google Scholar]
- 9.Alberts AW, Chen J, Kuron G, Hunt V, Huff J, Hoffman C, et al. Mevinolin: A highly potent competitive inhibitor of hydroxymethylglutaryl-coenzyme a reductase and a cholesterol-lowering agent. Proc Natl Acad Sci. 1980;77:3957–61. doi: 10.1073/pnas.77.7.3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tobert JA, Hitzenberger G, Kukovetz WR, Holmes IB, Jones KH. Rapid and substantial lowering of human serum cholesterol by mevinolin (MK-803), an inhibitor of hydroxymethylglutaryl-coenzyme A reductase. Atherosclerosis. 1982;41:61–5. doi: 10.1016/0021-9150(82)90070-3. [DOI] [PubMed] [Google Scholar]
- 11.Tobert JA, Bell GD, Birtwell J, James I, Kukovetz WR, Pryor JS, et al. Cholesterol-lowering effect of mevinolin, an inhibitor of 3-hydroxy-3-methylglutaryl-coenzyme A reductase, in healthy volunteers. J Clin Invest. 1982;69:913–9. doi: 10.1172/JCI110530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bilheimer DW, Grundy SM, Brown MS, Goldstein JL. Mevinolin and colestipol stimulate receptor-mediated clearance of low density lipoprotein from plasma in familial hypercholesterolemia heterozygotes. Proc Natl Acad Sci. 1983;80:4124–8. doi: 10.1073/pnas.80.13.4124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Illingworth DR, Sexton GJ. Hypocholesterolemic effects of mevinolin in patients with heterozygous familial hypercholesterolemia. J Clin Invest. 1984;74:1972–8. doi: 10.1172/JCI111618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thompson GR, Ford J, Jenkinson M, Trayner I. Efficacy of mevinolin as adjuvant therapy for refractory familial hypercholesterolaemia. Q J Med. 1986;60:803–11. [PubMed] [Google Scholar]
- 15.Havel RJ, Hunninghake DB, Illingworth DR, Lees RS, Stein EA, Tobert JA, et al. Lovastatin (mevinolin) in the treatment of heterozygous familial hypercholesterolemia. A multicenter study. Ann Intern Med. 1987;107:609–15. doi: 10.7326/0003-4819-107-5-609. [DOI] [PubMed] [Google Scholar]
- 16.Lovastatin Study Group II. Therapeutic response to lovastatin (mevinolin) in nonfamilial hypercholesterolemia. A multicenter study. JAMA. 1986;256:2829–34. doi: 10.1001/jama.1986.03380200067023. [DOI] [PubMed] [Google Scholar]
- 17.Lovastatin Study Group III. A multicenter comparison of lovastatin and cholestyramine therapy for severe primary hypercholesterolemia. JAMA. 1988;260:359–66. [PubMed] [Google Scholar]
- 18.Lovastatin Study Group IV. A multicenter comparison of lovastatin and probucol for treatment of severe primary hypercholesterolemia. Am J Cardiol. 1990;66:22b–30b. doi: 10.1016/0002-9149(90)90437-6. [DOI] [PubMed] [Google Scholar]
- 19.Tang JL, Armitage JM, Lancaster T, Silagy CA, Fowler GH, Neil HA. Systematic review of dietary intervention trials to lower blood total cholesterol in free-living subjects. BMJ. 1998;316:1213–20. doi: 10.1136/bmj.316.7139.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scandinavian Simvastatin Survival Study Group. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S) Lancet. 1994;344:1383–9. [PubMed] [Google Scholar]
- 21.Pedersen TR, Berg K, Cook TJ, Faergeman O, Haghfelt T, Kjekshus J, et al. Safety and tolerability of cholesterol lowering with simvastatin during 5 years in the Scandinavian Simvastatin Survival Study. Arch Intern Med. 1996;156:2085–92. [PubMed] [Google Scholar]
