

Abstract
NS5A is a key regulator of hepatitis C virus (HCV) life cycle including RNA replication, assembly, and translation. We and others have shown NS5A to augment HCV IRES-mediated translation. Further, Quercetin treatment and heat shock protein (HSP) 70 knockdown inhibit NS5A-driven augmentation of IRES-mediated translation and infectious virus production. We have also co-immunoprecipitated HSP70 with NS5A and demonstrated cellular colocalization leading to the hypothesis that the NS5A/HSP70 complex formation is important for IRES-mediated translation. Here, we have identified the NS5A region responsible for complex formation through in vitro deletion analyses. Deletion of NS5A domains II and III failed to reduce HSP70 binding, whereas domain I deletion eliminated complex formation. NS5A domain I alone also bound HSP70. Deletion mapping of domain I identified the C-terminal 34 amino acids (C34) to be the interaction site. Further, addition of C34 to domains II and III restored complex formation. C34 expression significantly reduced intracellular viral protein levels, in contrast to same size control peptides from other NS5A domains. C34 also competitively inhibited NS5A-augmented IRES-mediated translation, while controls did not. Triple-alanine scan mutagenesis identified an exposed beta-sheet hairpin in C34 to be primarily responsible for NS5A-augmented IRES-mediated translation. Moreover, treatment with a 10 amino acid peptide derivative of C34 suppressed NS5A-augmented IRES-mediated translation and significantly inhibited intracellular viral protein synthesis, with no associated cytotoxicity. Conclusion: These results support the hypothesis that the NS5A/HSP70 complex augments viral IRES-mediated translation, identify a sequence-specific hairpin element in NS5A responsible for complex formation, and demonstrate the functional significance of C34 hairpin-mediated NS5A/HSP70 interaction. Identification of this element may allow for further interrogation of NS5A-mediated IRES activity, sequence specific HSP recognition, and rational drug design.
Keywords: HSP70, NS5A, IRES, HCV, Protein binding
Introduction
Hepatitis C virus (HCV) infection has a worldwide prevalence of 3% and is the main indication for liver transplantation in developed countries for treatment of cirrhosis (1). In the United States, HCV is the most common chronic blood borne infection affecting 1.8% of the population and is the major etiologic factor responsible for the recent doubling of hepatocellular carcinoma (HCC) (2).
Current HCV therapy consists of pegylated interferon-α (PEG-IFN) and ribavirin which is suboptimal as 70–80% of US patients are infected with genotype 1, with sustained virologic response (SVR) of only 50–56% (3). Generally, therapy of all genotypes can be accompanied by adverse effects, and contraindications to therapy are not infrequent (4). Standard antiviral combination therapy with interferon has been shown to be an effective secondary prevention of HCC (5); however, 70–80% of HCV patients are not candidates for these therapies (6, 7). NS3/4A protease inhibitors in combination with PEG-IFN and ribavirin have recently been approved and show increased SVR, but also increased adverse events including anemia and gastrointestinal symptoms (8). Since these therapies still require interferon, a significant population of patients cannot receive this treatment. Current approaches for preventing HCV-related HCC in patients with contraindications to standard therapy are limited. For these reasons, there is the need to develop adjunct or replacement therapies that are less toxic and more efficacious in terms of higher SVR and HCC prevention.
HCV possesses a 10kb positive sense RNA genome that encodes 10 viral proteins (9). The non-structural protein 5A (NS5A) is a 56–59 kDa phosphoprotein that associates with the viral replicase complex. It has been implicated in the regulation of HCV genome replication, viral protein translation, and virion assembly (10–12). NS5A also appears to modulate hepatocyte cell signaling by 1) promoting cell survival (13), 2) facilitating the viral life cycle (10–12) and 3) interfering with the hepatocyte innate immune response (14).
The 5′ non-coding region (NCR) of HCV genome lacks a 5′ cap and instead possesses an internal ribosomal entry site (IRES) (15), a cis-acting element found in some host RNA transcripts as well as in viruses that allows ribosomal translation initiation to occur internally within a transcript in lieu of 5′ cap-dependent translation (16). NS5A has been implicated in modulating HCV IRES-mediated translation (11, 17).
We have previously identified, through co-immunoprecipitation, a complex of NS5A and heat-shock proteins (HSPs) composed of NS5A, HSP70, and HSP40 (cofactor of HSP70) and demonstrated their colocalization in Huh-7 cells (18). We further showed that both NS5A-augmented IRES-mediated translation and virus production are blocked by HSP70 knockdown as well as by HSP synthesis inhibitor Quercetin, with no associated cytotoxicity (18).
These findings led to the hypothesis that HCV utilizes an NS5A/HSP complex to facilitate IRES-mediated translation of its genome. Here we report the identification of the NS5A/HSP70 interaction site on NS5A and demonstrate that a soluble peptide derivative of this site inhibits NS5A-mediated IRES activity and viral infection.
Experimental Procedures
Plasmid constructs and cloning
HSP70 and HSP40 were amplified from Huh-7 cDNA using polymerase chain reaction (PCR), and NS5A was amplified from pMSCV-NS5A-FLAG described previously (18). pGEX-6P-2 plasmid (Amersham, 28-9546-50) was used to generate N-terminal GST fusion HSP70, HSP40, and NS5A as well as HSP70 nucleotide binding domain and substrate binding domain. pET-28b plasmid (Novagen, 69865-3) was used to generate C-terminal His6-tagged HSP70, HSP40, and NS5A. pTRACER-EF/Bsd A plasmid (Invitrogen, V889-01) was used to generate C-terminal V5-epitope-tagged NS5A, HSP70, and HSP40 and all NS5A subclones. The C34 construct as well as NS5A domain II and domain III peptides, NS3 peptide (P19), and C34 mutagenesis constructs (ASMs) were cloned into pCCL-CMV-IRES-EGFP plasmid. The HCV IRES reporter plasmid pNRLFC, and the GFP retroviral expression vector pMSCVGFP have been previously described (18). Please refer to Supplementary Experimental Procedures for more details.
Production and purification of recombinant proteins
GST-fusion proteins were expressed in bacteria and purified as previously described (19). His6-tagged proteins were expressed and purified using Ni-NTA Agarose bead slurry (Qiagen, 1018244) according to manufacturer’s instructions. V5-epitope-tagged proteins were expressed using TNT T7 Coupled Reticulocyte Lysate System (Promega, L4610).
GST pull-down assays
GST pull-down assays were performed as previously described (19). Pull-down targets were evaluated by western analysis.
Antibodies
HSP70 (Santa Cruz Biotech, C92F3A-5), HSP40 (Abcam, ab56589), NS5A (Abcam, ab20342), V5 epitope (Millipore, AB3792), and allophycocyanin-conjugated-anti-FLAG (PerkinElmer, AD0059F).
Cell culture
Cell lines 293T and Huh-7.5 were maintained in a humidified atmosphere containing 5% CO2 at 37°C in Dulbecco’s modified Eagle’s medium (Mediatech, 10-013-CM) supplemented with 10% fetal bovine serum (Omega Scientific, FB-01) and 2 mM L-glutamine (Invitrogen, 25030).
