

Abstract
Polymorphisms in the prion protein gene are known to affect prion disease incubation times and susceptibility in humans and mice. However, studies with inbred lines of mice show that large differences in incubation times occur even with the same amino acid sequence of the prion protein, suggesting that other genes may contribute to the observed variation. To identify these loci we analyzed 1,009 animals from an F2 intercross between two strains of mice, CAST/Ei and NZW/OlaHSd, with significantly different incubation periods when challenged with RML scrapie prions. Interval mapping identified three highly significantly linked regions on chromosomes 2, 11, and 12; composite interval mapping suggests that each of these regions includes multiple linked quantitative trait loci. Suggestive evidence for linkage was obtained on chromosomes 6 and 7. The sequence conservation between the mouse and human genome suggests that identification of mouse prion susceptibility alleles may have direct relevance to understanding human susceptibility to bovine spongiform encephalopathy (BSE) infection, as well as identifying key factors in the molecular pathways of prion pathogenesis. However, the demonstration of other major genetic effects on incubation period suggests the need for extreme caution in interpreting estimates of variant Creutzfeldt–Jakob disease epidemic size utilizing existing epidemiological models.
The appearance of the novel human prion disease variant Creutzfeldt–Jakob disease (vCJD) and the confirmation that it is caused by the bovine spongiform encephalopathy (BSE) prion strain, has led to concerns that a major epidemic of vCJD will evolve over the years ahead (1–3).
Prion diseases have prolonged incubation periods and coding polymorphisms in the prion protein (PrP) gene are known to affect incubation times and susceptibility in humans, mice, and sheep (4–8). In the human PrP gene (PRNP), a polymorphism occurs at codon 129 where either a methionine or valine may be encoded. Acquired and sporadic prion diseases occur mostly in homozygous individuals, and a protective effect of heterozygosity is also seen in some inherited cases (4, 5, 9). All cases of vCJD described to date have occurred in methionine homozygous individuals, a genotype shared by ≈40% of the British Caucasian population (10). In mice, two polymorphisms in the murine PrP gene (Prnp) have been described where Prnpa (Leu-108, Thr-189) and Prnpb (Phe-108, Val-189) are associated with short and long incubation times, respectively (6, 11–13).
Although the influence of PrP gene polymorphisms on susceptibility and incubation time is well established, other lines of evidence indicate that PrP amino acid differences are not the sole genetic influence (14–16). Comparison of several inbred lines of laboratory mice with the same Prnp genotype reveals major differences in incubation times to a defined prion strain (varying from 100 to 500 days), suggesting that other factors including additional genetic loci may contribute to the observed variation (11, 12, 17, 18). The identification of these loci in humans may allow identification of at risk individuals, allow more robust predictions of human epidemic parameters, and identify prion ligands and biochemical pathways that will allow a better understanding of prion pathogenesis and the development of rational therapeutics.
Direct identification of human quantitative trait loci (QTL) is both technically challenging and expensive because of the large sample sizes that are necessary to detect alleles of modest effect in randomly mating populations. The advantages of studying organisms in which breeding designs are under experimental control are well recognized; the availability of inbred lines of mice, the ability to generate large genetic crosses, and the similarity of the mouse and human genomes make mice an excellent model for identifying susceptibility loci.
Materials and Methods
Mice.
NZW/OlaHsd were obtained from Harlan U.K. Ltd. (Bicester, U.K.) and CAST/Ei mice were obtained from the Medical Research Council Mammalian Genome Center (Harwell, U.K.). The F1 generation was generated in two ways: male CAST/Ei × female NZW/OlaHsd and female CAST/Ei × male NZW/OlaHsd. The F2 generation was established by intercrossing the F1s in all four possible combinations (Table 2). A subcutaneous transponder tag identified mice individually.
Table 2.