- 22.Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet. 2002;360:7–22. [Google Scholar]
- 23.Shepherd J, Cobbe SM, Ford I, Isles CG, Lorimer AR, MacFarlane PW, et al. Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. N Engl J Med. 1995;333:1301–7. doi: 10.1056/NEJM199511163332001. [DOI] [PubMed] [Google Scholar]
- 24.Sacks FM, Pfeffer MA, Moye LA, Rouleau JL, Rutherford JD, Cole TG, et al. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. N Engl J Med. 1996;335:1001–9. doi: 10.1056/NEJM199610033351401. [DOI] [PubMed] [Google Scholar]
- 25.The Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. N Engl J Med. 1998;339:1349–57. doi: 10.1056/NEJM199811053391902. [DOI] [PubMed] [Google Scholar]
- 26.Downs JR, Clearfield M, Weis S, Whitney E, Shapiro DR, Beere PA, et al. Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: results of AFCAPS/TexCAPS. Air Force/Texas Coronary Atherosclerosis Prevention Study. JAMA. 1998;279:1615–22. doi: 10.1001/jama.279.20.1615. [DOI] [PubMed] [Google Scholar]
- 27.Serruys PW, de Feyter P, Macaya C, Kokott N, Puel J, Vrolix M, et al. Fluvastatin for prevention of cardiac events following successful first percutaneous coronary intervention. JAMA. 2002;287:3215–22. doi: 10.1001/jama.287.24.3215. [DOI] [PubMed] [Google Scholar]
- 28.Sever PS, Dahlöf B, Poulter NR, Wedel H, Beevers G, Caulfield M, et al. Prevention of coronary and stroke events with atorvastatin in hypertensive patients who have average or lower-than-average cholesterol concentrations, in the Anglo-Scandinavian Cardiac Outcomes Trial — Lipid Lowering Arm (ASCOT-LLA): a multicentre randomised controlled trial. Lancet. 2003;361:1149–58. doi: 10.1016/S0140-6736(03)12948-0. [DOI] [PubMed] [Google Scholar]
- 29.Chylack LT, Jr, Mantell G, Wolfe JK, Friend J, Rosner B. Lovastatin and the human lens; results of a two year study. The MSDRL Study Group. Optom Vis Sci. 1993;70:937–43. doi: 10.1097/00006324-199311000-00011. [DOI] [PubMed] [Google Scholar]
- 30.Harris ML, Bron AJ, Brown NA, Keech AC, Wallendszus KR, Armitage JM, et al. Absence of effect of simvastatin on the progression of lens opacities in a randomised placebo controlled study. Oxford Cholesterol Study Group. Br J Ophthalmol. 1995;79:996–1002. doi: 10.1136/bjo.79.11.996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schmidt J, Schmitt C, Hockwin O, Paulus U, von Bergmann K. Ocular drug safety and HMG-CoA-reductase inhibitors. Ophthalmic Res. 1994;26:352–60. doi: 10.1159/000267501. [DOI] [PubMed] [Google Scholar]
- 32.Laties AM, Shear CL, Lippa EA, Gould AL, Taylor HR, Hurley DP, et al. Expanded clinical evaluation of lovastatin (EXCEL) study results. II. Assessment of the human lens after 48 weeks of treatment with lovastatin. Am J Cardiol. 1991;67:447–53. doi: 10.1016/0002-9149(91)90002-3. [DOI] [PubMed] [Google Scholar]
- 33.Hippisley-Cox J, Coupland C. Unintended effects of statins in men and women in England and Wales: population based cohort study using the Q Research database. BMJ. 2010;340:c2197. doi: 10.1136/bmj.c2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.LaRosa J, He J, Vupputuri S. Effect of statins on risk of coronary disease: a meta-analysis of randomized controlled trials. JAMA. 1999;282:2340–6. doi: 10.1001/jama.282.24.2340. [DOI] [PubMed] [Google Scholar]