IRES reporter assay
293T cells were transfected with HCV IRES reporter plasmid and either pMSCV-NS5A-FLAG (bearing wild-type NS5A or one of the mutants) or pMSCV-GFP. pCCL-CMV-IRES-EGFP plasmid expressing one of the following peptides was also transfected where indicated: C34, NS5A domain II peptide, NS5A domain III peptide, NS3 peptide, or one of the C34 mutagenesis peptides. All transfections were done using Fugene6 (Roche, 11814443001). 48 hours post transfection, Renilla and Firefly luciferase activity was determined using Dual Luciferase Assay System (Promega, E1910).
Flow cytometry
293T cells were transfected with the construct expressing GFP, C34, NS5A domain II peptide, NS5A domain III peptide, or NS3 peptide. 48 hours post transfection, the cells were harvested and analyzed by flow cytometry analysis for the FLAG fusion epitope. Cells were washed with PBS three times, fixed with 1.5% paraformaldehyde, and permeabilized with 0.05% nonidet p40. Cells were stained with 1:500 dilution of allophycocyanin-conjugated-anti-FLAG antibody for 30 minutes and washed three times with PBS. Flow cytometry was performed using Becton Dickinson FACSCalibur cytometer and Cellquest v3.3 software.
Infectious virus production
pNRLFC was in vitro transcribed, and the purified RNA was electroporated into Huh-7.5 cells to generate infectious viral supernatant as previously described (20). Viral assays were done using the HCV reporter virus as described previously (18).
Peptide synthesis and characterization
Peptides were synthesized by the solid phase method using CEM Liberty automatic microwave peptide synthesizer (CEM Corporation), applying 9-fluorenylmethyloxycarbonyl (Fmoc) chemistry (21) and standard, commercially available amino acid derivatives and reagents (EMD Biosciences and Chem-Impex International). Rink Amide MBHA resin (EMD Biosciences) was used as a solid support. Peptides were cleaved from resin using modified reagent K (TFA 94% (v/v); phenol, 2% (w/v); water, 2% (v/v); TIS, 1% (v/v); EDT, 1% (v/v); 2 hours) and precipitated by addition of ice-cold diethyl ether. Reduced peptides were purified by preparative reverse-phase high performance liquid chromatography (RP-HPLC) to >95% homogeneity and their purity evaluated by matrix-assisted laser desorption ionization spectrometry (MALDI-MS) as well as by analytical RP-HPLC). Disulfide bond formation: Peptides were dissolved at a final concentration of 0.25 mg/ml in 50% DMSO:H2O and stirred overnight at room temperature. Subsequently peptides were lyophilized and re-purified on a preparative C18 SymmetryShield™ RP-HPLC column to >95% homogeneity. Their purity was evaluated by MALDI-MS as well as by analytical RP-HPLC. Analytical RP-HPLC: Analytical RP-HPLC was performed on a Varian ProStar 210 HPLC system equipped with ProStar 325 Dual Wavelength UV-Vis detector with wavelengths set at 220 nm and 280 nm (Varian Inc.). Mobile phases consisted of solvent A, 0.1% TFA in water, and solvent B, 0.1% TFA in acetonitrile. Analyses of peptides were performed with an analytical reversed-phase C18 SymmetryShield™ RP18 column, 4.6×250 mm, 5μm (Waters Corp.) applying linear gradient of solvent B from 0 to 100% over 100 min (flow rate: 1 ml/min).
Cell viability
Cell viability was determined using MTT Cell Proliferation assay (ATCC, 30–1010K).
Fluorescent microscopy
All images were taken by Olympus CKX41 fluorescent microscope via DP2-BSW v2.1.6207 software.
Quantitative reverse-transcriptase PCR
Huh-7.5 cells were treated with peptide and infected with Renilla reporter virus. 48 hours post infection, cells were harvested, and total RNA was extracted using RNeasy Mini Kit (Qiagen, 74104). cDNA was synthesized using iScript cDNA Synthesis Kit (Bio-Rad, 1708891). Quantitative PCR was performed using the Applied Biosystems 7500 Fast Real-Time PCR System with 2x SYBR Green Master Mix (Diagenode, GMO-SG2x-A300) in 25μL reactions. The real-time PCR cycling conditions were 50°C for 2 minutes and 95°C for 10 minutes, followed by 40 cycles at 95°C for 15 seconds, 60°C for 30 seconds and 72°C for 30 seconds each as well as a final dissociation stage of 95°C for 15 seconds and 60°C for 1 minute. The primers for the viral genome were derived from the 5′-non-coding region and were CTGGGTCCTTTCTTGGATAA and CCTATCAGGCAGTACCACA. HCV RNA levels were normalized to the housekeeping gene actin using the primers CCAACCGCGAGAAGATGA and CCAGAGGCGTACAGGGATAG.
Co-immunoprecipitation
Huh-7.5 cells were treated with 1μM of fluorescein isothiocyanate (FITC)-labeled HCV4 peptide. 24 hours later, cells lysates of FITC-HCV4 treated and control (untreated) cells were used in co-immunoprecipitation (co-IP) assays with antibody against FITC and IgG1 as control. Co-IPs were done using Protein G Plus-Agarose Immunoprecipitation Reagent (Santa Cruz Biotech, sc-2002) according to manufacturer’s instructions.
Statistical analysis
Error bars reflect standard deviation. P values were determined by student t test.
Results
The C-terminal region of NS5A domain I is necessary and sufficient for HSP70 binding
We have previously shown that NS5A forms a complex with HSP70 and HSP40 (encoded by HSPA1A and DNAJ2, respectively) (18). To determine whether NS5A (genotype 1a, clone H77) interacts directly with either or both of these HSPs, GST-fusion protein pull-down assays were performed using recombinant proteins purified from bacteria. GST-HSP70 interacted with NS5A, while GST-HSP40 showed minimal interaction (Fig. 1A). NS5A/HSP70 binding was further confirmed by using GST-NS5A as bait to successfully pull down HSP70 (Fig. 1B).
Fig. 1.

NS5A domain I directly binds HSP70. The upper part of all panels displays Western analysis of GST pull-down targets (antibodies are mentioned underneath). Arrows indicate presence of target polypeptides. The lower part(s) represent(s) Coomassie stains showing GST-fusion bait proteins. (A) and (B) NS5A directly binds HSP70. (C) Deletion of NS5A domain I abolishes its HSP70 interaction. (D) The membrane anchoring domain of NS5A is not required for HSP70 binding. (E) Only NS5A domain I binds HSP70.
NS5A consists of the four elements: membrane anchoring domain (MAD), domain I, domain II, and domain III (Fig. 2D). While MAD is generally considered to be part of domain I, throughout this paper, the term ‘domain I’ excludes MAD. To determine the individual domain(s) that bind(s) HSP70, each domain was specifically deleted, and the resulting proteins were expressed through in vitro coupled transcription/translation reactions. Each deletion mutant was then tested for HSP70 binding using GST pull-down assays with GST-HSP70 as bait. Deletion of NS5A domain I abolished HSP70 binding, while deletion of MAD, domain II, and domain III maintained this interaction (Fig. 1C and 1D). Furthermore, NS5A domain I, II, and III were individually expressed in vitro and tested for HSP70 binding. Only NS5A domain I bound HSP70 (Fig. 1E).
Fig. 2.


