

Abstract
Animal models that mimic human diabetic nephropathy are useful to identify key factors in pathogenesis of this disease as well as the development of new therapies. Several mouse models of diabetes have features of human diabetic nephropathy, yet none of these completely fulfill the Animal Models of Diabetes Complications Consortium criteria and completely reproduce pathological and functional features of the human disease. The Akita mouse carries a mutation in the insulin 2 gene and, to date, only survive as heterozygotes that develop spontaneous type 1 diabetes. Here we show that Akita mice with mutation of both insulin 2 alleles (Akita knockout (KO)) survive if crossed onto the Balb/c background. These mice develop hyperglycemia, more severe albuminuria and mesangial sclerosis compared to heterozygous mice on the same genetic background. Interestingly, crossing these AkitaKO mice with integrin α1KO mice, a model of exacerbated glomerulosclerosis after injury and also on the Balb/c background, resulted in a 16-fold increase in albuminuria, significant mesangial matrix expansion, nodular and diffuse glomerulosclerosis, and a 2-fold increase in glomerular basement membrane thickening when compared to non-diabetic mice. Moreover a significant decline in glomerular filtration was evident in the α1KOAkitaKO mice at 6 months of age. Thus, the integrin α1KOAkitaKO Balb/c mouse represents a promising model presenting with most features of human diabetic nephropathy.
Keywords: Streptozotocin, Balb/c, glomerular filtration rate, nodular glomerulosclerosis, Akita, integrins
INTRODUCTION
Diabetic nephropathy (DN) is now the leading cause of end-stage renal disease in the developed world and its incidence has increased by more than 50% in the past 10 years. It is initially characterized by glomerular hyperfiltration, glomerular and tubular epithelial hypertrophy, and microalbuminuria. In established DN, there is decreased renal function which is accompanied by glomerular basement membrane (GBM) thickening, glomerular sclerosis, and interstitial fibrosis1, 2.
Animal models of DN that closely recapitulate the features of human disease would be useful tools in understanding the pathogenesis and developing new treatment strategies for this devastating disorder. A good model should have the characteristics required by the Animal Models of Diabetes Complications Consortium (AMDCC, http://www.amdcc.org), which includes greater than a 50% decline in glomerular filtration rate (GFR) over the lifetime of the animal; greater than 10-fold increase in albuminuria; greater than a 50% increase in GBM thickness, and typical renal pathologic changes (mesangial matrix expansion; nodular glomerulosclerosis; mesangiolysis; arteriolar hyalinosis; tubulointerstitial fibrosis) 3, 4.
As a platform for developing models of diabetic complications, several mouse lines show promise. db/db mice, which carry a mutation of the leptin receptor gene, have been widely used as models of type 2 diabetes 5, although they only develop mild nephropathy 5, 6. Improvement to this model was made by crossing the db/db with mice lacking the endothelial cell nitric oxide synthase (eNOS) 7. db/db-eNOS−/− mice have hyperglycemia evident by approximately 6-8 weeks of age and exhibit an accelerated model of type 2 DN with increased systolic blood pressure, albuminuria, mesangial expansion, nodular glomerulosclerosis, thickening of the GBM, and decrease in GFR 7. More recently, it was shown that BTBR ob/ob mice, which carry a mutation of the leptin gene, develop progressive proteinuria, glomerular lesions similar to those of advanced human DN with thickening of the GBM, focal arteriolar hyalinosis, mesangiolysis, and focal mild interstitial fibrosis 8. Among the mouse model of type 1 diabetes, the C57Bl/6 eNOS−/− mouse is the best option for studying features of advanced DN. Upon injection with streptozotocin, the C57Bl/6 eNOS−/− mice develop nephropathy and produce lesions that closely mimic human glomerulopathy and tubulointerstitial disease 9. However, streptozotocin-mediated DN comes with pitfalls including drug toxicity and drug instability. Thus, animal models that develop spontaneous diabetes are preferable. The Akita mouse is one of the first described mouse models for spontaneous type 1 diabetes. These mice have a single nucleotide substitution in the insulin 2 gene (C96Y), originally identified as a spontaneous mutation in a colony of C57BL/6 mice 10. Heterozygosity for the mutation in male C57BL/6 mice (+/C96Y, here referred to as Akita-het) is associated with marked hyperglycemia, while homozygosity (C96Y/C96Y, here referred to as AkitaKO) leads to perinatal lethality. However, the renal injury in heterozygous mice is only moderate, which is consistent with the well accepted finding that C57BL/6 mice are more resistant to renal injures than other backgrounds 11, 12. Efforts have been made to improve this model by crossing the Akita mice into different genetic backgrounds11, 12, deleting the angiotensin converting enzyme-2 13 or the bradykinin receptors 14. Yet, while progress has been made, none of these crosses have yielded a robust mouse model that reproduces the pathological and functional features of human DN 3.
In an attempt to make the Akita a better model of DN, we crossed these mice onto the Balb/c background. Unlike other backgrounds, Akita mice with both insulin 2 alleles mutated (AkitaKO) survive on the Balb/c background. These mice developed more severe DN than heterozygous litters (Akitahet), which included mesangial expansion, glomerulosclerosis, and GBM thickening. However, no nodular glomerulosclerosis or significant GFR decline was observed in the AkitaKO mice. For this reason, we hypothesized that crossing the Balb/c AkitaKO with a mouse model of exacerbated glomerular injury might result in more severe DN. One such model is the integrin α1-null (α1KO) mouse, which we have previously shown to develop more severe glomerular injury following adriamycin-induced nephropathy 15 or streptozotocin-induced diabetes 16, especially on a Balb/c background. We therefore crossed the AkitaKO with integrin α1KO mice to determine whether the α1KOAkitaKO mouse developed more severe DN. We show that BALB/c α1KOAkitaKO mice show a 16-fold increase in albuminuria, severe glomerulosclerosis with nodules, a significant decline of GFR over their lifetime, and 2 fold thickening of the GBM when compared to non-diabetic Balb/c mice. Thus, the Balb/c integrin α1KOAkitaKO mice develop a series of renal abnormalities that closely resemble advanced human DN, making this strain particularly attractive for testing therapeutic interventions.
