

Case Report
A 40-year-old school teacher started experiencing headaches and vomiting from November 2011. The headaches were initially waxing and waning, being holocranial and moderately severe. They were usually accompanied by vomiting, which reduced the pain. Some events were more severe and were accompanied by bilateral transient blurring of vision during the peaks of headaches. He was unsure whether he got periods of total freedom pain in between the headaches. The headache and vomiting continued for 1 month, wherein he was seen by his family doctor and given symptomatic treatment, which did not help much.
Three weeks into the illness, on December 7 2011, at 3 pm, he felt his left arm and leg going weak for 2–3 min, for which he was hospitalized and investigated. The episode recurred briefly on the next day as well. At this stage, a computed tomography (CT) scan of the brain was done [Figure 1]. With the clinical features of headache and vomiting and the CT scan showing hyperdensities, a diagnosis of subarachnoid hemorrhage was considered. The patient underwent magnetic resonance (MR) angiography and digital subtraction angiography, which did not reveal any abnormality. He was asked to continue conservative measures at home as a case of non-aneurysmal subarachnoid hemorrhage.
Figure 1.
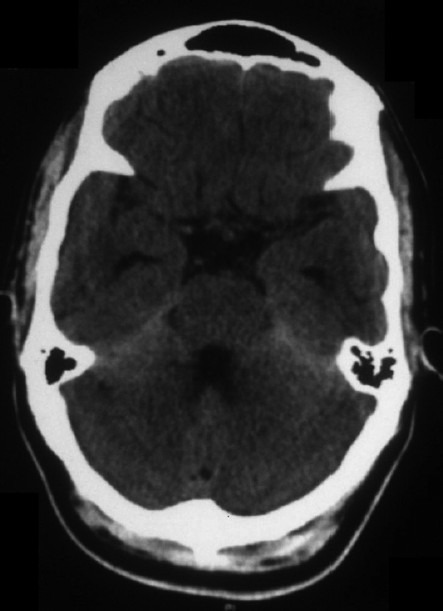
Plain computed tomography scan of the brain showing tentorial hyperdensitis
On January 4 2012, almost 1.5 months into the illness, he noticed painless chewing difficulty. Nasal regurgitation followed and his wife noticed slurring of speech. He went on to develop binocular vertical diplopia in the next few days. He was admitted again and investigated. This time, he underwent a cerebrospinal fluid (CSF) examination, which showed lymphocytic pleocytosis with mild elevation of proteins [Table 1]. With the subacute history of headaches and vomiting and the CSF report, a possibility of chronic lymphocytic meningitis was considered and the patient was started on anti-tubercular drugs with corticosteroids.
Table 1.
Cerebrospinal fluid findings
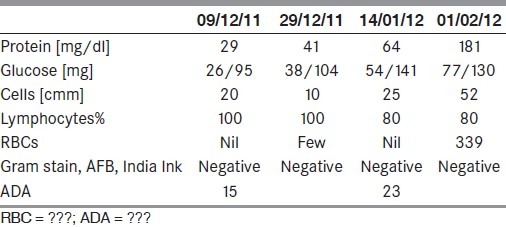
During these 2.5 months of illness, the patient also developed a significant weight loss of 10–12 kg, although his appetite was normal. He had not developed any fever in this period. There was no past or family history of tuberculosis and he did not indulge in high-risk sexual behavior. He was transferred to a tertiary reference center.
An examination performed on January 30 showed the general examination to be normal. In particular, they wear no neurocutaneous markers. He did not have any anemia, clubbing or lymphadenopathy and the examination of skull and spine was normal. The systems review was unremarkable. He did not have organomegaly. On neurologic examination, the patient was conscious, oriented and cooperated for the examination. The sense of smell was normal. Visual acuity, fields of vision and the examination of the optic fundi was normal. There were no features of papillaedema. The external ocular movements were complete. He had severe wasting of temporalis and masseter muscles [Figure 2]. He was unable to close his jaw or chew. There was sensory loss to pinprick and touch over the whole of the face on both sides in the distribution of all three divisions of the trigeminal nerves. Corneal and conjunctival reflexes were totally absent on both sides. His gag reflex was absent and he had bilateral weakness of the palatal movements. The sternomastoid was weak on the right side and tongue movements were normal. He also had wasting and weakness of the right supraspinatus and infraspinatus muscles. Examination of the trunk and lower limbs did not show any focal weakness. Deep tendon reflexes were normally elicited and there were no cerebellar signs. His plantars were bilaterally extensor.