Experimental crosses
| Cross | Breeding | Incubation time, days ± s.d. | ||
|---|---|---|---|---|
| F1 | ||||
| A | Male CAST/Ei × female NZW/OlaHsd | 169 ± 0 (n = 11) | ||
| B | Male NZW/OlaHsd × female CAST/Ei | 173 ± 8 (n = 28) | ||
| F2 | ||||
| 1 | Male cross A × female cross A | 154 ± 23 (n = 271) | Males 155 ± 22 (n = 136) | Females 154 ± 24 (n = 135) |
| 2 | Male cross A × female cross B | 157 ± 28 (n = 302) | Males 157 ± 29 (n = 139) | Females 157 ± 27 (n = 163) |
| 3 | Male cross B × female cross A | 162 ± 26 (n = 140) | Males 164 ± 28 (n = 61) | Females 160 ± 24 (n = 79) |
| 4 | Male cross B × female cross B | 159 ± 27 (n = 296) | Males 160 ± 25 (n = 137) | Females 159 ± 28 (n = 159) |
Inoculation and Phenotyping.
Chandler/RML mouse adapted scrapie was obtained from A. Aguzzi (Institute of Neuropathology, University of Zurich, Zurich) and passaged once in CD1 Swiss mice (Harlan U.K. Ltd.). The presence of the Prnpa allele was confirmed in CD1 Swiss mice by sequencing. Brains from these animals were used to generate a 1% homogenate in PBS, which was used as the inoculum for all subsequent experiments. Mice were anaesthetized with halothane/O2 and inoculated intracerebrally into the right parietal lobe with 30 μl of the inoculum. All mice were examined daily for the development of clinical signs of scrapie. At onset of signs, mice were examined more rigorously for neurological signs of disease. Animals were culled as soon as clinical scrapie was confirmed or if showing signs of distress. Criteria for clinical diagnosis of scrapie in mice were as described (6). Incubation time was measured by the number of days from inoculation to the onset of clinical scrapie.
Genotyping.
DNA was extracted from 1-cm tail biopsies by using a Promega DNA extraction kit and resuspended in 100 μl of TE (10 mM Tris⋅HCl/1 mM EDTA, pH 7.5). This stock DNA (0.5 μl of a 1:10 dilution) was used as template in a 5-μl PCR. All PCRs were carried out in 96-well plates either by using an 877 Integrated Thermal Cycler (Applied Biosystems) or a PTC-225 (MJ Research, Cambridge, MA). Fluorescently labeled primers were selected from the mouse MapPairs set (Research Genetics, Huntsville, AL), and additional primers were obtained from Sigma–Genosys. PCR conditions were determined empirically for each primer pair, but in general reactions were carried out in 3 mM MgCl2 with AmpliTaq-Gold (Applied Biosystems) in the buffer provided. Cycling conditions were as follows: 94°C for 10 min; 94°C for 30 s, 55°C for 30 s, 72°C for 30 s for 35 cycles; 72°C for 5 min. Alleles were detected on a 377 sequencer (Applied Biosystems) and analyzed by using genotyper software (Applied Biosystems).
Data Analysis.
Statistical analyses were carried out by using statview (SAS Institute, Cary, NC). Incubation times were log transformed for interval linkage analysis by using MAPMAKER/EXP 3.0 and MAPMAKER-QTL programs (19) to calculate logarithm of odds (lod) scores for free, dominant, recessive, and additive QTL models. Composite interval mapping (20, 21), which combines interval mapping with multiple regression, was performed with the qtl cartographer computer package (22, 23) in an analysis of the markers on chromosomes 2, 11, and 12. In the first stage of the analysis, the srmapqtl program, which uses the technique of forward stepwise regression, was used to construct a multilocus model. Markers were added to the model if they improved the overall fit at the P < 0.1 significance level. At the end of this procedure, all included markers were retested (by backward elimination) to check that they were still significantly retained in the multilocus model. The zmapqtl program was used to perform composite interval mapping, which allows for the presence of more than one QTL per chromosome under the assumption that the constitutive QTL do not interact epistatically. In composite interval mapping, the vector of trait values (Y) is fitted in a multiple regression model:
 |
where b* and d* are additive and dominance effects for a putative
QTL, x* and z* are indicator variable vectors of genotype probabilities
calculated by interval mapping techniques, B is a vector of effects and
X is the marker information matrix for the selected background markers,
and E is the random error vector. Maximum likelihood estimates of the
parameters are calculated by using an ECM (Expectation/Conditional
Maximization) algorithm (24). Under the null hypothesis,
H0: Y = XB + E, the residual variance will
be s . Under the alternative hypothesis,
H1: Y = x*b* + z*d* + XB + E, the residual
variance would be s