RESULTS
BALB/c Akita mice with both insulin 2 alleles mutated (AkitaKO) are viable and develop DN
In an attempt to generate better mouse models of DN, Akita mice (originally on the C57BL/6 background) were backcrossed onto the Balb/c background. Balb/c Akita with both insulin 2 alleles mutated (AkitaKO) can be generated (Supplemental Figs. 1A and 1B) and they survive up to 6 months of age, which contrasts with C57BL/6 mice which only survive as heterozygotes. When the AkitaKO mice were followed over time, they showed significant reduction in body weight at 4 and 6 months of age, while Akita-het mice maintained a body weight similar to that of non-diabetic mice (Fig. 1A). Although the AkitaKO mice can survive up to 6 months of age, ~ 20% had to be euthanized prior to this time because of failure to thrive. To determine if there were differences in renal function between the Akita-het and AkitaKO mice we measured GFR at the age of 4 and 6 months (Fig. 1B). Early stages of DN are often characterized by hyperfiltration, while a decline in GFR is a marker of disease progression to end stage kidney failure 17. The GFR was significantly elevated in Akita-het mice at 6 month; however, in the AkitaKO, increased GFR was already evident at 4 months and it was decreased by 6 months. When albuminuria was assessed by measuring albumin-to-creatinine (ACR) ratio, there was approximately a 3 fold increase in ACR in both 4 month Akita-het and AkitaKO mice compared to non-diabetic mice (Fig 1C). Increased albuminuria only persisted in the AkitaKO mice at 6 months of age (Fig 1C).
Figure 1. Physiological parameters of AkitaKO Balb/c mice.
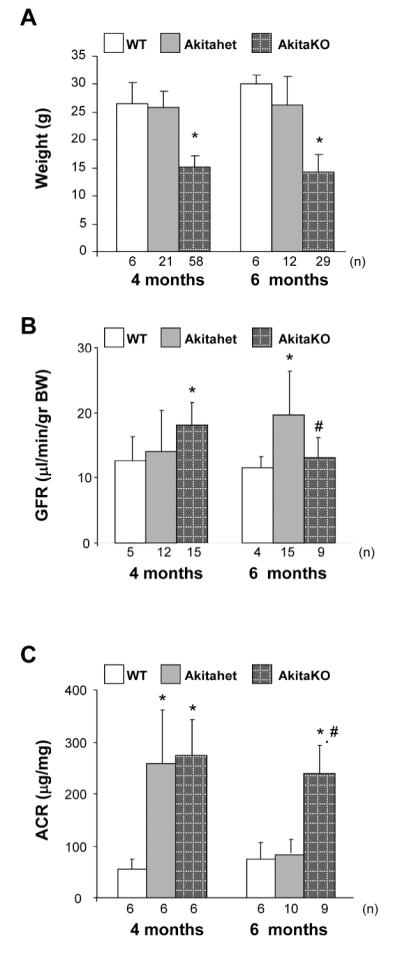
Body weight (A), GFR (B) and urine albumin excretion (C) in 4 and 6 months old wild type (WT), Akita-het and AkitaKO mice BALB/c mice. Values represent the mean +/− SD of the mice indicated. Differences between non-diabetic and Akita-het or AkitaKO mice (*); or Akita-het and AkitaKO within the same age group (#) were significant with p < 0.05.
When the kidneys were subjected to pathological examination, mesangial matrix expansion was present in both Akita-het and AkitaKO mice at the 4 month time point, although it was more pronounced in the latter group as assessed by a glomerulosclerosis score (Figs. 2A and 2B). The same degree of glomerular injury was present in both genotypes at six months of age and there was no evidence of tubular injury (Figs. 2C and 2D).
Figure 2. Histopathology of kidneys from AkitaKO Balb/c mice.

(A, C) Representative light micrographs of Periodic acid-Schiff-stained kidney sections from WT, Akita-het and AkitaKO mice at 4 (A) and 6 (C) months of age. (B, D) Mesangial sclerosis index was evaluated in kidneys from 4 (B) and 6 (D) months old non diabetic and diabetic mice in a blinded fashion and the percent of mesangial matrix occupying the glomerulus scored as 1 = 0-24%, 2 = 25-49%, 3 = 50-74% and 4 = >75%. Data are presented as the mean +/− SD of 120 glomeruli (30 glomeruli/mouse with a total of 4 mice evaluated). Both Akita-het and AkitaKO mice developed mild to moderate mesangial matrix expansion at 4 and 6 months of age compared with the control WT mice. * and # are as in Figure 1.
Taken together these data verify that Akita homozygote mice can be generated on a Balb/c background and they develop more accelerated DN than heterozygote mice on the same background. Despite this increased rate of disease development, the Akita homozygote mice only develop mild glomerular injury primarily characterized by mesangial expansion.