Figure 2.
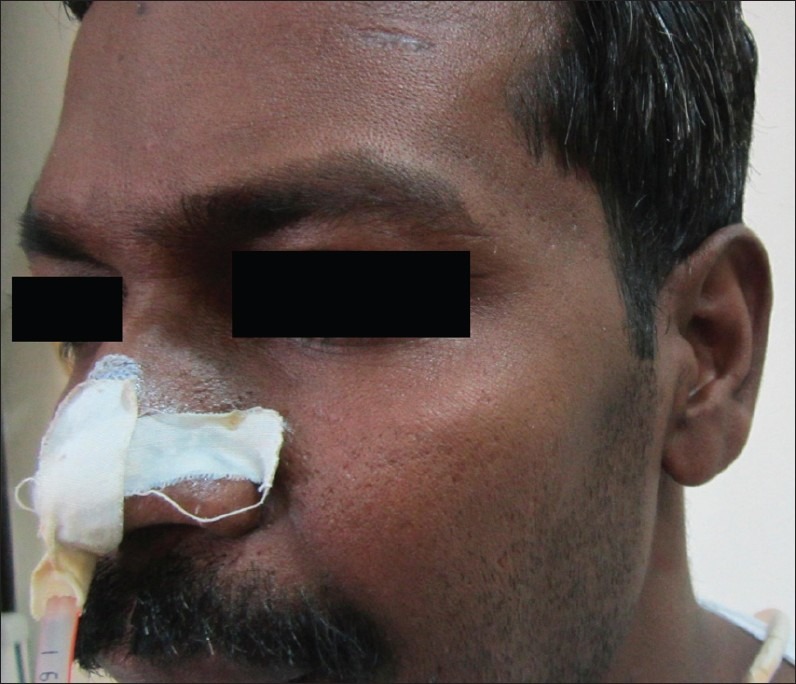
Wasting of temporalis and masseter muscles
In-charge clinical neurologist's comments
This patient has presented with subacute to chronic progressive neurological illness with some intervening periods of exacerbation and mild improvements. The initial presentation has been rather acute, with headache and vomitting as the main symptom, and at that point the thought of vascular diseases manifesting with subarachnoid hemorrhage was justified. The two episodes of transient neurological deficits on the left side are difficult to account for, as they would suggest a parenchymal right-sided dysfunction. In patients with sub arachnoid hemorrhage (SAH), events of vasospasm result in focal deficits, but these seem to be too transient and a little late in the illness. As the history unfolds, the headache and vomitting persist and cranial nerve dysfunctions appear. He has symptoms and signs of involvements of fifth, ninth and tenth cranial nerves on both sides. Besides the cranial nerves, there is evidence to suggest cervical root involvement resulting in weakness of spinatii. Thus, as the case advances, with headache, vomitting and multiple cranial nerve palsies, this process seems to be involving the meninges.
Various infective, inflammatory and infiltrative processes could affect the meninges in this subacute manner. Two clinical aspects need to be recounted at this stage. Firstly, the patient has had no fever throughout his illness, which would be against an infectious or inflammatory process. But, immunocompromised patients may not develop fever, and an underlying immunosupression like the retrovirus illness will need consideration. Secondly, the cranial nerves affected by this disease are also unusual. The common form of chronic meningits that we see in India, tuberculous meningits, and most other chronic infections tend to affect the lower cranial nerves and external ocular nerves in an asymmetrical manner. While tubercular meningitis is common, fungal and syphilitic meningitis also need to be discussed. Meningovascular syphilis tends to occur within the first 12 months after primary infection and is associated with headache and meningismus. It produces a basal meningitis, and cranial nerve palsies (seventh, sixth or second cranial nerve, in decreasing order of occurrence) are common.[1] Pachymeningitis affecting the brain and spinal cord is known to develop, typically 6–7 years after the primary infection. In our patient, there is no history of primary chancre and the evolution is rapid. Fungal meningitis affects immunocompromised individuals but, at times, can be seen otherwise. As the route of infection is often parameningeal (sinuses), the cranial nerves involved tend to be third, fourth and sixth. However, subacute fluctuating basal meningitis is well recognised in fungal infections and will have to be excluded.
