

Abstract
The tetrazine chromophore holds promise as an effective photochemical trigger to achieve structural release, directed at the determination of peptide/protein motions that occur early in the folding processes. The photochemistry of 3,6-di-cysteine-S,S-tetrazines was examined by femtosecond IR transient absorption spectroscopy. Excitation of the tetrazine chromophore by visible and near UV light in the end yields chemically inert, structurally unobtrusive photoproducts that are not expected to interfere with the conformational dynamics of peptides and proteins. Dicysteine S,S-tetrazine is suggested to undergo photocleavage via a photochemical pathway different than the parent molecule s-tetrazine, based on kinetic measurements that reveal a stepwise reaction pathway of photofragmentation, whereby the initial ring cleavage event occurs prior to the formation of the SCN groups.
1. Introduction
Investigation of the early kinetic events that govern conformational dynamics in peptides and proteins holds the promise of developing models to understand folding and misfolding pathways. Of particular interest is the characteristics of the basic motions of the peptide links that permit the peptide/protein to orient into structures that lead to the folded states. Many of these elementary, yet essential motions, occur on the ultrafast time scale. However few methods currently exist that are able to initiate, observe and/or assign the chemical dynamics of peptide linkages such as single bond rotations, hydrogen bond formation and breakage, in conjunction with the assembly of small structural units that are expected to occur on the picosecond to few nanosecond time scale. Such processes are of course completely hidden in data when the measurements involve thermal equilibrium distributions of the conformational states. To amplify such responses, introduction of spatial coherence into the measurement method is required. These elementary processes are a natural component of molecular dynamics simulations, whether quantum or classical. Acquisition of experimental knowledge would provide another class of tests for these theoretical predictions, one that in particular does not involve an average over a very large number of conformal states.
Short pulse induced photochemical reactions leading to initiation of peptide/protein movements, in conjunction with ultrafast spectroscopy to observe and characterize these events, would certainly augment our understanding of the folding/unfolding processes. Ideally incorporation of a phototrigger, or photo-initiator, should confine the peptide to be within a narrow initial distribution of conformers, akin to a line up of horses at the start of race. This initial condition can be evaluated by direct structure determination methods. The photolysis of a triggering chromophore is then intended to release the constraint with precise temporal control.[1–6] Ultrafast spectroscopic methods, such as infrared and two-dimensional infrared spectroscopy, combined with selective isotope substitution can be utilized to track the movements of any relevant chemical unit, as the peptide relaxes to the equilibrium distribution of configurations. Evolution of the protein dynamics after the ultra-fast phototriggering event would provide features of the free energy landscape of folding and misfolding, that are arrived at from a range of specific, known initial conditions. Gradual propagation on a multidimensional potential surface would create a distribution of states, whose average properties can be evaluated by time resolved spectroscopic methods.
The challenge is to develop a photochemical reaction system that has all the essential criteria required for use as a photochemical trigger of biological systems, including an optimum quantum yield, negligible side reactions, fast reactivity, biocompatibility, and accessibility to non-damaging light frequencies.[1,2] Moreover, the photochemical trigger with such features must be readily incorporated into the desired biological scaffold, with the assurance that the photochemical properties of the trigger construct are not significantly altered by the environment of the biomolecules upon incorporation. The triggering system also must not impair the structural evolution of the peptide or protein.
Several phototriggers have been developed,[5–10] but there remain significant challenges. For example, disulfide bonds in peptides can be employed as photo-triggers using high energy UV light to sever the S-S bonds of disulfides, thereby releasing the designed conformational constraints incorporated through built-in disulfides. Such experiments have been carried out in short helical peptides,[6,7] cyclic peptides and beta hairpins.[3,9] Experimental probes have included IR pulses and 2D IR methods.[9] Another example is the azobenzene construct, which undergoes fast, reversible photoisomerization. When incorporated into a peptide the cis-trans isomerization causes the system to switch between significantly different forms of constrained equilibrium conformations.[4,11,12]
Although all of these triggering methods offer the capability of measuring structure equilibration, certain aspects of the photochemistry are not ideal. Disulfide linkers require the use of damaging UV irradiation to dissociate homolytically the S-S bond into reactive sulfur radicals, which in turn can undergo geminate recombination, as well as side reactions with the peptide/protein sidechains. By varying the molecules attached to these linkers or by introducing radical scavengers, the rate and amount of germinate recombination may be controlled.[6] Triggers comprised of azobenzenes exploite a fast, reversible photo-isomerization to release structural movement of the two tethered ends. This linkage constraint can significantly limit the accessible conformational space available to the peptide or protein. Relaxation of the structure following isomerization has been suggested to mimic secondary structure reorganization in larger protein systems.[13]
Dicysteine S,S-tetrazine represents a new class of compounds that hold considerable potential as ultra-fast phototriggers.[1] First, the photoproducts of the photochemical release process for alkyl or aryl substituted s-tetrazines are the relatively inert and unobtrusive nitriles and nitrogen.[14,15] Second, the spectroscopy[16–20] of s-tetrazine has been intensively investigated both in solutions [15,16,21–27] and in the vapor phase by experiment and theory.[14,15,23,28–38] With a photoproduct yield close to unity in the vapor phase,[39] the photoreactions of s-tetrazines have been well established to occur on the ground state following internal conversion.[32,34–36]
While experiments performed on s-tetrazines in condensed phases have revealed quantum yield close to unity,[14,28,40] some experiments on dimethyltetrazine show a lower photochemical yield.[31,41] Although the overall yield decreases in the condensed phase[1,31,41] due to vibrational energy redistribution, sufficient yield (~22%) can be obtained by using S,S-tetrazines.[1] In the present study, we have explored the ultrafast kinetics and structures of the intermediates that derive upon the photolysis of dicysteine S,S-tetrazine. We demonstrate that a pathway of dissociation, which differs from that of simple alkyl substituted s-tetrazine is followed.[1] Nonetheless, based on our previous work, we would expect the photolysis of dicysteine S,S-tetrazine to yield molecular nitrogen and two thiocyanate groups, permitting the rate of the photolysis reaction to be probed directly through the SCN stretching vibrational mode (Figure 1). While SCN groups do in fact appear after the photolysis pulse, we now provide evidence of a precursor intermediate along the photochemical fragmentation reaction coordinate.