AkitaKO mice crossed with the integrin α1KO mice develop robust DN
In order to develop a more robust mouse model of DN than that seen in the AkitaKO mice on the Balb/c background, we crossed the AkitaKO mice with integrin α1-null (α1KO) mice on the same background. The rationale behind this cross is threefold: 1) integrin α1β1 is a negative regulator of collagen homeostasis and loss of this receptor leads to increased collagen synthesis 18; 2) mice lacking integrin α1β1 develop excessive fibrosis following injury 15, 16; and 3) integrin α1KO Balb/c mice develop more exacerbated glomerular injury compared to wild type mice following induction of type 1 diabetes 16. Integrin α1KOAkitaKO mice were successfully generated (Supplemental Figs. 1A and 1B) and these mice developed similar levels of hyperglycemia when compared to AkitaKO throughout the duration of the study (Supplemental Fig. 1C). Both AkitaKO and α1KOAkitaKO showed a significant drop in weight relative to non-diabetic wild type or integrin α1KO mice at both 4 and 6 months of age (Fig. 3A). Approximately 40% of the α1KOAkitaKO mice had to be euthanized before completion of the study because of failure to thrive.
Figure 3. Physiological parameters of integrin α1KOAkitaKO Balb/c mice.

Body weight (A), GFR (B) and urine albumin excretion (C) in 4 and 6 months old wild type (WT), AkitaKO and integrin α1KOAkitaKO mice BALB/c mice. Values represent the mean +/− SD of the mice indicated.
Differences between non-diabetic and AkitaKO or α1KOAkitaKO mice (*); or AkitaKO and α1KOAkitaKO within the same age group (#); or 4 months vs. 6 months old α1KOAkitaKO mice (**) were significant with p<005.
When GFR was measured in the mice at 4 and 6 months, there was an equal rise in GFR in the AkitaKO and α1KOAkitaKO mice at 4 months. At 6 months of age, GFR declined in both AkitaKO and α1KOAkitaKO mice, however this drop was only significant in the latter group with an overall ~50% decline compared to 4 months diabetic mice (Fig. 3B). No changes in GFR were observed between non-diabetic wild type and integrin α1KO mice throughout the study (Fig. 3B). Measurement of ACR in 4 and 6 months old mice revealed ~ 3 fold increase in albumin excretion in AkitaKO mice vs. ~ 16 fold increase in the α1KOAkitaKO mice compared to non-diabetic mice (Fig 3C).
Increased glomerular injury in diabetic integrin α1KOAkitaKO mice
We next defined the histopathological features of the kidneys of the α1KOAkitaKO mice at 4 and 6 months in order to define the severity of disease in these mice. There was significantly increased mesangial matrix expansion (Figs. 4A and 4C) and glomerulosclerosis score (Figs. 4B and 4D) in the α1KOAkitaKO when compared to the AkitaKO mice at both 4 and 6 months. Due to the severity of the glomerular injury in these mice we carefully defined the histological lesions found within the glomeruli of the α1KOAkitaKO mice. As shown in Figure 5, there was evidence of diffuse and nodular mesangial sclerosis as well as mesangiolysis seen under light microscopy. The mesangial expansion was confirmed on electron microscopy in both the AkitaKO and α1KOAkitaKO mice at 6 months; however it was much more severe in the latter group (Fig. 6A). Furthermore, there was GBM thickening in both genotypes compared to non-diabetic controls, which was significantly more severe in the α1KOAkitaKO mice (Figs. 6A and 6B). Collagen IV is the most abundant extracellular matrix (ECM) component in the glomerulus, and its upregulation has been described in many glomerular diseases, including DN 16. Analysis of 6 months old mice revealed increased glomerular collagen IV deposition in both AkitaKO and α1KOAkitaKO mice compared to non-diabetic mice, although it was more abundant in the latter group (Fig. 7A and 7B). Furthermore, excessive fibrillar collagen deposition in the α1KOAkitaKO mice was confirmed by trichrome staining in both diffuse and nodular mesangial lesions (Fig. 7C).
Figure 4. Histopathology of kidneys from α1KOAkitaKO Balb/c mice.

(A, C) Representative light micrographs of Periodic Acid Shiff-stained kidney sections from WT, integrin α1KO, AkitaKO and α1KOAkitaKO mice at 4 (A) and 6 (C) months of age. Both AkitaKO and α1KOAkitaKO mice developed mesangial matrix expansion at 4 months of age, although it was more severe in the latter group (A). The increased mesangial expansion was worse in the α1KOAkitaKO mice at 6 months of age and there was evidence of nodular glomerulosclerosis (arrow), which was not present in the AkitaKO mice (C). (B, D) Mesangial sclerosis index was evaluated in kidneys from the mice indicated above and the percentage of mesangial matrix occupying the glomerulus scored as described in Figure 1. Data are presented as the mean +/− SD of 120 glomeruli (30 glomeruli/mouse with a total of 4 mice evaluated). Although both AkitaKO and α1KOAkitaKO mice developed glomerular injury at 4 and 6 months of age, the degree of injury was significantly higher in the latter group. *, **, and # are as in Figure 3.
Figure 5. Characterization of the glomerular injury in the α1KOAkitaKO Balb/c mice.

Representative light micrographs of Jone’s (A, B), Periodic Acid Shiff (C), and Masson’s trichrome (D) stained glomeruli from 6 months old α1KOAkitaKO mice. Note the presence of nodular glomerulosclerosis (a), mesangial expansion (b), diffuse glomerulosclerosis (c), and mesangiolysis (d).
Figure 6. Increased mesangial matrix expansion and GBM thickening in α1KOAkitaKO Balb/c mice.

(A) Electron microscopy pictures of glomeruli of 6 month old WT, AkitaKO and α1KOAkitaKO mice. The upper panels demonstrate increased mesangial expansion (asterisk) in the AkitaKO mice, which is significantly worse in the α1KOAkitaKO mice. Scale bar, 2 μm. Obvious increase in the thickness of the GBM was evident in both AkitaKO and α1KOAkitaKO mice at 6 months (lower panel in A; scale bar, 2 μm) although it was more prominent in the latter group (B). Values in B represent the mean +/− SD of 30 measurements. Differences between (*) or AkitaKO vs. α1KOAkitaKO mice (#) were significant (p<0.05).