Our patient has lower cranial nerve palsies, but the most striking feature is the outstanding affectation of the trigeminal nerves. It is indeed uncommon to see such severe trigeminal nerve involvement in chronic infective meningitis. Diseases like sarcoidosis can affect the gasserian ganglia and lead to trigeminal neuropathy. Sarcoidosis of the central nervous system (CNS) results in basal meningitis and cranial neuropathies.[2] Facial nerve is affected in up to 50% of such patients, and involvement is often bilateral.[3] Parenchymal lesions preferentially occur in the brain stem and hypothalamus. Systemic involvement includes erythema nodosum and pulmonary symptoms. The present patient did not have other systemic manifestations of sarcoidosis, which is unusual for the diagnosis. Wegener's granulomatosis, another systemic vasculitis, commonly affects the central nervous system. The orbits are affected in approximately 50% of the cases, often leading to proptosis. Cranial nerve palsies can occur (most commonly affecting the second, sixth and seventh nerves) and pachymeningitis can develop.[4] Along with basal meningitis and cranial neuropathies, headaches, strokes and seizures are also seen. In this patient, there is no pulmonary, sinus or renal involvement, making the diagnosis of Wegener's granulomaatosis less likely.-[5] Sjogrens syndrome also tends to affect the fifth nerve, but the deficits are usually seen in the sensory system. Other systemic features of Sjogren's syndrome are not present in our patient. Infiltrative meningitis, like the carcinomatous meningitis, tends to involve the larger nerves and may involve the fifth nerves.
Our patient also has findings of motor radiculopathy in the cervical region on the right side. This suggests that the process has gone beyond the cranial meninges and has begun to affect the spinal meninges. These findings are more in keeping with the infiltration of meninges. To explain the extensor planters and the two transient events, we perhaps will need to consider compression of the brainstem by the meningial process or development of hydrocephalus. However, there is no examiantion evidence to point to either.
Thus, we seem to be dealing with a chronic meningeal process having brunt on the fifth nerve and affecting spinal segments, in the absence of fever, but with significant weight loss. Apart from tubercular meningitis, fungal meningitis and meningovasular syphillis, infiltrative processes like carcinomatous meningitis, and immunological disorders like sarcoidosis, wegeners granulomatosis will have to be carefully evaluated. He will also need a full systems review for the search of a primary mitotic process.
Investigations
Complete blood count, serum sodium 136 meq/L, potassium 4.6 meq/L, magnesium 2.2 mg%, blood urea nitrogen 14 mg%, creatinine 0.7 mg%, calcium 8.9 mg%, aspartate aminotransferase 26 U/L, alanine aminotransferase 18 IU/L, total bilirubin 1.1 mg% and direct bilirubin 0.4 mg%, alkaline phosphatase 55 IU/L, RBS 92 MG% were all normal. His serum angiotensin converting enzyme levels were 12 IU and ANA and ANCA were negative. HIV ELISA and HBsAg were non-reactive as was the VDRL.
The electrocardiogram, chest X-ray and urine and stool examinaions were within normal limits. Results of his CSF examination are tabulted in Table 1. It showed progressively increasing protein content with predominantly lymphocytic pleocytosis. There was reduction of CSF sugar, tending to be one-fourth to one-third of the simultaneous blood sugar levels.
Further course in the hospital
The patient's investigations suggested lymphocytic mennigitis. Thinking it to be of tuberculous origin on the basis of commonness, cranial neuropathies and ADA levels on the higher side of normal, the referring physician has decided to use antitubercular therapy (Rifampicin, INH, Pyrazinamide and ethambutol] alongwith 40 mg prednisolone daily.