Figure 1.

Photoreaction of dicysteine S,S-tetrazine upon 355 nm irradiation.
The central thrust of the work presented here is to set the stage to have the tetrazine trigger incorporated into peptides and proteins such that upon photolysis a change in their constrained conformation can be observed on an ultra-fast time scale. The chemistry utilized to insert tetrazine into peptide systems has been developed[1] such that tetrazine can be placed virtually anywhere within a peptide via two cysteine mutations at the desired positions of linkage. Towards this end, the dynamics of the carbonyl moieties of the cysteine residues in di-cysteine S,S-tetrazine will be directly monitored, employing ultrafast infrared spectroscopy in order to provide a proof of principle of the tetrazine trigger protocol.
2. Materials and Methods
2.1 Materials and Instrumentation
The peptide analogues employed in this study, prepared from 3,6-dichlorotetrazine by nucleophilic substitution, were fully characterized by 500 MHz NMR, high resolution mass spectrometry, and X-ray crystallography.[1] The test tetrazine analogs, as ~3–5 mM solutions in d4-methanol (Fisher), chloroform (Fisher) and methanol (Fisher), were used for fs transient absorption experiments. The optical density at the excitation wavelength of the samples ranges from 0.09–0.12 in a 400 μm path length CaF2 cell for femtosecond transient IR experiments. The excitation wavelength was 355 nm for the amide and ester carbonyl measurements and 400 nm for the SCN formation measurements unless otherwise specified. All FTIR spectra were taken on a Nicolet 6700 FTIR spectrometer.
The absorption spectra were collected on a Perkin–Elmer Lambda 25 UV-Vis spectrometer (Fremont, CA, USA). The excitation spectra at emission wavelength of 580 nm were obtained on a Fluorolog 3.10 spectrofluorometer (Jobin Yvon Horiba, Edison, NJ, USA), with a 1 cm quartz sample holder. To minimize self-quenching, the OD of each sample at the excitation wavelength was adjusted to be <0.1 below 300 nm.
2.2 Femtosecond IR Tranisent Absorption Method
Fourier-transform limited 70-fs pulses with center frequencies of 2150 cm−1 were used in the femtosecond IR transient absorption experiments for the samples in CHCl3/MeOH (95:5 v/v) mixture. Center frequencies of 1750 cm−1 and 1650 cm−1 were used for the femtosecond IR transient absorption experiments for the samples in d4-methanol. The energy of the 355 nm or 400 nm excitation pulse, 5 μJ, was attenuated by an iris. The beam radius of the IR probe pulse was 75% of the visible pump pulse radius at the sample cell. The visible beam passes through a hole in the center of the parabolas used to collect the infrared probe pulses and the overlap between the pump and probe pulses is achieved by first using 200 um pinhole at the sample holder, followed by maximizing the transient absorption signal of a Si wafer. The time zero was determined by scanning the time domain spectrum of the transient absorption of the Si wafer. The transient absorption of the carbonyl stretch of fluorenone is utilized as a standard to optimize further the overlap of the pump beam. Timing between the pump and probe pulses is varied by using two combined automated translation stages (Melles Griot, Sigma Koki Co., LTD) for fs-ns resolution. The transmitted probe pulse is focused on to the focal plane of a monochromator equipped with a 64 element mercury-cadmium-telluride array detector (InfraRed Associates, Stuart, FL). The appropriate monochromator grating was chosen, comprising either 50 lines per mm or 150 lines per mm groove. The sub-nanosecond time dependent spectra were obtained by sampling the frequency spectra (averaged over 16 scans, 1600 shots per scan) at various time points from 0–1.2 ns. The kinetic traces of the amide and ester carbonyl modes shown are collected over a 35 ps time range with a 200 fs interval.