Figure 7. Increased glomerular matrix deposition in α1KOAkitaKO Balb/c mice.

(A) Collagen IV staining of kidney sections from 6 months old WT, integrin α1KO, AkitaKO and α1KOAkitaKO mice. (B) The levels of collagen IV per glomerulus were quantified using Bioquant Image analysis software as described in Materials and Methods. Data represent the mean +/− SD of 120 glomeruli (20 glomeruli/mouse with 6 mice analyzed). * and # are as in Figure 3. (B) Representative light micrographs of Masson trichrome-stained glomeruli from 6 months old AkitaKO and α1KOAkitaKO mice. Note the presence of fibrillar collagen (blue) and the nodular pattern in the glomeruli of the α1KOAkitaKO mice only.
One of the features of the glomerular lesions of diabetic nephropathy is collapse of the glomerular tufts due to scar tissue consisting of ECM 19. To define the area occupied by the glomerular capillaries, we stained kidney frozen sections of 6 months old non-diabetic and diabetic mice with CD31, a well-defined marker of blood vessels. Compared to non-diabetic mice, there were significantly decreased CD31 positive structures in the glomeruli of both AkitaKO and α1KOAkitaKO mice and this reduction was much more evident in the latter group (Fig. 8).
Figure 8. Decreased glomerular capillary loops in α1KOAkitaKO Balb/c mice.

(A) CD31 staining of kidney sections from 6 months old wild type (WT), AkitaKO and α1KOAkitaKO mice. The glomerular area is marked by dotted lines. (B) The degree of vascularization per glomerulus was quantified using Scion Image as described in Materials and Methods. Data represent the mean +/− SD of 20 glomeruli (5 glomeruli/mouse with 4 mice analyzed). * and # are as in Figure 3.
Taken together these results demonstrate that α1KOAkitaKO mice develop more severe DN than AkitaKO mice, that is characterized by the development of excessive albuminuria, severe glomerulosclerosis, which is both diffuse and nodular, mesangiolysis, marked thickening of the GBM, excessive collagen deposition, significant decline in GFR and decreased glomerular capillary loops over the lifetime of the mouse.
No significant tubulointerstitial fibrosis is evident in diabetic integrin α1KOAkitaKO mice
One of the hallmarks of late stages of DN is the development of tubulointerstitial fibrosis. On examination by light microscopy of the kidneys, no evidence of severe tubulointerstitial fibrosis was seen (Figs. 2 and 4). However, we wanted to define subtle changes that might be present in the tubulointerstitium of the AkitaKO and α1KOAkitaKO mice. We therefore examined the tubulointerstitium by electron microscopy to define tubular morphology and tubular basement membrane thickness in the kidneys of 6 month old wild type, AkitaKO and α1KOAkitaKO mice (Fig 9A). No differences in any of the parameters analyzed were seen between the three genotypes. We next co-stained kidney paraffin sections for collagen I and Dolichos biflorus agglutinin [a glycoprotein that binds the apical aspect of CD cells, 20] to define whether there was increased collagen I in the tubulointerstitial compartment (Fig 9B). A small, but equal increase in collagen I staining was seen in both AkitaKO and α1KOAkitaKO mice when compared to non-diabetic wild type animals. When we immunostained with anti-α-smooth muscle actin antibodies to define whether there were increased interstitial fibroblasts or if tubular cells had acquired a more mesenchymal morphology, no differences were seen between the genotypes (Fig 9C). Finally, no differences in tubular integrity were seen in the different genotypes when kidneys were immunostained with anti-E-cadherin antibodies. Thus, other than a minimal increase in collagen I expression, there was negligible tubulointerstitial disease in the AkitaKO and α1KOAkitaKO mice at 6 months of age.
Figure 9. No significant evidence of tubulointerstitial fibrosis in α1KOAkitaKO Balb/c mice.

(A) Electron microscopy pictures of tubules of 6 month old WT, AkitaKO and α1KOAkitaKO mice. Scale bar, 2 μm. The values (in nm) represent the mean +/− SD of tubular basement membrane thickness of 30 measurements. (B) Paraffin kidney sections were co-stained with Dolichos biflorus agglutinin (DBA) and anti-collagen I (upper panel) or anti-α-smooth muscle action (α-SMA, lower panel). Merged images are shown. V = vessel (C) Paraffin kidney sections were co-stained with anti-mouse E-cadherin antibodies or DAPI. Merged images at both low and high magnification are shown.
DISCUSSION
The overall goal of this study was to use the Akita mouse as a platform to generate a mouse model that would fulfill many of the AMDCC criteria and reproduce most, if not all, the pathological and functional features of human DN. Whereas most Akita mice strains generated so far only survive as heterozygotes, we show that AkitaKO mice can be generated when crossed onto the BALB/c background. These mice develop more severe albuminuria and mesangial sclerosis than heterozygous mice. We also show that crossing the AkitaKO mice with integrin α1KO mice results in a 16-fold increase in albuminuria, diffuse and nodular mesangial sclerosis, mesangiolysis and a 2-fold increase in GBM thickening compared to non-diabetic mice. Finally, 6 months old α1KOAkitaKO mice had a significant decline (~ 50%) in glomerular filtration compared to 4 months old mice of the same genotype. Thus, we propose that the integrin α1KOAkitaKO mice Balb/c mouse have many of the features of human DN. In addition they fulfill many features of a good mouse model of DN as described by the AMDCC, which include a 10-fold increase in urinary albumin excretion, mesangiolysis, mesangial expansion, nodular glomerulosclerosis, hyalinosis, tubular interstitial fibrosis, 50% increase in GBM thickening, and 50 decline in GFR over the lifetime of the animal 3.