Initially, the patient showed some benefit in headaches and vomitting, but it soon recurred and he developed more difficulty in swallowing and chewing. A nasogastric tube had to be inserted. He also developed mild weakness of the left lateral rectus muscle. At this stage, an magnetic resonance imaging (MRI) was repeated and gadolineum was given. A fourth CSF was also performed on 1.2.12. CSF was difficult to obtain and the pressure was low. It also showed lymphocytic pleocytosis with raised proteins. The sample was sent for cryptococcal antigen and India Ink preperation, and tested negative. Fungal stains were negative. CSF cytospin was performed.
Radiologist's comments
The first available imaging is a plain CT scan of the brain. It revealed mild diffuse hyperdensity in the posterior fossa, especially along the tentorium [Figure 1], and mild prominence of the ventricular system. As these findings were not considered to be specific for any particular pathology, an MRI of the brain was advised. The MRI revealed mild diffuse T1 shortening, i.e. “bright” signal on T1W images, along the basal cisterns and cerebellar fissures. These regions were also slightly bright on FLAIR images. No significant parenchymal lesion was detected. The ventricles were mildly dilated. The T1 shortening was suspected to be due to subarachnoid hemorrhage. Hence, an MR angiogram was advised to rule out an aneurysm or arteriovenous malformation. The neck and intracranial MR angiogram was however found to be normal. MR venogram study was also normal. This was followed by a digital subtraction angiography (DSA) to further evaluate the intracranial vasculature in detail. The DSA study was absolutely normal.
A repeat MRI of the brain and screening of the whole spine with contrast was performed almost 1 month later at our hospital on a 3 Tesla system. Prior imaging studies were also reviewed. There was diffuse T1 shortening still seen along the basal cisterns, cortical sulci and cerebellar fissures [Figure 3a], which revealed marked homogenous and smooth post-contrast enhancement [Figure 3b]. This T1 brightness was unchanged on the fat-suppressed T1W sequence, ruling out the presence of fat. Both trigeminal nerves were also bright on T1W images, were thickened and showed homogenous post-contrast enhancement [Figure 4]. There was contiguous involvement of the spinal meninges, being most marked along the cauda equina nerve roots [Figure 5]. FLAIR images showed similar mild hyperintensity. On the T2 GRE images, there was no obvious hypointensity (“dark” signal). The diffusion-weighted images were unremarkable. Mild ventricular prominence was still noted.
Figure 3.
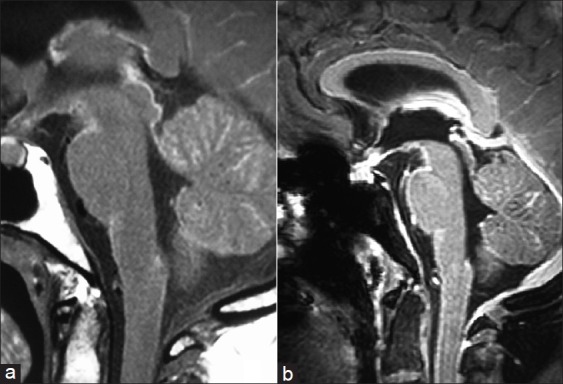
Diffuse T1 shortening along the basal cisterns and cerebellar fissures (a) with marked homogenous and smooth post-contrast enhancement (b)
Figure 4.
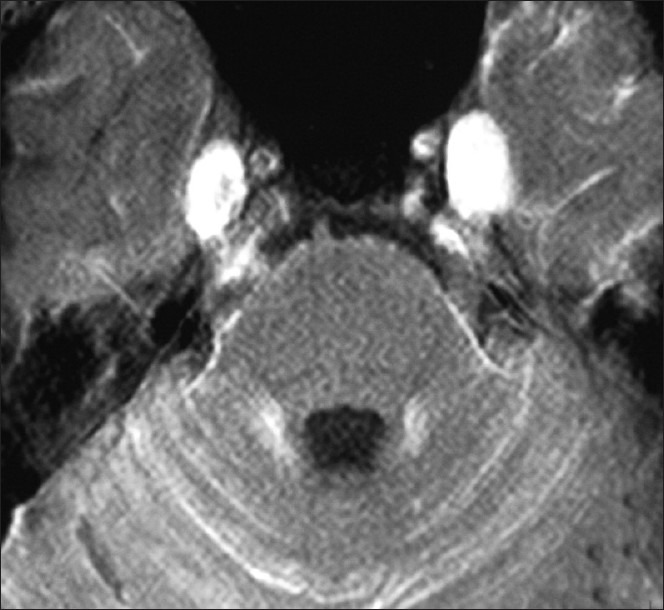
Striking thickening and post-contrast enhancement of the trigeminal nerves
Figure 5.