3. Results
3.1 Photochemical Yield Dependence
The UV-Vis absorption spectra of the dicysteine S,S-tetrazine reveals several distinct transitions allowing for excitation over the range of wavelengths, 310 to 555 nm (Figure 2). The 3,6-substitution of sulfur atoms introduces a new band centered at 410 nm, attributed either to a separate n→pi* transition or charge transfer band involving sulfur, having a peak extinction similar to that at 542 nm. The yield of emitting state formation for the dicysteine S,S-tetrazine was determined to be dependent on the excitation wavelength, as seen from the fluorescence excitation spectrum in Figure 2. Interestingly, introduction of a sulfur substitution between the tetrazine ring and the peptide results in an increase of the photolysis yield, compared to the non-sulfur counterpart, consistent with the fluorescence loss deduced from the difference between the absorption and excitation spectra in Figure 2. All thio-containing derivatives exhibit this phenomenon.[1] The fluorescence quantum yield of the relaxed excited state was estimated from rise time of the triplet-triplet absorption kinetics to be 0.02. The photolysis yield has been determined to be 22%.[1]
Figure 2.

Comparison of UV-Vis absorption (blue) and excitation (brown) spectra of dicysteine S,S-tetrazine normalized to unity at 532 nm. (Inset) Femtosecond IR transient absorption spectra of dicysteine S,S-tetrazine in SCN region at 1 ns delay upon 400 nm (red) and 355 nm (black) excitation.
3.2 Timescale and Spectral Change of the Carbonyls Upon Photocleavage of Di-cysteine S,S-Tetrazine
Both the ester and amide carbonyl transitions of di-cysteine S,S-tetrazine are sensitive to the photocleavage, as illustrated by the FTIR spectrum of the tetrazine model system, before and after 355 nm irradiation (Figure 3C). In order to display the undisturbed, unphotolyzed, and the pure photolyzed material spectra separately (Figure 3C), two independent FTIR spectra were measured. These separately determined measurements avoided the presence of any unphotolyzed sample in the photolyzed case. However, the solvent subtraction is somewhat arbitrary and these spectra need to be scaled to fit well with the transient signals. The photolyzed sample contained two thiocyanate substituted cysteines by 1H and 13C NMR spectroscopy. The transient IR absorption spectrum of the carbonyl ester modes (1710–1790 cm−1) of the di-cysteine S,S-tetrazine were measured at different time delays (Figure 3A). The dashed red lines in Figure 3A and 3B represent a steady state photolysis measurement of the in situ difference spectrum between an unphotolyzed and photolyzed sample. The femtosecond pulse initiated transient difference spectra were essentially the same after a 50 ps or 150 ps time delay, and very similar to the FTIR difference spectra shown as dashed red lines in Fig. 3A and Fig. 3B. The transient difference spectrum consists of two bleach signals, at 1730 cm−1 and 1754 cm−1, due to removal of the parent molecules and the two absorption peaks at 1765 cm−1 and 1745 cm−1 from the ester absorption after the molecule is photolyzed. The transient spectra after time delays of 150 ps, and even as early as 50 ps, match the normalized, stationary FTIR difference spectrum after complete photolysis (Figure 3A). The transient optical density, as well as the band shapes of the transient spectra, does not change after 50 ps. A similar observation was noted for the transient IR absorption spectrum of the carbonyl amide I modes (1600–1700 cm−1) of the di-cysteine S,S-tetrazine dipeptide measured at the same time delays (50 ps and 150 ps), between which no change is apparent (Figure 3B). The transient difference spectrum consists of one bleach signal found at 1647 cm−1 and one absorption band at 1664 cm−1. The transient spectrum after 50 ps time delay was very similar to the normalized FTIR difference spectrum obtained from the measured difference between unphotolyzed sample and the same sample after complete photolysis (Figure 3B).
Figure 3.

Femtosecond IR transient absorption of (A) carbonyl ester region and (B) amide carbonyl region of dicysteine S,S-tetrazine in CD3OD at (green) 50 ps and (black) 150 ps after excitation. The stationary FTIR difference spectrum of the sample before and after photolysis is shown as a dashed red line in (A) and (B). (C) FTIR spectrum of amide and ester carbonyl region of unphotolyzed (red) and photolyzed (blue) samples after solvent subtraction.
Examination of the spectra of the ester carbonyls on the ultrafast timescale by taking several time delays prior to the 50 ps delay (Figure 4A) revealed that the bleaching of the initial signal at 1754 cm−1 reaches a maximum before 5 ps, followed by a decrease in the signal over 10 picoseconds. Even at the earliest measured time delay of 200 fs, the bleaching resulting from the disappearance of the initial carbonyl absorption is already present. This initial bleaching of the ester carbonyl absorption occurs in less than 250 fs and is followed by a recovery to a fixed value (Figure 4B). The recovery time of this initial bleach was fit to a single exponential and the time constant determined to be 10 ps. This time constant matches the 10 ps rise time of the transient absorption band of the photoproduct carbonyl ester observed at 1763 cm−1 (Figure 4C). In Figure 4D, the transient spectra of several different data sets immediately following time zero show no presence of a new absorption band of the intermediate state resulting from the disappearance of the initial ester carbonyl absorption. A slightly different observation was made for the carbonyl region of the amide I band of the model compound. At a time delay of 4 ps (Figure 5A), both the bleach signal observed at 1647 cm−1 and the transient absorption at 1664 cm−1 have already reached a maximum value. By following the kinetics of these two peaks from 0 to 35 ps by 200 fs intervals(Figure 5B and 5C), the bleach signal and the rise of the new absorption signal were observed to occur within 200 fs. After this initial fast process, kinetic analysis of both signals demonstrates that the amide signals recover to a fixed value over the next few ps. Both traces were fit to a single exponential to yield a time constant of 10 ps. It should be noted that the bleach signal observed at the earliest times for the amide (Figure 5A) reveal a slight shift to higher frequency.