The C-terminal region of NS5A domain I is necessary and sufficient for its interaction with the nucleotide binding domain of HSP70. The upper part of all panels displays Western analysis of GST pull-down targets using antibody against the V5 epitope. Arrows indicate presence of target polypeptides. The lower part(s) represent(s) Coomassie stains showing GST-fusion bait proteins. (A) The N-terminal region of NS5A domain I is not necessary for HSP70 binding. (B) The C-terminal 34 amino acids of NS5A domain I (C34) are necessary for HSP70 interaction. (C) C34 region is sufficient for HSP70 binding. (D) Schematic of the structure of NS5A. (E) Summary of all NS5A subclones used in GST pull-down analyses in Fig. 1 and 2. (+) and (−) indicate interaction and lack of interaction with HSP70, respectively. (F) NS5A domain I directly interacts with the nucleotide binding domain of HSP70, but not with the substrate binding domain.
To determine which region of NS5A domain I binds HSP70, N-terminal and C-terminal deletions of domain I were expressed in vitro and tested for HSP70 interaction using GST pull-down assays. Deletions of 34 and 68 amino acids at the N terminus of NS5A domain I did not have any effect on its HSP70 binding (Fig. 2A). However, deletion of 34 amino acids from the C terminus abolished the HSP70-binding capability of NS5A domain I (Fig. 2B). Deleting an additional 34 amino acids at the C terminus showed the same result (Fig. 2B).
To demonstrate that the C-terminal region of NS5A domain I is indeed responsible for HSP70 binding, the C-terminal 34 amino acids of domain I were added to the NS5A domain I deletion mutant. This addition restored the HSP70 binding capability of NS5A domain I deletion mutant in subsequent GST pull-down assays (Fig. 2C). Results of all HSP70 binding assays are summarized in Fig. 2E.
NS5A directly interacts with the nucleotide binding domain of HSP70
HSP70 binds in a non-specific and sequence-independent manner to hydrophobic peptides through its C-terminal substrate binding domain whereas its N-terminal nucleotide-binding domain (NBD) interacts with regulatory proteins (22). GST-fusion HSP70-NBD and HSP70-SBD were tested for interaction with NS5A domain I. Only HSP70-NBD interacted with NS5A domain I (Fig. 2F).
The C-terminal 34 amino acid peptide of NS5A domain I suppresses NS5A-augmented IRES-mediated translation
After identifying the C-terminal region of NS5A as the HSP70 binding site in vitro, we sought to determine whether this region has any functional significance. NS5A is reported to increase IRES-mediated translation in a cell culture-based bicistronic reporter system (11, 18) (Fig. 3A). This reporter system consists of Firefly luciferase open reading frame (ORF) driven by HCV IRES and Renilla luciferase ORF under the control of a 5′ cap (Fig. 3A). The ratio of Firefly to Renilla luciferase activity is used to measure IRES-mediated translation levels.
Fig. 3.


The C-terminal 34 amino acids of NS5A domain I (C34) regulate NS5A-augmented IRES-mediated translation and intracellular viral protein production. (A) Schematic of the bicistronic reporter construct used to measure IRES-mediated translation. Firefly luciferase (FLuc) and Renilla luciferase (RLuc) are driven by HCV IRES and a 5′ cap, respectively. Each reporter has a stop codon to allow independent expression. Firefly to Renilla ratios reflect changes in IRES-mediated translation. For all experiments, the IRES reporter, either NS5A or GFP, and a peptide construct (if indicated) were transfected into cells. (B) C34 suppresses the NS5A-driven increase in IRES-mediated translation. (C) Flow cytometry of cells transfected with the peptide constructs reveals that all peptides (FLAG-tagged) are expressed at equivalent levels. An allophycocyanin (APC)-conjugated-anti-FLAG antibody was used. (D) Renilla luciferase assay demonstrates that C34 significantly inhibits intracellular viral protein production. Huh-7.5 cells were transfected with the indicated peptide construct and 48 hours later infected with the reporter virus for three hours. Cells were harvested 24 hours post infection, and Renilla activity was measured. * indicates P<0.05.
The C-terminal 34 amino acid peptide corresponding to amino acids 165–198 of NS5A domain I (hereafter referred to as C34) was tested for altering IRES-mediated translation. Peptides of identical length from NS5A domains II and III and a 19 amino acid peptide (P19) from NS3 (23) were also tested. C34 was found to block NS5A-augmented IRES-mediated translation in cell culture, while NS5A domain II and III peptides and the NS3 peptide did not (Fig. 3B).
All peptides (designed to bear the FLAG tag at their C termini) were determined to be expressed at similar levels through flow cytometry of transfected cells with antibody against the FLAG tag (Fig. 3C).
C34 blocks intracellular viral protein synthesis
Expression of C34 in the HCV cell culture (HCVcc) system using the HCV Renilla reporter virus significantly reduced intracellular viral protein production as measured by Renilla luciferase reporter activity (Fig. 3D). In addition, the P19 peptide from NS3 showed modest reduction in intracellular virus in agreement with previous studies (23) (Fig. 3D). In contrast, expression of equal sized peptides from other areas of NS5A failed to have an effect on infection (Fig. 3D). C34 expression had no effect on cell viability as measured by MTT assay (data not shown).
Mutation of an exposed antiparallel beta-sheet hairpin in C34 inhibits NS5A augmentation of IRES-mediated translation
Scanning triple alanine substitution mutants of C34 were generated to determine the amino acids important for NS5A/HSP70 complex-mediated IRES translation. The IRES reporter, NS5A, and C34 mutants and wild-type were transfected into cells, and IRES activity was measured. The most significant mutations affecting NS5A activity localized to amino acids 171–179 (ASM3, 4, and 5) (Fig. 4A).
Fig. 4.



The hairpin structure at the C terminus of NS5A domain I regulates HCV NS5A-driven IRES activity and intracellular virus production. (A) Triple-alanine scan mutagenesis (ASM) across the C34 region indicates that amino acids 171–179 of NS5A domain I are primarily involved in augmenting IRES-mediated translation. (B) Mutations of the C34 hairpin in full-length NS5A impair its ability to augment IRES-mediated translation. (C) MTT viability assay showing the cytotoxicity of the HCV4 synthetic peptide derivative of C34 hairpin (Table 1). (D) HCV4 blocks NS5A-augmented IRES-mediated translation. IRES basal activity in the absence of NS5A was normalized to one for each concentration of peptide. There was a slight decrease in IRES basal activity with increasing peptide treatment (approximately 8% at 1μM). (E) HCV4 significantly suppresses intracellular viral protein translation. Huh-7.5 cells were treated with the indicated concentrations of HCV4 or the arginine tag control peptide (Table 1) and immediately infected with the Renilla reporter virus for three hours. Cells were assayed 24 hours post infection for Renilla activity. * indicates P<0.05.