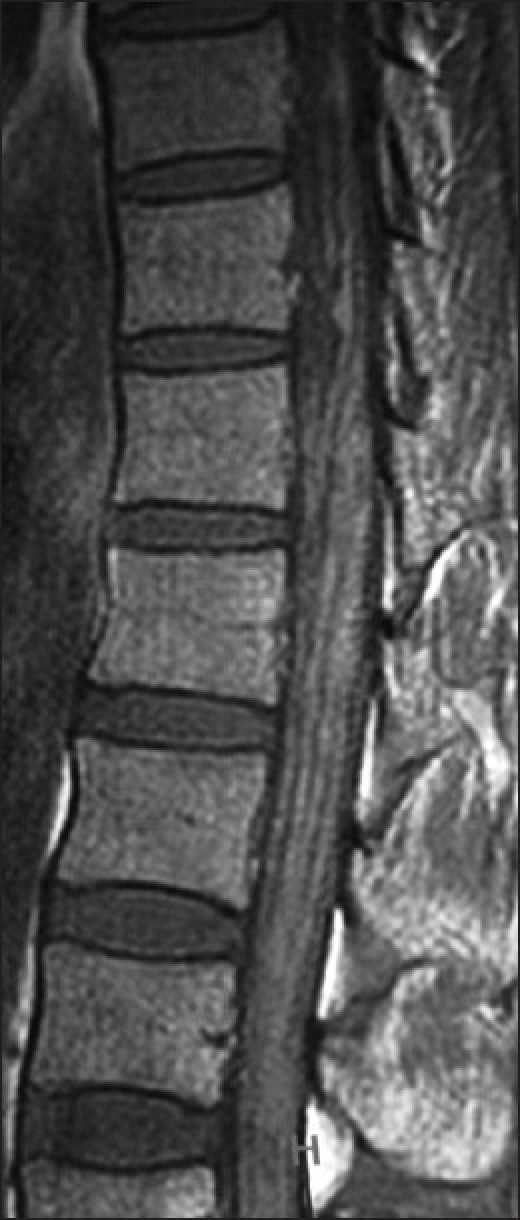
Involvement of the spinal meninges along the cauda equine nerve roots
Based on these findings, we can conclude that we were dealing with primary leptomeningeal pathology and that it was hyperintense on T1W images. The differential diagnosis of leptomeningeal diseases considered were subarachnoid hemorrhage, meningitis, metastases, neurosarcoidosis, Sturge-Weber syndrome, Lyme disease and melanosis. Causes of T1 shortening are presence of lipid (fat and sebaceous material), blood (methemoglobin), paramagnetic material like gadolinium (contrast enhancement), manganese, magnesium and copper; melanin, dystrophic calcium, protein (high concentration like mucus) and slow flowing blood (flow-related enhancement). Among the leptomeningeal pathologies, subarachnoid hemorrhage was ruled out as the T1 brightness had remained unchanged in serial MR scans and there was no abnormality detected on GRE images. It should be noted that hemorrhage evolves with time and only subacute blood products (methemoglobin) appear bright on T1W images. Further resolution results in formation of ferritin and hemosiderin, which are hypointense (“dark”) on T2 GRE images. The MRA and DSA were also normal. All the other pathologies, except melanosis, did not exhibit such marked, diffuse and smooth T1 hyperintensity. Presence of fat was ruled out on the fat-suppressed sequence. The features were also not consistent with the rest of the T1 bright lesions other than melanin. Hence, a diagnosis of primary leptomeningeal melanomatosis was considered. However, for further evaluation, CT scan of chest, abdomen and pelvis was performed to look for any primary malignancy or any other pathology. It was found to be normal.