Figure 4.
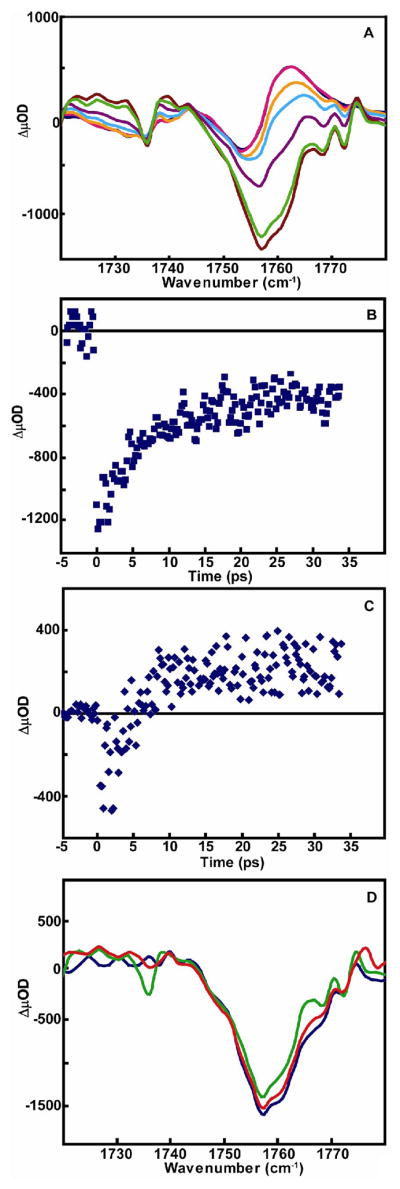
(A) Femtosecond IR transient absorption of carbonyl ester region of dicysteine S,S- tetrazine in CD3OD at (brown) 5 ps, (green) 6 ps, (purple) 10 ps, (light blue) 15 ps, (orange) 20ps, (pink) 40 ps, (dark blue) 50 ps. Kinetic trace of (B) bleach signal from ester carbonyl at 1753 cm−1 and (C) transient absorption of photoproduct carbonyl at 1763 cm−1 from 0–35 ps. (D) Three independent femtosecond IR transient absorption data sets of the carbonyl ester region immediately following time zero.
Figure 5.

(A) Femtosecond IR transient absorption of carbonyl amide I region of dicysteine tetrazine in CD3OD at (blue) 4 ps, (purple) 10 ps, (red) 30 ps, (cyan) 50 ps. Kinetic trace of (B) bleach signal from amide I carbonyl at 1647 cm−1 and (C) transient absorption of photoproduct carbonyl at 1664 cm−1 from 0–35 ps.
3.2 Spectral Evolution and Yield Dependence of –SCN formation
The rates of formation of the thiocyanate groups were measured for the tetrazine model system by monitoring the formation of the stretching mode of the SCN, formed upon photolysis employing the IR transient absorption. The transient absorption spectrum corresponding to the SCN stretch (see Figure 6) at 2263 cm−1 was probed as a function of time delays up to 1 ns. Figure 6 (dashed black) also displays the stationary FTIR spectrum after complete photolysis. The thiocyanate transient was fit to a single exponential growth and the time constant was determined to be 177 ps for the dicysteine S,S-tetrazine. The wavelength dependence of the quantum yields of the -SCN transient, for 400 nm and 355 nm excitations, were obtained by maintaining the same sample OD and laser energy at the corresponding excitation wavelength (inset to Figure 2). The signal strength of the transient absorption band after 355 nm excitation is approximately twice that observed using 400 nm, in agreement with a loss in fluorescence shown in Figure 2. The SCN experiments were also performed in methanol but not reported here. The formation kinetics of the SCN was very similar to those discussed here.
Figure 6.

Femtosecond IR transient absorption of the SCN region following the photolysis of dicysteine S,S-tetrazine at 25 ps (blue), 75 ps (purple), 200 ps (red), and 1 ns (cyan) time delays in chloroform/MeOH (95%/5%) mixture. FTIR absorption band (dashed black) of SCN stretching frequency after steady-state irradiation of dicysteine S,S-tetrazine at 400 nm.
4. Discussion
4.1 Photochemistry of S,S-Tetrazines
The UV-vis absorption spectra of s-tetrazines[24,36] exhibit a main band at 542 nm resulting from a n → π* transition. Thio substitution leads to an additional band centered at 410 nm, which has been attributed to another n → pi* transition,[24,26] or a charge-transfer band involving sulfur. The mechanism of the photodissociation of simple s-tetrazines involves a crossing of a high barrier on the electronic ground state surface[34,35] so that following internal vibrational relaxation, cooling of the excited modes quenches the photochemistry. When additional substituents are attached to the tetrazine, the number of modes reached by internal conversion is greatly increased and the photochemical yield is reduced. However, the placement of a sulfur atom at the 3 and 6 positions of the tetrazine core results in an increased yield. A new photochemical pathway appears to be opened by sulfur substitution.[1] Evidence of this new pathway can be seen from the wavelength dependence of the quantum yield as discussed below. To investigate further the photolysis of these compounds, femtosecond IR transient absorption measurements were undertaken.