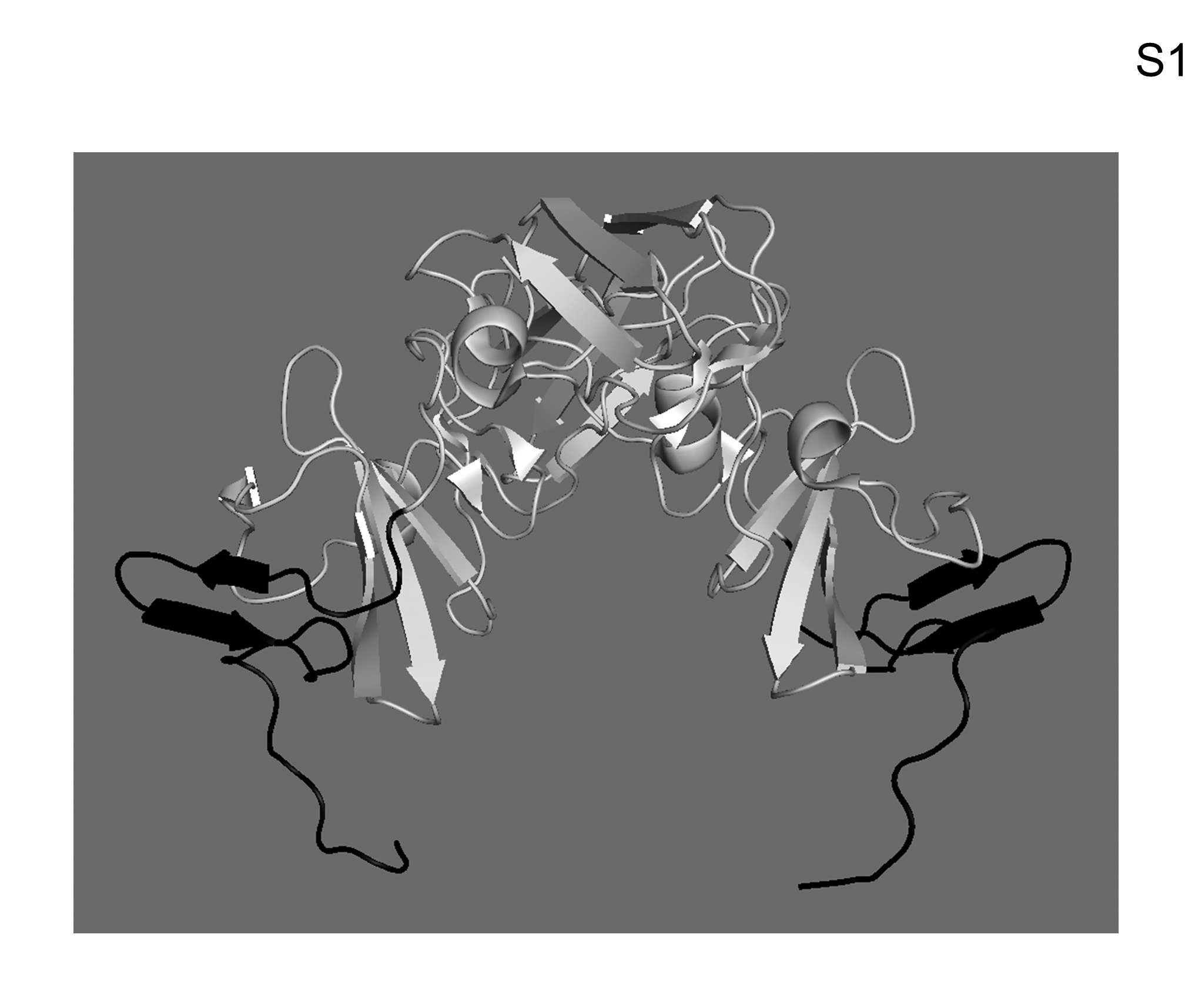
The C34 residues (strain H77) correspond to amino acids 169–202 of the NS5A variant used in the crystal structure of dimeric NS5A domain I (24); however, the crystal structure itself terminates at amino acid 198 and, therefore, includes the first 30 amino acids of C34 (Fig. S1). Amino acids 171–179 correspond to residues 175–183 of the crystal structure. This region maps to an exposed anti-parallel beta-sheet hairpin (Fig. S1) external to the claw-like structure of NS5A domain I dimer that binds single-stranded RNA (24).
Mutations of the C34 hairpin in full-length NS5A impair its ability to augment IRES-mediated translation
To verify the role of C34 hairpin in NS5A-augmented IRES-mediated translation, we introduced mutations in the hairpin region of full-length NS5A. Four triple alanine substitution mutants (corresponding to the ASM3, 4, 5, and 7 constructs) were generated and tested for their effect on IRES-mediated translation. All three mutations in C34 hairpin significantly reduced IRES-mediated translation levels compared to wild-type NS5A (Fig. 4B). The ASM7 mutation, downstream of the hairpin, however, did not significantly effect IRES-mediated translation (Fig. 4B).
A 10 amino acid peptide corresponding to the C34 hairpin blocks NS5A-augmented IRES-mediated translation
The crystal structure of NS5A domain I reveals the hairpin to be the only moiety in C34 with secondary structure (24) (Fig. S1). To determine whether the hairpin alone (as opposed to the entire C34) would be sufficient for IRES activity, a modified peptide of 10 amino acid length corresponding to the C34 hairpin was generated (HCV4 peptide) (Table 1). Two cysteine residues were added to each end to allow for disulfide bond formation and to achieve a conformation similar to the C34 hairpin. An arginine tag (Table 1) was added to the N-terminal cysteine to allow for efficient uptake by cells without any transfection reagent. MTT assays demonstrated toxicity at 100μM and minimal toxicity at 10μM, while no cytotoxicity was observed at any lower concentrations (Fig. 4C). A 48 hour MTT assay was also performed with HCV4 and a similar toxicity profile was obtained (data not shown).
Table 1.
| H77 Amino acid position | 169 | 170 | 171 | 172 | 173 | 174 | 175 | 176 | 177 | 178 | 179 | 180 | Cyclization | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal structure | Val | Thr | Phe | Leu | Val | Gly | Leu | Asn | Gln | Tyr | Leu | Val | n/a | ||
| H77 NS5A | Val | Ser | Phe | Arg | Val | Gly | Leu | His | Glu | Tyr | Pro | Val | n/a | ||
| HCV2 peptide | Tyr | Phe | Val | Pro | His | Glu | Ser | Gly | Arg | Val | Val | Leu | -CONH2 | No | |
| HCV3 peptide | Cys | Ser | Phe | Arg | Val | (D)Pro | Cha | His | Glu | Tyr | Pro | Cys | -CONH2 | -S-S- Bridge | |
| HCV4 peptide | R-(Ahx-R)6-Ahx-Ahx- | Cys | Ser | Phe | Arg | Val | (D)Pro | Cha | His | Glu | Tyr | Pro | Cys | -CONH2 | -S-S- Bridge |
| Arginine tag | R-(Ahx-R)6-Ahx-Ahx- | n/a | |||||||||||||
Next, cells were treated with HCV4 peptide and transfected with the IRES reporter construct and either NS5A or GFP. The peptide blocked NS5A-augmented IRES-mediated translation in a dose dependent manner as shown in Fig. 4D.