Our studies agree with the observation that the Akita mouse is a promising model of spontaneous type 1 DN 11. This mouse carries a mutation in the insulin 2 gene and heterozygotes mice develop spontaneous type 1 diabetes 10. The Akita-het mouse presents sustained and robust hyperglycemia, enhanced levels of albuminuria, and mesangial pathology. However, the extent of proteinuria and mesangial pathology is relatively modest in the original line of C57BL/6 Akita-het mice 11. As the genetic background has a strong influence on the characteristics of kidney injury associated with DN 21, novel lines of inbred Akita-het mice were generated resulting in increased albuminuria in these mice 12. However, parameters such as GFR and pathologic glomerular changes still did not meet the AMDCC criteria 3.
In this study we successfully generated Balb/c Akita mice with either one or both insulin-2 genes mutated, namely the Balb/c Akita-het or AkitaKO mice. Whereas Balb/c mice appear to be relatively resistant to diabetic kidney disease 11, 22, Balb/c Akita-het mice develop a moderate increase of GFR, 2-fold increase in albuminuria, and glomerulosclerosis within 6 months of age. This result agrees with the finding obtained with Akita-het on the C57BL/6 background 12. Balb/C AkitaKO mice have more pronounced proteinuria (3 fold increased over non diabetic mice), mesangial expansion and glomerulosclerosis and while their GFR is significantly increased in 4 months old mice, a ~22% decline is observed at 6 months when the mice usually die. Although promising, these mice do not completely meet the AMDCC criteria which require 50% decline in GFR over the lifetime and greater than a 10 fold increase in albuminuria.
In order to generate a more robust mouse model of type 1 DN, we crossed the AkitaKO mice onto the integrin α1-null background. Integrin α1β1 is a major collagen IV receptor, highly expressed by glomerular cells, that plays a key role in negatively regulating the degree of glomerulosclerosis 15, 16, 18. We have shown that integrin α1β1 is a negative regulator of glomerular collagen synthesis by negatively regulating EGF receptor activation, generation of pro-fibrotic ROS and consequent collagen levels 23, 24. More recently, we showed that integrin α1β1 also exerts its anti-fibrotic role by regulating both the level and phosphorylation state of caveolin-1 24, 25, a scaffolding protein that alters EGF receptor activation. Thus, integrin α1β1 protects mice from glomerular injury by controlling many cell signaling pathways that regulate ROS generation and collagen synthesis. Consistent with these finding, Balb/c integrin α1KO mice develop severe albuminuria, glomerulosclerosis, thickening of the GBM, and altered GFR following injection with streptozotocin 16. Here we show that Balb/C AkitaKO mice crossed with the integrin a1KO mice background, show 16-fold increase in albuminuria, significant mesangial matrix expansion, glomerulosclerosis, nodular glomerulosclerosis, mesangiolysis and 2-fold increase in GBM thickening when compared to non-diabetic mice. Moreover, these mice present hyper-filtration at 4 months of age followed by a ~ 50% decline by 6 months. At 6 months of age GFR in the α1KOAkitaKO mice reached the levels of non-diabetic α1KO mice. Whether these mice could develop further decline in GFR is at this point in time difficult to determine, as they do not survive longer than 6 months. Interestingly, neither the AkitaKO nor the α1KOAkitaKO mice developed significant tubulointerstitial disease, despite the severe glomerular lesions and heavy proteinuria. While this is similar to many mouse models of diabetic nephropathy, it contrasts with the eNOS−/− mice, the db/db mice crossed onto the eNOS−/− background, and the Akita diabetic mice crossed onto the bradykinin receptor null mice where both glomerular and tubulointerstitial changes occur 7, 9, 14, 26. The reason for the lack of tubulointerstitial changes in our model is not known, but because some models do indeed develop tubulointerstitial disease, it is likely dependent on the model or the genetic background of the mice. Nevertheless, these findings do bring into question whether the level of proteinuria corresponds to the severity of interstitial disease in the setting of diabetic nephropathy.
Our studies are a continuation of the quest to generate a better mouse model of DN by investigating animals that have undergone genetic manipulation. One of the best described examples of these mice is the eNOS−/− mouse. When this mouse on the C57BL/6 background is rendered diabetic by the injection of streptozotocin, it develops more severe DN than wild type controls 9. Diabetic eNOS−/− mice developed a 10-fold increase in albuminuria with low-dose and a 40-fold increase with high-dosage streptozotocin, and they also developed significant increases in mesangial expansion, focal sclerosis and tubulointerstitial disease. Although a promising model of DN, diabetes in these mice is induced by streptozotocin, a drug with high toxicity and high instability. In order to obviate streptozotocin treatment, the C57BL/6 eNOS−/− mice were also crossed onto the C57BL/6 Akita mice. The C57BL/6 eNOS−/−/Akita mice die before 5 months of age; however outbred C57/B6×Sv129 eNOS-het/Akita or eNOS−/−/Akita mice develop DN characterized by mesangial expansion, nodular glomerulosclerosis, proteinuria and altered GFR 27. The renal characteristics observed in these outbred mice are very similar to the ones seen in the integrin α1KOAkitaKO mice on the pure Balb/c background.
Interestingly the eNOS−/− mice have been also used to worsen DN in type 2 diabetes induced by crossing the eNOS−/− mice with C57BLKS-db/db mice 7. These mice exhibit marked albuminuria, progressive increase in serum creatinine, and decrease in GFR 7. They also show mesangial expansion, GBM thickening, focal nodular glomerulosclerosis and mesangiolysis, as well as arteriolar hyalinosis 7. These results, together with the ones reported in this study, suggest that crossing the integrin α1KO mouse onto the C57BLKS-db/db mouse might also result in a robust mouse model of type 2 diabetes.