The IR transient absorption peaks at 1765 cm−1 and 1745 cm−1 result from the ester absorption after the molecule has been photolyzed, while the bleach signals at 1730 cm−1 and 1755 cm−1 are due to the removal of parent molecules. The rise time of the photoproduct absorption band at 1765 cm−1 was determined to be 10 ps, which is the same as the recovery time following the initial ultrafast removal of the parent molecule ester carbonyl. The recovery includes recreation of the initial ground state and formation of the photolyzed product. These results indicate that photolysis has begun on <250 fs timescale. The initial ultrafast bleach of the parent carbonyl at 1753 cm−1 (Figure 4D) further suggests that the carbonyl is involved in a transient interaction with the tetrazine ring that quickly removes that carbonyl double bond absorption out of the spectral range, akin to the processes of ring formation observed in dimethoxybenzoin photochemistry.[42–44] The vibrational frequency shifts of the ester carbonyl are not seen, only the populations of the initial and recovered states are observed. Another possible interpretation is that the new CO absorption band of the intermediate state might be anomalously wide and comparable with the noise. We have found no structural reason why this would be the case. While it is also possible that heating of the surroundings can occur after the energy is absorbed by the tetrazine, it would have to result in a strongly red shifted or unusually broad CO absorption band. The expected shifts due to heating are ca. 10–30 cm−1.[9,45] No red shifted absorption band is observed in the transient spectrum. Neither would conventional solvent shifts would not likely move the carbonyl absorptions out of the spectral range of the present experiments. The recovery of this carbonyl bleach results in a carbonyl absorption band at the location of that found in the final photoproduct. The photocleavage of the tetrazine appears to be connected with the interaction of the tetrazine with the ester carbonyl en route to the intermediate that ultimately generates the final photoproducts involving the –SCN groups. Some possible intermediate structures have been discussed in prior literature for simple tetrazine dissociation.[32,37,46] This is speculative and requires further synthetic and photochemical study to become established. Although this ester carbonyl is suggested to be involved in the dissociation for the compound under study, the same effects are expected to occur in longer peptide systems having different backbone structures.
The amide carbonyls also exhibit a response due to the excitation pulse. The bleaching of the initial state signal is at 1647 cm−1 while the absorption bands at 1665 cm−1 are characteristic of the photoproduct. After an initial fast jump of the signals (<200 fs), the bleach and absorption signals decay with a timescale of 10 ps. The ultrafast dynamics observed for the amide carbonyls suggest that these transitions are sensitive to the changes induced by the initial photocleavage of the tetrazine concomitant with the disappearance of the ester carbonyl absorption (The triplet state was measured to rise from 0 with a time constant of 10 ns (data not shown) and it is apparently not involved in ps processes). The spectrum after the 10 ps recovery matches the difference spectrum of before and after irradiation (Figure 3 and 5), giving further support that the model dicysteine tetrazine undergoes photolysis by as early as 10 ps. Since no changes are observed in both the amide and ester carbonyl bands after a timescale of 50 ps, these results would suggest that the photochemistry is complete by that time. We will however show that this cannot be the case.
The thiocyanate vibrational mode of the photoproduct was monitored at different delays between the optical pump and infrared probe. The spectrum of the transient absorption of the dicysteine tetrazine in the SCN region reveals the presence a peak at 2163 cm−1 as early as 25 ps, with little change in spectral lineshape up to 1 ns (Figure 6). This signal corresponds to the 2163 cm−1 band observed in stationary FTIR spectra after irradiation for long periods (Figure 6 –dashed black). It should be noted that shape of the transient absorption spectra of the SCN band is very slightly distorted by the detector resolution. The observed thiocyanate signal was 120 μOD at the 2163 cm−1 peak at 1 ns time delay for the dicysteine S,S-tetrazine, which is about 4 times the magnitude of the carbonyl signals resulting from the formation of the photoproduct after 50 ps. Assuming that two SCN groups are produced upon photolysis, this comparison is precisely the ratio of their peak extinctions and indicates that the photocleavage has reached completion.
The appearance rates of the photoproduct absorption bands provide further information about the photolysis. Since both the thiocyanate growth time of 177 ps and the carbonyls rise time of 10 ps for the di-cysteine S,S-tetrazine are much less than the singlet lifetime in the nanosecond regime, the photochemistry of the model system must produce the SCN group from a tetrazine state that is more highly excited than the n → π* transition at 542 nm.
Further support for this picture comes from the fluorescence excitation spectra (Figure 2). The decrease in quantum yield at shorter wavelengths suggests that the photochemistry arises from the unrelaxed states following excitation. The signal strength observed upon 355 nm excitation was indeed twofold larger than at 400 nm (inset to Figure 2) demonstrating that both these initial states lead to photocleavage. Although the transient spectra in the SCN region do not provide spectral evidence for the presence of any intermediate bands as early 25 ps, the photocleavage appears to occur prior to the SCN becoming fully formed based on the transient traces arising from the carbonyl transitions. Thus, we posit that the photocleavage occurs through a stepwise process in which the tetrazine rapidly forms an intermediate which dissociates to generate the SCN groups on a 200 ps time scale. The double bond of the ester carbonyl also appears to undergo a subpicosecond process.