- 35.Pedersen TR, Wilhelmsen L, Faergeman O, Strandberg TE, Thorgeirsson G, Troedsson L, et al. Follow-up study of patients randomized in the Scandinavian simvastatin survival study (4S) of cholesterol lowering. Am J Cardiol. 2000;86:257–62. doi: 10.1016/s0002-9149(00)00910-3. [DOI] [PubMed] [Google Scholar]
- 36.Bruckert E, Hayem G, Dejager S, Yau C, Begaud B. Mild to moderate muscular symptoms with high-dosage statin therapy in hyperlipidemic patients: the PRIMO study. Cardiovasc Drugs Ther. 2005;19:403–14. doi: 10.1007/s10557-005-5686-z. [DOI] [PubMed] [Google Scholar]
- 37.Norman DJ, Illingworth DR, Munson J, Hosenpud J. Myolysis and acute renal failure in a heart-transplant recipient receiving lovastatin. N Engl J Med. 1988;318:46–7. doi: 10.1056/NEJM198801073180110. [DOI] [PubMed] [Google Scholar]
- 38.Pierce LR, Wysowski DK, Gross TP. Myopathy and rhabdomyolysis associated with lovastatin–gemfibrozil combination therapy. JAMA. 1990;264:71–5. [PubMed] [Google Scholar]
- 39.Gruer P, Vega J, Mercuri M, Dobrinska M, Tobert J. Concomitant use of cytochrome P450 3A4 inhibitors and simvastatin. Am J Cardiol. 1999;84:811–5. doi: 10.1016/s0002-9149(99)00442-7. [DOI] [PubMed] [Google Scholar]
- 40.Ballantyne CM, Corsini A, Davidson MH, Holdaas H, Jacobson TA, Leitersdorf E, et al. Risk for myopathy with statin therapy in high-risk patients. Arch Intern Med. 2003;163:553–64. doi: 10.1001/archinte.163.5.553. [DOI] [PubMed] [Google Scholar]
- 41.How a statin might destroy a drug company. Lancet. 2003;361:793–3. [PubMed] [Google Scholar]
- 42.Raine JM. Withdrawal of cerivastatin (Libobay) by Bayer PLC. (Department of Health, Medicines Control Agency, London. 2001 [Google Scholar]
- 43.Magarian GJ, Lucas LM, Colley C. Gemfibrozil-induced myopathy. Arch Intern Med. 1991;151:1873–4. [PubMed] [Google Scholar]
- 44.Backman J, Kyrklund C, Neuvonen M, Neuvonen P. Gemfibrozil greatly increases plasma concentrations of cerivastatin. Clin Pharmacol Ther. 2002;72:685–91. doi: 10.1067/mcp.2002.128469. [DOI] [PubMed] [Google Scholar]
- 45.Arad Y, Ramakrishnan R, Ginsberg HN. Effects of lovastatin therapy on very-low-density lipoprotein triglyceride metabolism in subjects with combined hyperlipidemia: evidence for reduced assembly and secretion of triglyceride-rich lipoproteins. Metabolism. 1992;41:487–98. doi: 10.1016/0026-0495(92)90206-p. [DOI] [PubMed] [Google Scholar]
- 46.Raal FJ, Pilcher GJ, Illingworth DR, Pappu AS, Stein EA, Laskarzewski P, et al. Expanded-dose simvastatin is effective in homozygous familial hypercholesterolaemia. Atherosclerosis. 1997;135:249–56. doi: 10.1016/s0021-9150(97)00168-8. [DOI] [PubMed] [Google Scholar]
- 47.Steinberg D. The cholesterol wars: the skeptics vs. the preponderance of evidence. New York: Elsevier Academic press; 2007. [Google Scholar]
- 48.The Lipid Research Clinics Coronary Primary Prevention Trial results. I. Reduction in incidence of coronary heart disease. JAMA. 1984;251:351–64. doi: 10.1001/jama.1984.03340270029025. [DOI] [PubMed] [Google Scholar]