The HCV4 peptide blocks intracellular virus production
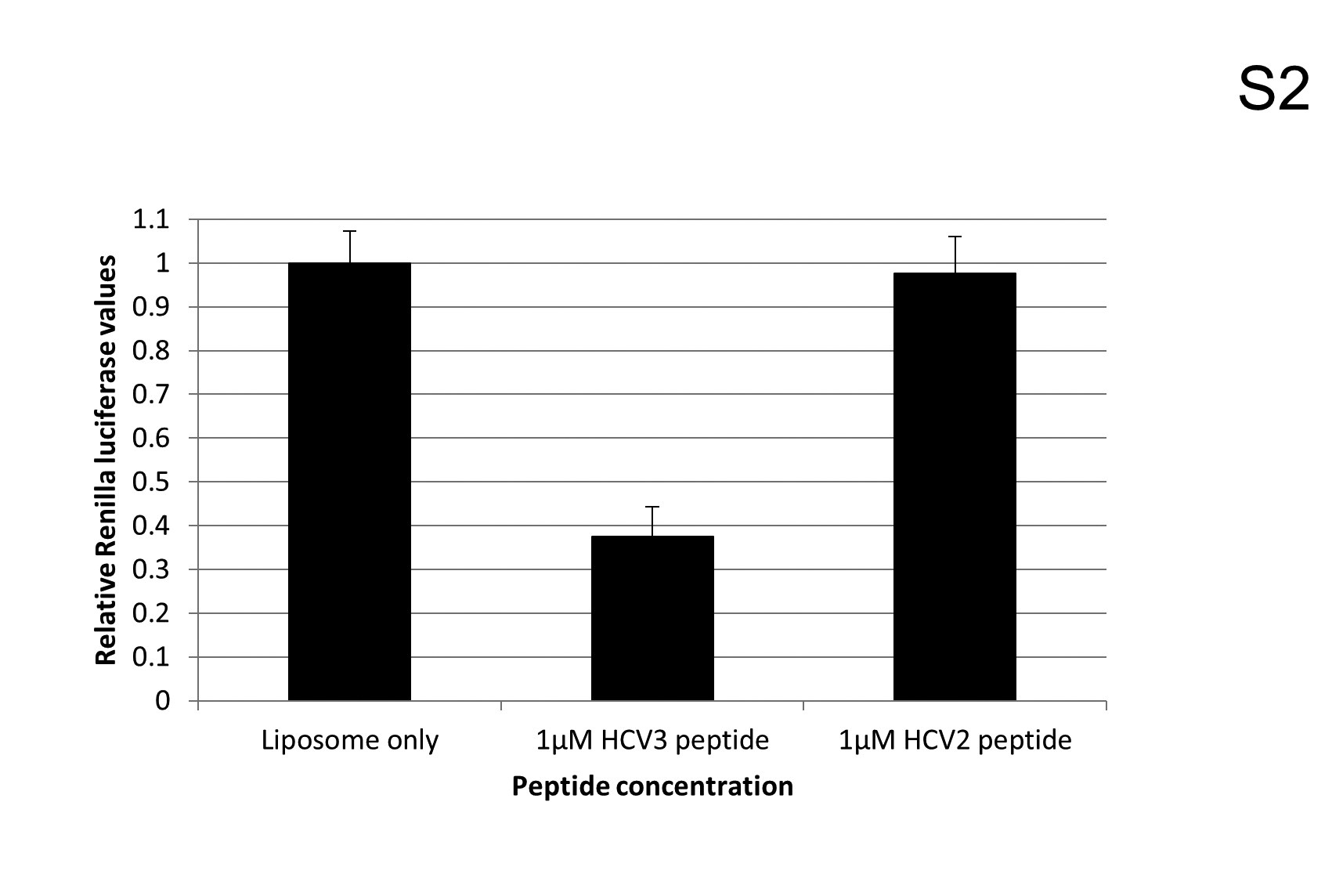
We next sought to assess the antiviral activity of the HCV4 peptide. Huh-7.5 cells were treated with HCV4 in the HCVcc system and immediately infected with the HCV Renilla reporter virus. The HCV4 peptide dramatically suppressed intracellular viral protein production even in picomolar doses, with an estimated IC50 of 500pM, while the arginine tag control peptide (Table 1) by itself did not show any effect (Fig. 4E). Cellular uptake of the peptide was confirmed by fluorescent microscopy of huh-7.5 cells treated with fluorescein isothiocyanate (FITC)-labeled HCV4 (Fig. 5). Further, a non-arginine tagged hairpin peptide (HCV3) (Table 1) transferred into cells using liposomes at 1μM also suppressed viral protein production compared to the sequence-scrambled control peptide (HCV2) (Table 1) (Fig. S2).
Fig. 5.

The HCV4 peptide enters huh-7.5 cells efficiently. Cells were treated with 500nM FITC-HCV4 and washed three times with PBS after 40 minutes and imaged. (A) and (C) Fluorescent images of FITC-HCV4 treated and untreated cells, respectively. (B) and (D) phase-contrast images of the same cells as in panels A and C, respectively.
We further hypothesized that the potent translation inhibitory effect of HCV4 should also result in a significant inhibition of infectious virion secretion and viral RNA replication. Levels of infectious virion secretion were measured by infecting naïve huh-7.5 cells with the supernatant of infected cells. Fig. 6A shows that viral secretion is blocked with HCV4 treatment. Furthermore, quantitative reverse-transcriptase PCR analysis of HCV4-treated cells revealed a significant decrease in viral RNA replication levels (Fig. 6B).
Fig. 6.

HCV4 peptide treatment inhibits infectious virion secretion and viral RNA replication. Huh-7.5 cells were treated with HCV4 and immediately infected with Renilla reporter virus. 48 hours later, the supernatant (A) was used to infect naïve cells. 48 hours post infection, the cells were harvested and assayed for Renilla luciferase activity. (B) Huh-7.5 cells were treated with HCV4 and infected as in part B. 48 hours later, cell lysates were used to quantify viral RNA levels.
The HCV4 peptide blocks NS5A/HSP70 complex formation in vitro and binds to HSP70 in vivo
To determine if disruption of the NS5A/HSP70 complex is the mechanism by which the C34 hairpin might inhibit IRES activity and virus production, we tested the ability of the HCV4 peptide to block NS5A/HSP70 complex formation in vitro. GST pull-down assays were performed as before using GST-HSP70 as bait and NS5A domain I. increasing concentrations of HCV4 was able to competitively block pull-down of NS5A domain I, while the HCV2 sequence scrambled control peptide (Table 1) did not (Fig. 7A). To determine if the HCV4 peptide interacts with HSP70 in vivo, FITC-labeled HCV4 was introduced into huh-7.5 cells followed by immunoprecipitation with anti-FITC antibody or IgG isotype control. Western analysis demonstrated HCV4/HSP70 complex formation (Fig. 7B).
Fig. 7.


The HCV4 peptide blocks NS5A/HSP70 binding in vitro and binds HSP70 in vivo. (A) GST pull-down assays showing that increasing concentrations of the HCV4 peptide competitively blocks pull-down of HSP70 with GST-NS5A while the sequence-scrambled control peptide (Table 1) does not. (B) HSP70 co-immunoprecipitates with HCV4. Huh-7.5 cells were treated with 1μM FITC-HCV4 and harvested 24 hours later for co-immunoprecipitation (co-IP). Co-IP targets were analyzed through Western analysis with antibody against HSP70 demonstrating that the peptide binds HSP70 in vivo. (C) A proposed model of NS5A-augmented IRES-mediated translation. Our data supports the hypothesis that the NS5A/HSP70 complex formation is important for NS5A-augmented IRES-mediated translation. (D) Soluble C34 hairpin peptide derivative inhibits NS5A/HSP70 binding resulting in the inhibition of NS5A-augmented IRES-mediated translation.