4.2 Utility of S,S-Tetrazine as a Photochemical Trigger for Peptide Systems
The formation of secondary peptide/protein structure covers a range of timescales from 50 ns to a few microseconds.[47–52] Yet, several experiments have suggested the presence of even faster processes, which either could not be time resolved, or for which little structural information has been extracted.[48,50,53–55] Molecular dynamics have revealed that peptides can undergo significant structural changes within 1 ns or less.[52,56–59] These timescales are ideal for tetrazine triggers with a fast photocleavage in the picosecond time regime. The tetrazine phototrigger can be incorporated into basic units of protein structure such as alpha helices, β-hairpins, helix-turn-helix motifs, and loops of fast folding proteins.[60,61] Many challenges that simulations face comprising metastable states, non-native contacts, and rugged energy landscapes [62–66] can be uncovered through optical triggering, coupled with methods that have structural resolution, such as 2D IR spectroscopy. For example, the propagation or ‘zipping time’ of a helical peptide has been suggested to occur over 300 ps,[50] while simulations have ascertained the formation of the helical turn occurs within 0.1–1 ns.[58] Tetrazine phototriggers hold the promise of capturing the elementary peptide movements not accessible by many equilibrium techniques and in turn bridge the gap between timescale of 200 ps to tens of nanoseconds.
The spectral changes in ester or amide carbonyls described in detail above demonstrate how these transitions might be utilized to follow structural changes in a peptide backbone. Transient signals from isotopically substituted residues that are backbone structure sensitive can be expected to have a cross section on a similar scale as the carbonyls of the model system. Thus, these experiments represent a proof of principle. The changes in the amide and ester frequencies in the present case are attributed both to local structure changes and solvent reorganization around the carbonyl and to intrinsic frequency differences of the parent and photoproduct molecules. The model amide and ester carbonyls are intended to mimic the situation in future experiments where in the constraint will be positioned in a longer peptide backbone. The dynamics of the protein backbone movements would then be expected on an even longer timescale. The kinetics resulting from the rearrangement of the backbone structure and variations in the coupling associated with those changes in structure will be seen through the carbonyls interacting with one another, which in the present example are not occurring to any significant extent. The experiments of the model compound reported here capture the release of the tetrazine constraint, which can act as the initiation of the folding of a peptide or protein.
5. Conclusions
We have demonstrated that the photochemical yield of S,S-tetrazines in the condensed phase is maintained at a level of >20%, when thioalkyl-groups are substituted symmmetically for hydrogens on the tetrazine ring. Importantly, the sulfur substitution opens a new photchemical pathway that is quite different from that of simple s-tetrazines. Like the photo-production of HCN and nitrogen, with s-tetrazine, the photoreaction of dicysteine S,S-tetrazine produces thiocyanates, albeit through an intermediate that is produced with a 10 picosecond time constant, that forms the -SCN groups with a time constant of 177 ps. This new photochemical pathway initiates on the excited state surface whereas s-tetrazine photochemistry occurs on the ground state. The results suggest that the reaction is stepwise wherein the photocleavage entails an initial ultrafast step that competes favorably with energy relaxation to the fluorescent state. The initial process causes the removal of the ester carbonyl double bond in <250 fs. The dicysteine S,S-tetrazine construct thus has great potential as a photochemical trigger for fast tracking of secondary structure changes of peptides and proteins during the early events in protein/peptide folding.
Highlights.
New photochemistry by symmetric sulfur substitution of s-tetrazines.
Stepwise photocleavage of cysteine S,S-tetrazine tracked by femtosecond IR.
Peptide structural changes can be initiated by S,S-Tetrazine photochemistry.