. Under the alternative hypothesis,
H1: Y = x*b* + z*d* + XB + E, the residual
variance would be s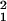 . The proportion of variance
explained by a QTL at a test location conditioned on the background
markers can be defined as r2 =
(s
. The proportion of variance
explained by a QTL at a test location conditioned on the background
markers can be defined as r2 =
(s −
s
−
s )/s2, where
s2 is the trait variance. Alternatively, the
proportion of the total variance explained by the QTL and the
background markers (r
)/s2, where
s2 is the trait variance. Alternatively, the
proportion of the total variance explained by the QTL and the
background markers (r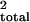 ) is defined as
) is defined as
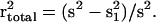 |
Results and Discussion
In mice, prion disease incubation time can be treated as a continuous or quantitative trait that ranges from around 100 to over 500 days and is known to be influenced by many factors, including route of infection, dose, prion strain, levels of PrP expression, and genetic susceptibility (25). To identify QTL for prion disease incubation time we generated an F2 intercross between two strains of mice, CAST/Ei and NZW/OlaHsd, with significantly different incubation times (Table 1) when inoculated intracerebrally with Chandler/RML scrapie prions. Sequencing the ORF of Prnp from the mouse colonies used in the cross confirmed that both parental strains were Prnpa/a and had no other coding differences. Incubation times were recorded for a total of 1,009 F2 animals. As detailed in Table 2, the F2 intercross was set up in four ways to test for the presence of epigenetic effects. Male and female F2 progeny (crosses 3 and 4) from cross-B fathers (male NZW/OlaHsd × female CAST/Ei) have a significantly longer incubation time (P = 0.01) than animals (crosses 1 and 2) derived from cross-A fathers (male CAST/Ei × female NZW/OlaHsd) as determined by two-way ANOVA. This effect is independent of maternal origin and genetic locus, therefore all linkage data shown represent a combination of all crosses (Table 3). Animals that died at inoculation, from intercurrent illness, or without showing clinical signs of scrapie were excluded from the analysis. However, this did not alter the expected genotype ratios for each locus examined. The mean incubation time (in days) for the F2 animals was 158 ± 26 with a minimum of 99 and maximum of 274. The upper limit of this range (274 days) is substantially greater than the CAST/Ei parental incubation time (188 ± 12), suggesting a greater number of “long” incubation time QTL in these animals as a result of independent assortment of “long” and “short” alleles from both the NZW/OlaHsd and CAST/Ei parents. The distribution of the F2 log transformed incubation times had a skewness of 0.86 and a kurtosis of 0.99.
Table 1.
Incubation times
| Strain | Incubation time, days ± s.d. |
|---|---|
| NZW/OlaHsd | 108 ± 4 (n = 38) |
| CAST/Ei | 188 ± 12 (n = 16) |
| F1 | 172 ± 7 (n = 34) |
| F2 | 158 ± 26 (n = 1009) |
| Males 158 ± 26 (n = 473) | |
| Females 157 ± 26 (n = 536) |
Data shown represent the combined data for all crosses.
Table 3.
Results of genome scan in F2 intercross (MapMaker analysis)
| Nearest marker | Position | Peak lod score | % Variance | Genotype mean, days ± sd (n)
|
||
|---|---|---|---|---|---|---|
| N/N | C/N | C/C | ||||
| D2Mit107 | 61.2 | 8.2 | 3.7 | 165 ± 27 (242) | 158 ± 26 (502) | 152 ± 24 (265) |
| D2Mit194 | 66.7 | 4.8 | 2.2 | 163 ± 27 (238) | 158 ± 27 (459) | 152 ± 25 (245) |
| D2Mit266 | 98.4 | 4.2 | 2.1 | 158 ± 27 (163) | 154 ± 24 (572) | 163 ± 27 (195) |
| D11Mit36 | 43.7 | 57.6 | 24.4 | 143 ± 19 (250) | 155 ± 21 (442) | 177 ± 29 (275) |
| D11Mit179 | 49.2 | 56.1 | 25.0 | 143 ± 19 (235) | 154 ± 21 (453) | 176 ± 19 (235) |
| D12Mit97 | 42.6 | 5.2 | 2.4 | 153 ± 27 (267) | 160 ± 26 (388) | 164 ± 27 (226) |
| D12Mit28 | 47 | 6.8 | 3.1 | 150 ± 26 (239) | 159 ± 25 (507) | 162 ± 27 (261) |
| D12Mit141 | 51.4 | 5.5 | 2.7 | 152 ± 29 (235) | 158 ± 24 (476) | 163 ± 27 (265) |
| D6Mit123 | 17.5 | 3.9 | 1.8 | 152 ± 24 (240) | 160 ± 27 (476) | 161 ± 26 (271) |
| D7Mit250 | 28.4 | 3.6 | 2.1 | 152 ± 25 (220) | 161 ± 28 (465) | 159 ± 25 (227) |
Position is given as cM from centromere (data from Whitehead Institute/Massachusetts Institute of Technology Center for Genome Research). % variance is calculated by mapmaker. Significant lod scores >4.3; suggestive lod scores >2.8 (26). sd, standard deviation; n, number of animals; N/N, NZW/OlaHsd homozygote; C/C, CAST/Ei homozygote; C/N, heterozygote.