Discussion
We and others have previously reported the NS5A/HSP70 interaction (18, 25). However, direct interaction between NS5A and HSPs was not demonstrated due to the use of cellular systems. Here we report a direct interaction between NS5A and HSP70 using purified recombinant proteins and in vitro pull-down assays. This suggests that no other viral or cellular protein is necessary for the interaction between NS5A and HSP70.
Deletion studies of NS5A domain I narrowed down the HSP70-binding site to the C terminus of NS5A domain I (C34). Recently it was shown that NS5A amino acids 221–302 may be responsible for NS5A/HSP70 interaction (26). This region includes most of the N terminus of NS5A domain II and some of the linker peptide between domains I and II. While this region does not directly bind HSP70, as shown by our in vitro interaction studies using purified proteins, it may represent an indirect interaction. In another report, a region of NS5A including the entire domain II and the first few amino acids of domain III was shown to inhibit NS5A-augmented IRES-mediated translation (17). Both of these reports point to a common region in NS5A (i.e. domain II), and thus, it may be possible that NS5A domain II have a negative effect on IRES-mediated translation through (an) adaptor protein(s). However, further biochemical studies are needed to verify (an) indirect interaction(s).
Our data shows that the C34 hairpin is the site of NS5A/HSP70 interaction. This is supported by the fact that the HCV4 peptide is able to block pull-down of HSP70 with NS5A in vitro as well as bind HSP70 in vivo (Fig. 7A and 7B). Furthermore, the C34 hairpin is the only region of C34 with a secondary structure, based on the crystal structure (24). It has been reported that HSP27 also binds NS5A at residues 1–181 (27). It is important to note that the C34 hairpin structure lies exactly at the C terminus of this region. Thus, it may be possible that, in addition to HSP70, other HSPs either interact with the C34 hairpin or are also in complex with HSP70. Recently an alternate crystal structure of dimeric NS5A domain I was published which displays the two domain I units to be closer together closing the proposed RNA-binding cleft (28). Analysis of this structure reveals the two C34 hairpins to be also exposed.
HSP70 binds non-specifically to a large number of hydrophobic peptide sequences (nascent or denatured) through its C-terminal substrate-binding domain (SBD) allowing the client peptide to attain its native conformation (29). The N-terminal nucleotide binding domain (NBD) of HSP70 does not interact with peptides directly. Rather, it binds ATP and hydrolyzes it to ADP to induce conformational changes in SBD to facilitate its function (29). NS5A hydrophobicity plot reveals that C34 is not significantly hydrophobic, and the C34 hairpin is, in fact, hydrophilic (data not shown). Our findings reveal a specific and sequence-dependent interaction between HSP70 and NS5A as there are many regions in NS5A domain I as well as in other domains that are significantly more hydrophobic than C34. This specific interaction is further verified by our discovery that only HSP70-NBD binds NS5A directly. HSP70-NBD is known to interact with proteins that regulate the HSP70 chaperone activity. These proteins include BAG5, co-chaperones of J-domain proteins (such as HSP40), and HSP110 all of which bind HSP70-NBD (29–32). Thus, interactions with NBD of HSP70 are an important regulatory mechanism, and it may be possible that NS5A modulates HSP70 activity as well resulting in increased IRES activity.
NS5A domains II and III have been implicated in viral genome replication and virion assembly (10, 12), and domains I, II, and III have been shown to bind viral RNA (33). None of the NS5A domains, however, have been previously ascribed to regulation of viral IRES-mediated translation. In this study, we have shown that NS5A domain I plays an important role in regulation of IRES-mediated translation through the C34 beta-sheet hairpin structure. We have shown that C34 expression and treatment with the C34 hairpin peptide derivative (HCV4) block NS5A-augmented IRES-mediated translation. This effect is sequence specific as peptides of equal length from NS5A domains II and III fail to produce similar results. We have previously shown that Quercetin treatment and HSP70 knock-down block NS5A-augmented IRES-mediated translation (18). Taken together, our data implicates the C34 hairpin of NS5A domain I in the regulation of viral IRES-mediated translation.
Our current model of NS5A-augmented IRES-mediated translation consists of a complex of NS5A, HSP70, HSP40, and probably additional, yet unidentified, factors such as the ribosome which may stabilize the translation complex (Fig. 7C and 7D). An RNA-binding cleft between the two claw-like domain I units has been predicted in the crystal structure (24) (Fig. S1). It has also been reported that HSP70 (specifically the HSPA1A isoform used in this study) is able to bind to the 40S ribosomal subunit (34). Our finding that NS5A directly binds HSP70, therefore, provides a possible link between the NS5A dimers and the ribosome both of which interact with the RNA template. This NS5A/HSP70 interaction may be responsible for stabilizing the components of translation machinery on the RNA template during translation initiation and/or elongation.
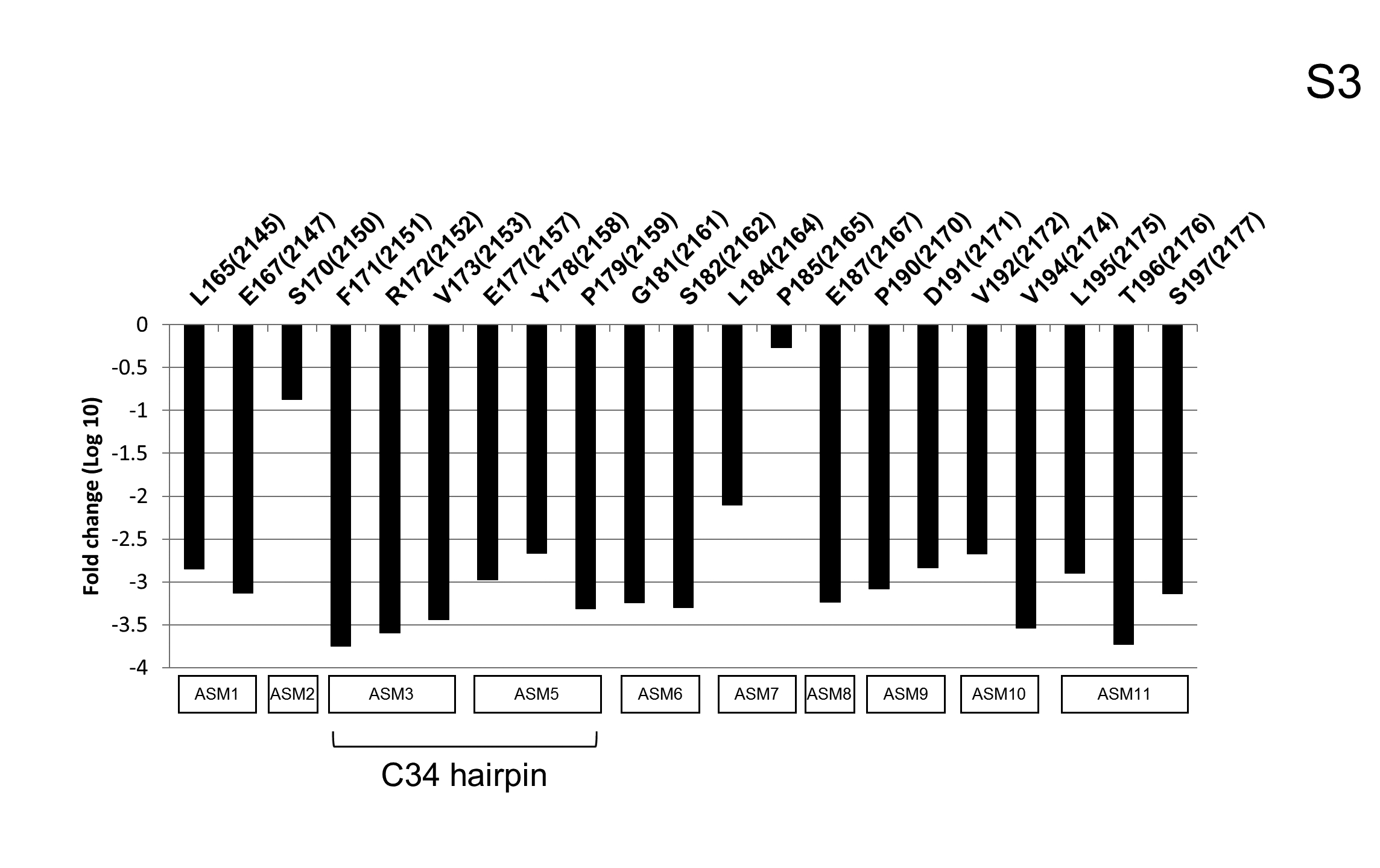
The significance of C34 region and the hairpin within is further underlined by the mutation analysis of HCV genome (20). As shown in Fig. S3, the majority of insertion mutants in C34 and all mutants within the hairpin are detrimental for the virus. Quite interestingly, mutation of the region immediately upstream of the hairpin and the region a few amino acids downstream are well tolerated (Fig. S3). This data supports our hypothesis that the C34 hairpin is critical for viral proliferation.