CSF cyto spin performed on concentrated CSF was now available and showed clusters of large round to polyhedral cells with moderate cytoplasm containing abundant melanin pigment obscuring the nucleus [Figure 6]. With the information of MRI and CSF, a biopsy of the meninges was performed.
Figure 6.
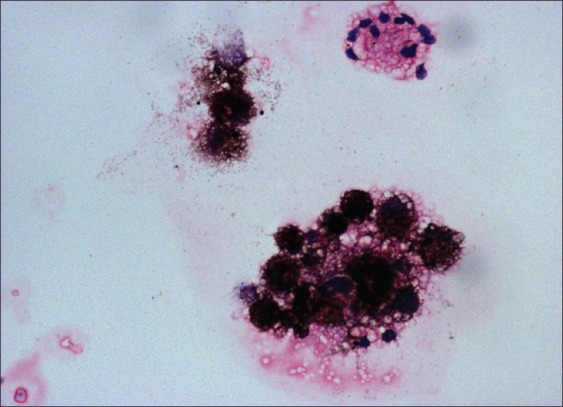
Cerebrospinal fluid cytospin showing melanin containing cells
Surgical findings
The working plan was to biopsy the meninges from the right tentorial area and, if necessary, extend the procedure to obtain tissue from the enlarged gassarian ganglion. On opening the skull vault, the exposed dura appeared bluish. As the dura was cut, arachnoid was seen to be uniformly blackish. Arachnoid tissue was taken and sent for histopathological study. The diagnosis considered on table was of melanocyte infiltration of meninges.
Histopathologists findings
At examination of the gross specimen, diffuse darkening and thickening of the leptomeninges was noted. Microscopy showed dispersed and perivascular aggregates of bland oval to spindle-shaped cells with coarse dark pigmentation seen in majority of the cellular cytoplasm of all types of cells, including stromal cells and macrophages [Figure 7]. This cellular morphology was simlar to what was found on CSF cytospin study.
Figure 7.
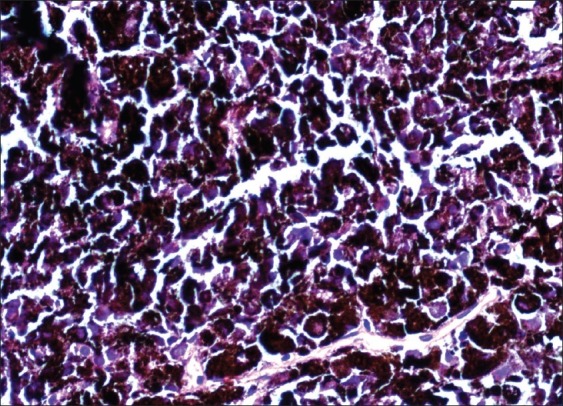
Aggregates of bland oval to spindle-shaped cells with coarse dark pigmentation seen in majority of cellular cytoplasm. × 40
As histology confirmed a melanocytic infiltration, a search for primary melanoma was performed. He was re-examined carefully for cutaneous melanomas, but he did not have any. Positron emission tomography CT scan was done, which did not show any increased activity besides the surgical site. Thus, the process was primarily affecting the craniospinal meninges alone.
Final Diagnosis
Primary leptomeningeal melanomatosis with multiple cranial nerve palsies medical oncologist's opinion
Primary melanocytic lesions of the CNS arise from melanocytes located within the leptomeninges, and this group includes diffuse melanocytosis and meningeal melanomatosis, melanocytoma and malignant melanoma. Primary melanin-containing lesions of the CNS must be differentiated from metastatic melanoma because these lesions require different workup and also differ in therapy.