Acknowledgments
The research was supported by NIH (GM12592) and the ultrafast laser Research Resource (NIH P41RR001348).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tucker MJ, Courter JR, Chen JX, Atasoylu O, Smith AB, Hochstrasser RM. Angewandte Chemie-International Edition. 2010;49:3612–16. doi: 10.1002/anie.201000500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rock RS, Hansen KC, Larsen RW, Chan SI. Chemical Physics. 2004;307:201–08. [Google Scholar]
- 3.Kolano C, Helbing J, Bucher G, Sander W, Hamm P. Journal of Physical Chemistry B. 2007;111:11297–302. doi: 10.1021/jp074184g. [DOI] [PubMed] [Google Scholar]
- 4.Bredenbeck J, Helbing J, Sieg A, Schrader T, Zinth W, Renner C, Behrendt R, Moroder L, Wachtveitl J, Hamm P. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:6452–57. doi: 10.1073/pnas.1036583100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bredenbeck J, Helbing J, Behrendt R, Renner C, Moroder L, Wachtveitl J, Hamm P. Journal of Physical Chemistry B. 2003;107:8654–60. [Google Scholar]
- 6.Lu HSM, Volk M, Kholodenko Y, Gooding E, Hochstrasser RM, DeGrado WF. Journal of the American Chemical Society. 1997;119:7173–80. [Google Scholar]
- 7.Volk M, Kholodenko Y, Lu HSM, Gooding EA, DeGrado WF, Hochstrasser RM. Journal of Physical Chemistry B. 1997;101:8607–16. [Google Scholar]
- 8.Hansen KC, Rock RS, Larsen RW, Chan SI. Journal of the American Chemical Society. 2000;122:11567–68. [Google Scholar]
- 9.Kolano C, Helbing J, Kozinski M, Sander W, Hamm P. Nature. 2006;444:469–72. doi: 10.1038/nature05352. [DOI] [PubMed] [Google Scholar]
- 10.Ihalainen JA, Bredenbeck J, Pfister R, Helbing J, Chi L, van Stokkum IHM, Woolley GA, Hamm P. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:5383–88. doi: 10.1073/pnas.0607748104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Behrendt R, Renner C, Schenk M, Wang FQ, Wachtveitl J, Oesterhelt D, Moroder L. Angewandte Chemie-International Edition. 1999;38:2771–74. doi: 10.1002/(sici)1521-3773(19990917)38:18<2771::aid-anie2771>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 12.Sporlein S, Carstens H, Satzger H, Renner C, Behrendt R, Moroder L, Tavan P, Zinth W, Wachtveitl J. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:7998–8002. doi: 10.1073/pnas.122238799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ihalainen JA, Paoli B, Muff S, Backus EHG, Bredenbeck J, Woolley GA, Caflisch A, Hamm P. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:9588–93. doi: 10.1073/pnas.0712099105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hochstrasser RM, King DS. Journal of the American Chemical Society. 1975;97:4760–2. [Google Scholar]
- 15.Hochstrasser RM, King DS, Smith AB., III Journal of the American Chemical Society. 1977;99:3923–33. [Google Scholar]
- 16.Mason SF. Advances in Molecular Spectroscopy, Proceedings of the International Meeting on Molecular Spectroscopy. 1962;1:466–70. [Google Scholar]
- 17.Mason SF. Journal of the Chemical Society. 1959:1269–74. [Google Scholar]
- 18.Mason SF. Journal of the Chemical Society. 1959:1263–8. [Google Scholar]
- 19.Spencer GH, Jr, Cross PC, Wiberg KB United States Department of Commerce, Office of Technical Services. PB Report. 1960;146:746. 25. [Google Scholar]
- 20.Spencer GH, Jr, Cross PC, Wiberg KB. Journal of Chemical Physics. 1961;35:1925–38. [Google Scholar]
- 21.Guckel F, Maki AH, Neugebauer FA, Schweitzer D, Vogler H. Chemical Physics. 1992;164:217–27. [Google Scholar]
- 22.Morter CL, Wu YR, Levy DH. Journal of Chemical Physics. 1991;95:1518–29. [Google Scholar]
- 23.Levy DH. Journal of the Chemical Society-Faraday Transactions Ii. 1986;82:1107–21. [Google Scholar]
- 24.Waluk J, Spanget-Larsen J, Thulstrup EW. Chemical Physics. 1995;200:201–13. [Google Scholar]
- 25.Thulstrup EW, Spanget-Larsen J, Gleiter R. Molecular Physics. 1979;37:1381–95. [Google Scholar]
- 26.Sandstrom J. Acta Chemica Scandinavica. 1961;15:1575–82. [Google Scholar]
- 27.Scheiner AC, Schaefer HF., III Journal of Chemical Physics. 1987;87:3539–56. [Google Scholar]
- 28.Hochstrasser RM, King DS. Journal of the American Chemical Society. 1976;98:5443–50. [Google Scholar]
- 29.Bjorklund GC, Burland DM, Alvarez DC. Journal of Chemical Physics. 1980;73:4321–8. [Google Scholar]
- 30.Glownia JH, Riley SJ. Chemical Physics Letters. 1980;71:429–35. [Google Scholar]
- 31.Paczkowski M, Pierce R, Smith AB, III, Hochstrasser RM. Chemical Physics Letters. 1980;72:5–9. [Google Scholar]
- 32.Scheiner AC, Scuseria GE, Schaefer HF., III Journal of the American Chemical Society. 1986;108:8160–62. doi: 10.1021/ja00284a023. [DOI] [PubMed] [Google Scholar]
- 33.Scuseria GE, Scheiner AC, Lee TJ, Rice JE, Schaefer HF., III Journal of Chemical Physics. 1987;86:2881–90. [Google Scholar]