It is known that in the context of chronic HCV infection, a number of viral proteins including NS5A are able to dramatically alter host gene expression to evade the innate immune response to viral infection resulting in the low efficacy of the currently available pegylated interferon-α (PEG-IFN) and ribavirin treatment. Our findings demonstrate the significance of the NS5A/HSP70 interaction and the role of HSPs in HCV life cycle, in particular, IRES-mediated viral protein translation. Therefore, the C34 modified hairpin peptide may be a candidate for HCV therapy. Furthermore, considering the potency of the C34 hairpin peptide in suppressing viral translation levels, treatment with this peptide may significantly improve the efficacy of PEG-IFN and ribavirin treatment in patients resistant to these compounds or allow for IFN-free therapy in combination with other antiviral agents, which may be beneficial for patients unable to receive IFN therapy.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
Financial support:
NIH 1R01DK090794-01A1, SWF.
NIH AI084090, AD.
California Center for Antiviral Drug Discovery grant, University of California Office of the President, MRPI # 143226, AD.
The authors thank David Dawson and Mike Kovochich for helpful discussions, Charles Rice for providing huh-7.5 cells, and Anne Kovochich, George Yeh, Jeffrey Calimlim, and the Flow Cytometry Core Laboratory at UCLA for technical assistance.
List of abbreviations
- HCV
hepatitis C virus
- HCC
hepatocellular carcinoma
- PEG-IFN
pegylated interferon-α
- SVR
sustained virologic response
- NS5A
non-structural protein 5A
- NCR
non-coding region
- IRES
internal ribosomal entry site
- HSP
heat shock protein
- PCR
polymerase chain reaction
- His6
hexa-histidine
- ASM
alanine scan mutagenesis
- GST
glutathione S-transferase
- Fmoc
9-fluorenylmethyloxycarbonyl
- RP-HPLC
reverse-phase high performance liquid chromatography
- MALDI-MS
matrix-assisted laser desorption ionization spectrometry
- FITC
fluorescein isothiocyanate
- IP
immunoprecipitation
- ORF
open reading frame
- C34
C-terminal 34 amino acids of NS5A domain I
- HCVcc
HCV cell culture
- NBD
nucleotide binding domain
- SBD
substrate binding domain
- FLuc
Firefly luciferase
- RLuc
Renilla luciferase
- APC
allophycocyanin
Contributor Information
Ronik Khachatoorian, Email: RnKhch@ucla.edu.
Vaithilingaraja Arumugaswami, Email: VArumugaswami@mednet.ucla.edu.
Piotr Ruchala, Email: PRuchala@mednet.ucla.edu.
Santanu Raychaudhuri, Email: SRaychau@ucla.edu.
Eden M. Maloney, Email: EMaloney@ucla.edu.
Edna Miao, Email: MiaoEdna@ucla.edu.
Asim Dasgupta, Email: Dasgupta@ucla.edu.
Samuel W. French, Email: SFrench@mednet.ucla.edu.
References
- 1.Shepard CW, Finelli L, Alter MJ. Global epidemiology of hepatitis C virus infection. The Lancet infectious diseases. 2005;5:558–567. doi: 10.1016/S1473-3099(05)70216-4. [DOI] [PubMed] [Google Scholar]
- 2.El-Serag HB. Hepatocellular carcinoma: an epidemiologic view. Journal of clinical gastroenterology. 2002;35:S72–78. doi: 10.1097/00004836-200211002-00002. [DOI] [PubMed] [Google Scholar]
- 3.Gambarin-Gelwan M, Jacobson IM. Optimal dose of peginterferon and ribavirin for treatment of chronic hepatitis C. Journal of viral hepatitis. 2008;15:623–633. doi: 10.1111/j.1365-2893.2008.01018.x. [DOI] [PubMed] [Google Scholar]
- 4.McHutchison JG, Patel K. Future therapy of hepatitis C. Hepatology. 2002;36:S245–252. doi: 10.1053/jhep.2002.36795. [DOI] [PubMed] [Google Scholar]
- 5.El-Serag HB. Hepatocellular carcinoma: recent trends in the United States. Gastroenterology. 2004;127:S27–34. doi: 10.1053/j.gastro.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 6.Butt AA, Skanderson M, McGinnis KA, Kwoh CK, Justice AC. Real-life Rates of treatment completion for HCV. Hepatology. 2007;46:364A. [Google Scholar]
- 7.Falck-Ytter Y, Kale H, Mullen KD, Sarbah SA, Sorescu L, McCullough AJ. Surprisingly small effect of antiviral treatment in patients with hepatitis C. Annals of internal medicine. 2002;136:288–292. doi: 10.7326/0003-4819-136-4-200202190-00008. [DOI] [PubMed] [Google Scholar]
- 8.Ciesek S, Manns MP. Hepatitis in 2010: the dawn of a new era in HCV therapy. Nature reviews Gastroenterology & hepatology. 2011;8:69–71. doi: 10.1038/nrgastro.2010.219. [DOI] [PubMed] [Google Scholar]
- 9.Lindenbach BD, Rice CM. Unravelling hepatitis C virus replication from genome to function. Nature. 2005;436:933–938. doi: 10.1038/nature04077. [DOI] [PubMed] [Google Scholar]
- 10.Tellinghuisen TL, Foss KL, Treadaway JC, Rice CM. Identification of residues required for RNA replication in domains II and III of the hepatitis C virus NS5A protein. Journal of virology. 2008;82:1073–1083. doi: 10.1128/JVI.00328-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.He Y, Yan W, Coito C, Li Y, Gale M, Jr, Katze MG. The regulation of hepatitis C virus (HCV) internal ribosome-entry site-mediated translation by HCV replicons and nonstructural proteins. The Journal of general virology. 2003;84:535–543. doi: 10.1099/vir.0.18658-0. [DOI] [PubMed] [Google Scholar]