The genome screen was carried out in two stages. In the first stage, ≈400 F2 animals were genotyped with 137 fluorescently labeled primers (Research Genetics, Huntsville, AL) covering the whole genome with an average intermarker distance of 11 cM. The largest intermarker interval was 27.3 cM. Linkage analysis was carried out on this data set by using the MAPMAKER/EXP 3.0 and MAPMAKER-QTL programs (19). Regions that gave lod scores of greater than 2 were followed up in stage 2 with a more extensive screen. In this stage, a further 600 F2 animals were genotyped with markers selected from the first screen. In addition, all 1,009 mice were genotyped with an additional 20 markers selected from the Mouse Genome Database to increase the mapping resolution in regions of interest. Incubation times were log transformed (to ensure homoscedascity) for interval linkage analysis with MAPMAKER-QTL, where the QTL models were not constrained. The thresholds for significant (genome-screen P-value <0.05) and suggestive linkage were taken as lod scores of 4.3 and 2.8, respectively (26). Chromosomes 2, 11, and 12 show regions of highly significant linkage (Table 3 and Fig. 1), whereas suggestive linkage is seen on chromosomes 6 and 7 (Table 3).
Figure 1.

Lod score plots obtained for 1,009 F2 animals on (a) chromosome 2, (b) chromosome 11, and (c) chromosome 12. All plots represent output from MAPMAKER-QTL showing the results from the unconstrained genetic model. The vertical axis shows the lod scores and the horizontal axis displays the relative positions of the markers along the chromosome from centromere (Left) to telomere (Right) in cM as determined by MAPMAKER. The dashed horizontal line shows the lod score (4.3), which represents significant linkage (26).
The data for chromosomes 2, 11, and 12 was analyzed further in a
composite interval mapping analysis by using the computer package
qtl cartographer. This approach tests for a putative QTL in
an interval while using other (linked or unlinked) markers as
covariates in a multivariate model to reduce the residual variance with
the result that the efficiency of the linkage test can be improved.
Forward stepwise regression analysis (srmapqtl) identified
seven markers contributing significantly to overall model goodness of
fit (Table 4). Composite interval mapping
results (zmapqtl), controlling for the seven markers
identified in the previous step and examining the complete maps for
chromosomes 2, 11, and 12, are presented in Table
5. Multiple linked QTL were identified on
all three chromosomes; the three chromosomes jointly explained 82% of
the total variance of log-incubation time (r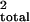 ,
Table 5).
,
Table 5).
Table 4.
Markers identified in stepwise regression
| Chromosome | Marker | Position | F-ratio | Degrees of freedom (denominator) |
|---|---|---|---|---|
| 2 | D2Mit182 | 0.01 | 5.42 | 996 |
| 2 | D2Mit106 | 31.51 | 6.27 | 998 |
| 2 | D2Mit107 | 31.81 | 57.79 | 1,004 |
| 2 | D2Mit194 | 39.51 | 3.04 | 994 |
| 11 | D11Mit36 | 40.31 | 307.63 | 1,006 |
| 11 | D11Mit127 | 55.41 | 22.32 | 1,000 |
| 12 | D12Mit28 | 25.01 | 41.71 | 1,002 |
QTL position is calculated by mapmaker and is given in cM. The significance of the partial regression coefficients (to model additive and dominance effects) for each marker are evaluated as they stepwise increment the multiple regression model. The numerator degrees of freedom is 2 for each marker. The denominator degrees of freedom is 1,009 (number of animals) − 1 − 2 multiplied by the number of markers included in the regression equation at each step. All F-ratios are significant at the 5% level.