- 34.Windisch VL, Smith AB, III, Hochstrasser RM. Journal of Physical Chemistry. 1988;92:5366–70. [Google Scholar]
- 35.Zhao XS, Miller WB, Hintsa EJ, Lee YT. Journal of Chemical Physics. 1989;90:5527–35. [Google Scholar]
- 36.Scuseria GE, Schaefer HF., III Journal of Physical Chemistry. 1990;94:5552–4. [Google Scholar]
- 37.King DS, Denny CT, Hochstrasser RM, Smith AB., III Journal of the American Chemical Society. 1977;99:271–3. [Google Scholar]
- 38.McDonald JR, Brus LE. Journal of Chemical Physics. 1973;59:4966–71. [Google Scholar]
- 39.Meyling JH, Vanderwe Rp, Wiersma DA. Chemical Physics Letters. 1974;28:364–72. [Google Scholar]
- 40.Hochstrasser RM, King DS, Nelson AC. Chemical Physics Letters. 1976;42:8–12. [Google Scholar]
- 41.Burland DM, Carmona F, Pacansky J. Chemical Physics Letters. 1978;56:221–26. [Google Scholar]
- 42.Ma CS, Kwok WM, An HY, Guan XG, Fu MY, Toy PH, Phillips DL. Chemistry-a European Journal. 2010;16:5102–18. doi: 10.1002/chem.200902891. [DOI] [PubMed] [Google Scholar]
- 43.Pirrung MC, Ye T, Zhou Z, Simon JD. Photochemistry and Photobiology. 2006;82:1258–64. doi: 10.1562/2006-02-07-RA-798. [DOI] [PubMed] [Google Scholar]
- 44.Ma CS, Kwok WM, Chan WS, Zuo P, Kan JTW, Toy PH, Phillips DL. Journal of the American Chemical Society. 2005;127:1463–72. doi: 10.1021/ja0458524. [DOI] [PubMed] [Google Scholar]
- 45.To TT, Heilweil EJ, Duke CB, Ruddick KR, Webster CE, Burkey TJ. Journal of Physical Chemistry A. 2009;113:2666–76. doi: 10.1021/jp8068909. [DOI] [PubMed] [Google Scholar]
- 46.Oxley JC, Smith JL, Zhang J. Journal of Physical Chemistry A. 2000;104:6764–77. [Google Scholar]
- 47.Huang C-Y, Getahun Z, Zhu Y, Klemke JW, DeGrado WF, Gai F. Proc Natl Acad Sci U S A. 2002;99:2788–93. doi: 10.1073/pnas.052700099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kubelka J, Henry ER, Cellmer T, Hofrichter J, Eaton WA. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:18655–62. doi: 10.1073/pnas.0808600105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kubelka J, Hofrichter J, Eaton WA. Current Opinion in Structural Biology. 2004;14:76–88. doi: 10.1016/j.sbi.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 50.Thompson PA, Munoz V, Jas GS, Henry ER, Eaton WA, Hofrichter J. Journal of Physical Chemistry B. 2000;104:378–89. [Google Scholar]
- 51.Werner JH, Dyer RB, Fesinmeyer RM, Andersen NH. Journal of Physical Chemistry B. 2002;106:487–94. [Google Scholar]
- 52.Zhou YQ, Karplus M. Nature. 1999;401:400–03. doi: 10.1038/43937. [DOI] [PubMed] [Google Scholar]
- 53.Culik RM, Serrano AL, Bunagan MR, Gai F. Angewandte Chemie-International Edition. 2011;50:10884–87. doi: 10.1002/anie.201104085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Leeson DT, Gai F, Rodriguez HM, Gregoret LM, Dyer RB. Proc Natl Acad Sci U S A. 2000;97:2527–32. doi: 10.1073/pnas.040580397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Serrano AL, Tucker MJ, Gai F. Journal of Physical Chemistry B. 2011;115:7472–78. doi: 10.1021/jp200628b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Duan Y, Kollman PA. Science. 1998;282:740–44. doi: 10.1126/science.282.5389.740. [DOI] [PubMed] [Google Scholar]
- 57.Hummer G, Garcia AE, Garde S. Phys Rev Lett. 2000;85:2637–40. doi: 10.1103/PhysRevLett.85.2637. [DOI] [PubMed] [Google Scholar]
- 58.Hummer G, Garcia AE, Garde S. Proteins-Structure Function and Genetics. 2001;42:77–84. [PubMed] [Google Scholar]
- 59.Lazaridis T, Karplus M. Science. 1997;278:1928–31. doi: 10.1126/science.278.5345.1928. [DOI] [PubMed] [Google Scholar]
- 60.Piana S, Lindorff-Larsen K, Shaw DE. Biophysical journal. 2011;100:L47–L49. doi: 10.1016/j.bpj.2011.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Eaton WA, Munoz V, Thompson PA, Henry ER, Hofrichter J. Accounts of Chemical Research. 1998;31:745–53. [Google Scholar]
- 62.Bowman GR, Pe VS. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:10890–95. doi: 10.1073/pnas.1003962107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Faisca PFN, Nunes A, Travasso RDM, Shakhnovich EI. Protein Science. 2010;19:2196–209. doi: 10.1002/pro.498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Freddolino PL, Harrison CB, Liu YX, Schulten K. Nature Physics. 2010;6:751–58. doi: 10.1038/nphys1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Piana S, Sarkar K, Lindorff-Larsen K, Guo MH, Gruebele M, Shaw DE. Journal of Molecular Biology. 2011;405:43–48. doi: 10.1016/j.jmb.2010.10.023. [DOI] [PubMed] [Google Scholar]
- 66.Auer S, Miller MA, Krivov SV, Dobson CM, Karplus M, Vendruscolo M. Physical Review Letters. 2007;99 doi: 10.1103/PhysRevLett.99.178104. [DOI] [PubMed] [Google Scholar]