- 12.Hughes M, Griffin S, Harris M. Domain III of NS5A contributes to both RNA replication and assembly of hepatitis C virus particles. The Journal of general virology. 2009;90:1329–1334. doi: 10.1099/vir.0.009332-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peng L, Liang D, Tong W, Li J, Yuan Z. Hepatitis C virus NS5A activates the mammalian target of rapamycin (mTOR) pathway, contributing to cell survival by disrupting the interaction between FK506-binding protein 38 (FKBP38) and mTOR. The Journal of biological chemistry. 2010;285:20870–20881. doi: 10.1074/jbc.M110.112045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kriegs M, Burckstummer T, Himmelsbach K, Bruns M, Frelin L, Ahlen G, Sallberg M, et al. The hepatitis C virus non-structural NS5A protein impairs both the innate and adaptive hepatic immune response in vivo. The Journal of biological chemistry. 2009;284:28343–28351. doi: 10.1074/jbc.M109.038877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang C, Sarnow P, Siddiqui A. Translation of human hepatitis C virus RNA in cultured cells is mediated by an internal ribosome-binding mechanism. Journal of virology. 1993;67:3338–3344. doi: 10.1128/jvi.67.6.3338-3344.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pacheco A, Martinez-Salas E. Insights into the biology of IRES elements through riboproteomic approaches. Journal of biomedicine & biotechnology. 2010;2010:458927. doi: 10.1155/2010/458927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kalliampakou KI, Kalamvoki M, Mavromara P. Hepatitis C virus (HCV) NS5A protein downregulates HCV IRES-dependent translation. The Journal of general virology. 2005;86:1015–1025. doi: 10.1099/vir.0.80728-0. [DOI] [PubMed] [Google Scholar]
- 18.Gonzalez O, Fontanes V, Raychaudhuri S, Loo R, Loo J, Arumugaswami V, Sun R, et al. The heat shock protein inhibitor Quercetin attenuates hepatitis C virus production. Hepatology. 2009;50:1756–1764. doi: 10.1002/hep.23232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.French SW, Dawson DW, Chen HW, Rainey RN, Sievers SA, Balatoni CE, Wong L, et al. The TCL1 oncoprotein binds the RNase PH domains of the PNPase exoribonuclease without affecting its RNA degrading activity. Cancer letters. 2007;248:198–210. doi: 10.1016/j.canlet.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 20.Arumugaswami V, Remenyi R, Kanagavel V, Sue EY, Ngoc Ho T, Liu C, Fontanes V, et al. High-resolution functional profiling of hepatitis C virus genome. PLoS pathogens. 2008;4:e1000182. doi: 10.1371/journal.ppat.1000182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fields GB, Noble RL. Solid phase peptide synthesis utilizing 9-fluorenylmethoxycarbonyl amino acids. International journal of peptide and protein research. 1990;35:161–214. doi: 10.1111/j.1399-3011.1990.tb00939.x. [DOI] [PubMed] [Google Scholar]
- 22.Wegele H, Muller L, Buchner J. Hsp70 and Hsp90--a relay team for protein folding. Reviews of physiology, biochemistry and pharmacology. 2004;151:1–44. doi: 10.1007/s10254-003-0021-1. [DOI] [PubMed] [Google Scholar]
- 23.Gozdek A, Zhukov I, Polkowska A, Poznanski J, Stankiewicz-Drogon A, Pawlowicz JM, Zagorski-Ostoja W, et al. NS3 Peptide, a novel potent hepatitis C virus NS3 helicase inhibitor: its mechanism of action and antiviral activity in the replicon system. Antimicrobial agents and chemotherapy. 2008;52:393–401. doi: 10.1128/AAC.00961-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tellinghuisen TL, Marcotrigiano J, Rice CM. Structure of the zinc-binding domain of an essential component of the hepatitis C virus replicase. Nature. 2005;435:374–379. doi: 10.1038/nature03580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Randall G, Panis M, Cooper JD, Tellinghuisen TL, Sukhodolets KE, Pfeffer S, Landthaler M, et al. Cellular cofactors affecting hepatitis C virus infection and replication. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:12884–12889. doi: 10.1073/pnas.0704894104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen YJ, Chen YH, Chow LP, Tsai YH, Chen PH, Huang CY, Chen WT, et al. Heat shock protein 72 is associated with the hepatitis C virus replicase complex and enhances viral RNA replication. The Journal of biological chemistry. 2010;285:28183–28190. doi: 10.1074/jbc.M110.118323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Choi YW, Tan YJ, Lim SG, Hong W, Goh PY. Proteomic approach identifies HSP27 as an interacting partner of the hepatitis C virus NS5A protein. Biochemical and biophysical research communications. 2004;318:514–519. doi: 10.1016/j.bbrc.2004.04.052. [DOI] [PubMed] [Google Scholar]
- 28.Love RA, Brodsky O, Hickey MJ, Wells PA, Cronin CN. Crystal structure of a novel dimeric form of NS5A domain I protein from hepatitis C virus. Journal of virology. 2009;83:4395–4403. doi: 10.1128/JVI.02352-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mayer MP, Bukau B. Hsp70 chaperones: cellular functions and molecular mechanism. Cellular and molecular life sciences: CMLS. 2005;62:670–684. doi: 10.1007/s00018-004-4464-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arakawa A, Handa N, Ohsawa N, Shida M, Kigawa T, Hayashi F, Shirouzu M, et al. The C-terminal BAG domain of BAG5 induces conformational changes of the Hsp70 nucleotide-binding domain for ADP-ATP exchange. Structure. 2010;18:309–319. doi: 10.1016/j.str.2010.01.004. [DOI] [PubMed] [Google Scholar]
- 31.Polier S, Dragovic Z, Hartl FU, Bracher A. Structural basis for the cooperation of Hsp70 and Hsp110 chaperones in protein folding. Cell. 2008;133:1068–1079. doi: 10.1016/j.cell.2008.05.022. [DOI] [PubMed] [Google Scholar]
- 32.Vembar SS, Jin Y, Brodsky JL, Hendershot LM. The mammalian Hsp40 ERdj3 requires its Hsp70 interaction and substrate-binding properties to complement various yeast Hsp40-dependent functions. The Journal of biological chemistry. 2009;284:32462–32471. doi: 10.1074/jbc.M109.000729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Foster TL, Belyaeva T, Stonehouse NJ, Pearson AR, Harris M. All three domains of the hepatitis C virus nonstructural NS5A protein contribute to RNA binding. Journal of virology. 2010;84:9267–9277. doi: 10.1128/JVI.00616-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cornivelli L, Zeidan Q, De Maio A. HSP70 interacts with ribosomal subunits of thermotolerant cells. Shock. 2003;20:320–325. doi: 10.1097/01.shk.0000082443.66379.d9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.