In conclusion, our study suggests that integrin α1KOAkitaKO mice, with their significant decline in GFR, together with the higher than 10 fold increase in albuminuria, and higher than 50% increase in GBM thickening offer a mouse model that carries key features of human DN, making this strain particularly attractive for testing therapeutic interventions.
MATERIALS AND METHODS
Animals
C57BL/6 Akita mice were purchased from The Jackson Laboratory (Bar Harbor, ME). Inbred BALB/c Akita mice were generated by backcrossing the Akita mutation onto the BALB/c background for 10 generations. Transmission of the Akita mutation (C96Y) and successful generation of the Balb/c AkitaKO mice was assessed by PCR analysis of tail genomic DNA (Supplemental Figs. 1A and 1B). To generate the integrin α1KOAkitaKO mice, BALB/c integrin α1KO mice 16 were crossed with BALB/c-AkitaKO mice and F1 siblings (α1hetAkitahet) crossed among themselves to obtain the genotype required. Successful generation of the Balb/c integrin α1KOAkitaKO mice was assessed by PCR analysis of tail genomic DNA (Supplemental Figs. 1A and 1B). Mice were housed in AALAC-accredited animal facility following NIH guidelines. Because of the limited hyperglycemia observed in diabetic female mice, only male mice were studied.
Genotyping
The protocol for the Akita genotyping was obtained from The Jackson Laboratory. Briefly, following PCR amplification of genomic DNA using the sense 5′-TGCTGATGCCCTGGCCTGCT-3′ and antisense 5′-TGGTCCCACATATGCACATG-3′ primers, the PCR products were digested with Fnu4HI enzyme (NEB, R0178s) at 37C overnight and run onto 2% agarose gels (Sigma, St. Louis, MO) for detection of the wild-type (140 bp) and/or the mutant (280 bp) allele. The protocol for genotyping integrin α1-null mice was as previously described 28. Briefly, following PCR amplification of genomic DNA using the wild type 5′GTTGTTCTATTTTTGTAGTTAAC3′; knockout 5′GGGGAACTTCCTGACTAG 3′; and common 5′AATCCTCCATTCGGGTTGGTG 3′ primers, the PCR products were run onto 2% agarose gels of detection of the wild type (103bp) and/or the knockout (273bp) allele. Supplemental Figs. 1A and 1B shows the genotypes of mice used in this study.
Weight and blood glucose measurements
Body weight was measured weekly and expressed in grams. Blood glucose levels were measured monthly between 8:30-10:30 AM using an OneTouch® glucometer (LifeScan, Inc.) and expressed as mg/dL. Approximately 5 μl of blood were collected in conscious mice via tail vein puncture.
Urinary albumin measurement
For the analysis of albuminuria, 24 hour-urine collection was performed in 4 and 6 months old non-diabetic and diabetic mice and the concentration of urine albumin and creatinine was measured using the ELISA Albuwell M test kit (Exocel Inc., Philadelphia, PA). The ratio of albumin/creatinine was expressed as g/mg.
Measurement of glomerular filtration rate (GFR)
FITC-inulin clearance was measured in conscious mice as previously described 22, 29. Briefly, FITC-inulin (5% in PBS) was injected retro-orbitally in 4 and 6 months old non-diabetic and diabetic mice. At 3, 7, 10, 15, 35, 55, and 75 min after the FITC-inulin injection, blood samples were obtained from the lateral saphenous vein 30. Fluorescence intensity was measured at 485 excitation/520 emission using a FLUOstar Omega plate reader (BMG Labtech). Plasma fluorescence was fitted to a two-phase exponential decay using nonlinear regression (GraphPad Prism), and GFR was calculated as using standard formulas and is reported as microliters per minute per gram body weight.
Renal histology
Kidneys were harvested from 4 and 6 months old non-diabetic and diabetic mice for histological analysis. Kidneys were immediately fixed in 4% formaldehyde and then embedded in paraffin. Sections of paraffin-embedded kidneys were either subjected to periodic-acid Schiff (PAS), Masson’s trichrome, or Jone’s staining. The mesangial sclerosis index (MSI) was evaluated in a blinded fashion as previously described 31. Briefly, the percentage of mesangial matrix occupying each glomerulus was rated as 1 = 0-24%, 2 = 25-49%, 3 = 50-74% and 4 = >75%. Kidneys from 6 mice per genotype with a total of 120 glomeruli were analyzed and the mesangial sclerosis index expressed as mean +/− SD.
Immunofluorescence and Immunohistochemistry
Paraffin kidney sections were co-stained with Dolichos biflorus agglutinin and rabbit anti-mouse collagen I antibodies (1:100; Biodesign, Saco, ME) or rabbit-anti-mouse α-smooth muscle actin (1:100, Sigma) followed by rhodamine-conjugated goat anti-rabbit IgG (1:200; Jackson Immunoresearch, West Grove, PA). Paraffin kidney sections were also stained with rabbit anti-mouse E-cadherin (1:100, Sigma) or rabbit anti-mouse collagen IV antibodies (1:100; Biodesign, Saco, ME), followed by rhodamine-conjugated or horseradish peroxidase-conjugated goat anti-rabbit IgG (1:200; Jackson Immunoresearch, West Grove, PA). The levels of glomerular collagen IV deposition were analyzed using Bioquant Image analysis software (Bioquant Image Analysis Corp, Nashville, TN). Collagen IV was expressed as percentage of area occupied by collagen IV positive structures per glomerulus. Kidneys from 6 mice per genotype with a total of 120 glomeruli were evaluated.