Melanin is normally present within cells at certain locations of the neuraxis, viz. melanocytes of the leptomeninges and neurons of the substantia nigra, locus ceruleus and roof of the fourth ventricle. Melanocytes are seen normally within the leptomeninges and are more concentrated at the base of the brain and on the ventral surface of the cervical spinal cord. They are of neural crest origin.[6]
Primary leptomeningeal melanomatosis is a rare, aggressive neoplasm of the CNS that arises from melanocytes within the leptomeninges and carries a poor prognosis. It is also referred to as a meningeal variant of primary malignant melanoma.[7] The first case of diffuse leptomeningeal melanomatosis was described by Virchow in 1859. Primary leptomeningeal melanomatosis is more common in adults (peak prevalence, 4th decade of life) than in children, and patients present with variable signs and symptoms including seizures, signs of increased intracranial pressure, psychiatric disturbances, cranial nerve palsies and spinal cord compression.[8] These lesions are not common and limited information is available on the definitive treatment of primary meningeal melanomatosis. As it is believed to be an aggressive lesion, it is generally treated with chemotherapy. We decided to start daily doses of thalidomide 200 mg and temazolimide 75 mg/m2 body surface area. At 1-month follow-up, the patient continued to deteriorate and ocualar cranial nerves were also affected.
Fisher's rule[9]
“Resist the temptation to prematurely place a case or disorder into a diagnostic cubbyhole that fits poorly. Allowing it to remain unknown stimulates continuing activity and thought.”
Acknowledgment
We thank Dr Prasanna Kasegaonkar for his help.
Footnotes
Source of Support: Nil
Conflict of Interest: Nil
References
- 1.Luxon L, Lees AJ, Greenwood RJ. Neurosyphilis today. Lancet. 1979;1:90–3. doi: 10.1016/s0140-6736(79)90074-6. [DOI] [PubMed] [Google Scholar]
- 2.Zajicek JP, Scolding NJ, Foster O, Rovaris M, Evanson J, Moseley IF, et al. Central nervous system sarcoidosis diagnosis and management. QJM. 1999;92:103–17. doi: 10.1093/qjmed/92.2.103. [DOI] [PubMed] [Google Scholar]
- 3.Nowak DA, Widenka DC. Neurosarcoidosis: A review of its intracranial manifestation. J Neurol. 2001;248:363–72. doi: 10.1007/s004150170175. [DOI] [PubMed] [Google Scholar]
- 4.Newman NJ, Slamovits TL, Friedland S, Wilson WB. Neuro-ophthalmic manifestations of meningocerebral inflammation from the limited form of Wegener's granulomatosis. Am J Ophthalmol. 1995;120:613–21. doi: 10.1016/s0002-9394(14)72208-1. [DOI] [PubMed] [Google Scholar]
- 5.Spranger M, Schwab S, Meinck HM, Tischendorf M, Sis J, Breitbart A, et al. Meningeal involvement in Wegener's granulomatosis confirmed and monitored by positive circulating antineutrophil cytoplasm in cerebrospinal fluid. Neurology. 1997;48:263–5. doi: 10.1212/wnl.48.1.263. [DOI] [PubMed] [Google Scholar]
- 6.Smith AB, Rushing EA, Smirniotopoulos Pigmented Lesions of the Central Nervous System: Radiologic-Pathologic Correlation. Radiographics. 2009;29:1503–24. doi: 10.1148/rg.295095109. [DOI] [PubMed] [Google Scholar]
- 7.Gaetani P, Martelli A, Sessa F, Zappoli F, Rodriguez Y, Baena R. Diffuse leptomeningeal melanomatosis of the spinal cord: A case report. Acta Neurochir. 1993;121:206–11. doi: 10.1007/BF01809277. [DOI] [PubMed] [Google Scholar]
- 8.Sagiuchi T, Ishii K, Utsuki S, Asano Y, Tsukahara S, Kan S, et al. Increased uptake of technetium-99m hexamethyl propyleneamine oxime related to primary leptomeningeal melanoma. AJNR Am J Neuroradiol. 2002;23:1404–6. [PMC free article] [PubMed] [Google Scholar]
- 9.Campbell WW, DeJong RN, Haere AF. 6th ed. USA: Lippincott Williams & Wilkins, Baltimore; 2005. Dejong's the neurological examination; p. 622. [Google Scholar]