Frozen kidney sections were stained with rat anti-mouse CD31 antibodies (1:500, Santa Cruz, CA) followed by rhodamine-conjugated goat anti-rat IgG (1:200, Jackson Immunoresearch). The degree of vascularization, expressed as percentage of area occupied by CD31-positive structures/glomerulus, was evaluated using Scion Imaging Software (Frederick, MD) 32 using 5 glomeruli/mouse and 4 mice/genotype.
Electron Microscopy
Portions of renal cortex from 4 and 6 months old non-diabetic and diabetic mice were fixed in 2.5% glutaraldehyde in phosphate buffer. Samples were postfixed in OsO4, dehydrated in ethanol, and embedded in resin. Glomeruli and tubules were then selected from toluidineblue-stained thick sections and cut for thin sections. Ultrastructural assessment of glomerular and tubular pathologic changes was performed using a Morgagni transmission electron microscope (FEI, Eindhoven, The Netherlands). Glomerular and tubular basement membranethickening was assessed by point to point measurements using a 2X2K camera (Advanced MicroscopyTechniques Corp., Danvers, MA, USA) with the associated digital imaging software. Ten different segments of glomerular or tubular basement membrane per mouse from three mice were measured with a total of 30 measurements in each group.
Statistical analysis
The values for each parameter within a group are expressed as mean ± SD. For comparisons between two experimental groups, an unpaired t-test was used to assess statistical significance. For comparisons among multiple groups, ANOVA with Tukey’s multiple comparisons test was used. p < 0.05 was considered statistically significant.
Supplementary Material
ACKNOWLEDGMENTS
These studies were in part supported by a Merit Review from the Department of Veterans Affairs (AP, RZ, RCH) and the NIH grants: 2P01DK065123 (AP and RZ); DK075594, DK65123 and DK083187 (RZ); DK051265 and DK062794 (RCH); DK061018 (AMDCC) (RCH, AP); AHA established investigator award (RZ); the O’Brien P30DK79341-01 (AP, RZ, RCH).
Footnotes
DISCLOSURE
All the authors declared no competing interests.
REFERENCES
- 1.Schrijvers BF, De Vriese AS, Flyvbjerg A. From hyperglycemia to diabetic kidney disease: the role of metabolic, hemodynamic, intracellular factors and growth factors/cytokines. Endocr Rev. 2004;25:971–1010. doi: 10.1210/er.2003-0018. [DOI] [PubMed] [Google Scholar]
- 2.van Dijk C, Berl T. Pathogenesis of diabetic nephropathy. Rev Endocr Metab Disord. 2004;5:237–248. doi: 10.1023/B:REMD.0000032412.91984.ec. [DOI] [PubMed] [Google Scholar]
- 3.Brosius FC, 3rd, Alpers CE, Bottinger EP, et al. Mouse models of diabetic nephropathy. J Am Soc Nephrol. 2009;20:2503–2512. doi: 10.1681/ASN.2009070721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Breyer MD, Bottinger E, Brosius FC, 3rd, et al. Mouse models of diabetic nephropathy. J Am Soc Nephrol. 2005;16:27–45. doi: 10.1681/ASN.2004080648. [DOI] [PubMed] [Google Scholar]
- 5.Tesch GH, Lim AK. Recent insights into diabetic renal injury from the db/db mouse model of type 2 diabetic nephropathy. Am J Physiol Renal Physiol. 2011;300:F301–310. doi: 10.1152/ajprenal.00607.2010. [DOI] [PubMed] [Google Scholar]
- 6.Sharma K, McCue P, Dunn SR. Diabetic kidney disease in the db/db mouse. Am J Physiol Renal Physiol. 2003;284:F1138–1144. doi: 10.1152/ajprenal.00315.2002. [DOI] [PubMed] [Google Scholar]
- 7.Zhao HJ, Wang S, Cheng H, et al. Endothelial nitric oxide synthase deficiency produces accelerated nephropathy in diabetic mice. J Am Soc Nephrol. 2006;17:2664–2669. doi: 10.1681/ASN.2006070798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hudkins KL, Pichaiwong W, Wietecha T, et al. BTBR Ob/Ob mutant mice model progressive diabetic nephropathy. J Am Soc Nephrol. 2010;21:1533–1542. doi: 10.1681/ASN.2009121290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kanetsuna Y, Takahashi K, Nagata M, et al. Deficiency of endothelial nitric-oxide synthase confers susceptibility to diabetic nephropathy in nephropathy-resistant inbred mice. Am J Pathol. 2007;170:1473–1484. doi: 10.2353/ajpath.2007.060481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yoshioka M, Kayo T, Ikeda T, et al. A novel locus, Mody4, distal to D7Mit189 on chromosome 7 determines early-onset NIDDM in nonobese C57BL/6 (Akita) mutant mice. Diabetes. 1997;46:887–894. doi: 10.2337/diab.46.5.887. [DOI] [PubMed] [Google Scholar]
- 11.Gurley SB, Clare SE, Snow KP, et al. Impact of genetic background on nephropathy in diabetic mice. Am J Physiol Renal Physiol. 2006;290:F214–222. doi: 10.1152/ajprenal.00204.2005. [DOI] [PubMed] [Google Scholar]
- 12.Gurley SB, Mach CL, Stegbauer J, et al. Influence of genetic background on albuminuria and kidney injury in Ins2(+/C96Y) (Akita) mice. Am J Physiol Renal Physiol. 2010;298:F788–795. doi: 10.1152/ajprenal.90515.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wong DW, Oudit GY, Reich H, et al. Loss of angiotensin-converting enzyme-2 (Ace2) accelerates diabetic kidney injury. Am J Pathol. 2007;171:438–451. doi: 10.2353/ajpath.2007.060977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kakoki M, Sullivan KA, Backus C, et al. Lack of both bradykinin B1 and B2 receptors enhances nephropathy, neuropathy, and bone mineral loss in Akita diabetic mice. Proc Natl Acad Sci U S A. 2010;107:10190–10195. doi: 10.1073/pnas.1005144107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen X, Moeckel G, Morrow JD, et al. Lack of integrin alpha1beta1 leads to severe glomerulosclerosis after glomerular injury. Am J Pathol. 2004;165:617–630. doi: 10.1016/s0002-9440(10)63326-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zent R, Yan X, Su Y, et al. Glomerular injury is exacerbated in diabetic integrin alpha1-null mice. Kidney Int. 2006;70:460–470. doi: 10.1038/sj.ki.5000359. [DOI] [PubMed] [Google Scholar]
- 17.Jerums G, Premaratne E, Panagiotopoulos S, et al. The clinical significance of hyperfiltration in diabetes. Diabetologia. 2010;53:2093–2104. doi: 10.1007/s00125-010-1794-9. [DOI] [PubMed] [Google Scholar]
- 18.Gardner H, Broberg A, Pozzi A, et al. Absence of integrin alpha1beta1 in the mouse causes loss of feedback regulation of collagen synthesis in normal and wounded dermis. J Cell Sci. 1999;112(Pt 3):263–272. doi: 10.1242/jcs.112.3.263. [DOI] [PubMed] [Google Scholar]
- 19.Futrakul N, Futrakul P. Vascular homeostasis and angiogenesis determine therapeutic effectiveness in type 2 diabetes. Int J Vasc Med. 2011;2011:971524. doi: 10.1155/2011/971524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Holthofer H, Schulte BA, Spicer SS. Expression of binding sites for Dolichos biflorus agglutinin at the apical aspect of collecting duct cells in rat kidney. Cell Tissue Res. 1987;249:481–485. doi: 10.1007/BF00217319. [DOI] [PubMed] [Google Scholar]
- 21.Breyer MD, Qi Z, Tchekneva EE, et al. Insight into the genetics of diabetic nephropathy through the study of mice. Curr Opin Nephrol Hypertens. 2008;17:82–86. doi: 10.1097/MNH.0b013e3282f49cc9. [DOI] [PubMed] [Google Scholar]
- 22.Qi Z, Fujita H, Jin J, et al. Characterization of susceptibility of inbred mouse strains to diabetic nephropathy. Diabetes. 2005;54:2628–2637. doi: 10.2337/diabetes.54.9.2628. [DOI] [PubMed] [Google Scholar]
- 23.Chen X, Abair TD, Ibanez MR, et al. Integrin alpha1beta1 controls reactive oxygen species synthesis by negatively regulating epidermal growth factor receptor-mediated Rac activation. Mol Cell Biol. 2007;27:3313–3326. doi: 10.1128/MCB.01476-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen X, Whiting C, Borza C, et al. Integrin alpha1beta1 regulates epidermal growth factor receptor activation by controlling peroxisome proliferator-activated receptor gamma-dependent caveolin-1 expression. Mol Cell Biol. 2010;30:3048–3058. doi: 10.1128/MCB.00892-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Borza CM, Chen X, Mathew S, et al. Integrin {alpha}1{beta}1 promotes caveolin-1 dephosphorylation by activating T cell protein-tyrosine phosphatase. J Biol Chem. 2010;285:40114–40124. doi: 10.1074/jbc.M110.156729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakagawa T, Sato W, Glushakova O, et al. Diabetic endothelial nitric oxide synthase knockout mice develop advanced diabetic nephropathy. J Am Soc Nephrol. 2007;18:539–550. doi: 10.1681/ASN.2006050459. [DOI] [PubMed] [Google Scholar]
- 27.Wang CH, Li F, Hiller S, et al. A modest decrease in endothelial NOS in mice comparable to that associated with human NOS3 variants exacerbates diabetic nephropathy. Proc Natl Acad Sci U S A. 2011;108:2070–2075. doi: 10.1073/pnas.1018766108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gardner H, Kreidberg J, Koteliansky V, et al. Deletion of integrin alpha 1 by homologous recombination permits normal murine development but gives rise to a specific deficit in cell adhesion. Dev Biol. 1996;175:301–313. doi: 10.1006/dbio.1996.0116. [DOI] [PubMed] [Google Scholar]
- 29.Qi Z, Whitt I, Mehta A, et al. Serial determination of glomerular filtration rate in conscious mice using FITC-inulin clearance. Am J Physiol Renal Physiol. 2004;286:F590–596. doi: 10.1152/ajprenal.00324.2003. [DOI] [PubMed] [Google Scholar]
- 30.Hem A, Smith AJ, Solberg P. Saphenous vein puncture for blood sampling of the mouse, rat, hamster, gerbil, guinea pig, ferret and mink. Lab Anim. 1998;32:364–368. doi: 10.1258/002367798780599866. [DOI] [PubMed] [Google Scholar]
- 31.Yu L, Border WA, Anderson I, et al. Combining TGF-beta inhibition and angiotensin II blockade results in enhanced antifibrotic effect. Kidney Int. 2004;66:1774–1784. doi: 10.1111/j.1523-1755.2004.00901.x. [DOI] [PubMed] [Google Scholar]
- 32.Pozzi A, Moberg PE, Miles LA, et al. Elevated matrix metalloprotease and angiostatin levels in integrin alpha 1 knockout mice cause reduced tumor vascularization. Proc Natl Acad Sci U S A. 2000;97:2202–2207. doi: 10.1073/pnas.040378497. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.