Table 5.
Composite interval mapping results
| Chromosome | Closest marker | QTL position | Lod score | r2 | r
|
|---|---|---|---|---|---|
| 2 | D2Mit107 | 31.81 | 8.15 | 0.036 | 0.073 |
| 2 | D2Mit194 | 39.51 | 4.51 | 0.018 | 0.054 |
| 2 | D2Mit266 | 97.31 | 4.60 | 0.001 | 0.044 |
| 11 | D11Mit36 | 40.31 | 56.41 | 0.212 | 0.255 |
| 11 | D11Mit179 | 47.01 | 53.97 | 0.161 | 0.197 |
| 12 | D12Mit97 | 14.01 | 5.86 | 0.033 | 0.061 |
| 12 | D12Mit28 | 25.01 | 7.55 | 0.041 | 0.070 |
| 12 | D12Mit141 | 33.21 | 5.68 | 0.031 | 0.060 |
QTL position is calculated by mapmaker and is given
in cM. r2 and r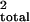 are two different
estimates of the variance attributed to a particular QTL as determined
by zmapqtl.
are two different
estimates of the variance attributed to a particular QTL as determined
by zmapqtl.
The composite interval analysis (20, 21) suggests that multiple linked QTL underlie the linked regions identified in the interval mapping (mapmaker) analysis, which searches for a single QTL at a time. Maximum lod scores were observed in several intervals of markers not identified in stepwise regression; however, the stepwise regression analysis was intended only to identify potential background contributions. Modest significant effects were observed for three linked QTL on chromosomes 2 and 12, but two linked QTL on chromosome 11 were found to account for over 45% of the total variance observed for this trait. This analysis did not explore potential epistasis or gene-environment interactions, which may have contributed to the overall r2 for individual QTL. These results will be useful to guide the design and interpretation of advanced crosses and/or congenic lines that we plan to construct to fine-map and positionally clone the loci that underlie the three linked regions.
Prnp and its paralogue Prnd both map within the 95% confidence interval for the peak lod score of 8.2 on chromosome 2 (Fig. 1a), thus providing excellent candidates for the QTL—although there are no amino acid differences between CAST/Ei and NZW/OlaHsd for either PrP or Dpl (data not shown), suggesting that regulatory regions may be important (27). Restriction fragment length polymorphisms around the Prnp locus have been described (12) where CAST/Ei is haplotype Prnpe and NZW/OlaHsd is Prnpa, suggesting that differences exist between the two strains in this region. This is not particularly surprising because CAST/Ei and NZW/OlaHsd are only distantly related (28). Polymorphisms have been identified in the promoter of Prnp, which have been implicated in regulating levels of expression (29, 30). We have identified single nucleotide polymorphisms in the promoter of Prnp between CAST/Ei and NZW/OlaHsd, but their significance remains to be determined (data not shown). Expression levels of PrP are known to correlate inversely with incubation time in knockout and transgenic mice (31, 32). PrPc levels were measured in the brains of 8-week-old CAST/Ei and NZW/OlaHsd parents, but in preliminary experiments no substantial differences were detected (data not shown). The genotype means for D2Mit107 are 165 and 152 days for NZW/OlaHsd and CAST/Ei homozygotes, respectively, which is the reverse of what would be expected based on the parental incubation times and that observed with all other linked loci (Tables 1 and 3). This is consistent with the observation that some F2 animals have an incubation time greater than either of the parental strains and reflects the fact that both NZW/OlaHsd and CAST/Ei have both “long” and “short” incubation time alleles, which segregate independently in the F2 generation. The genotype means for D2Mit107 (Table 3) also show significant differences for all genotypes (P < 0.005), suggesting an additive pattern of inheritance.
Of the total variance observed in this cross, 45% maps to two loci on chromosome 11. Numerous potential candidate genes map to this region of chromosome 11; however, further work will be required to refine the mapping data .The genotype means for markers on chromosome 11 (Table 3) show significant differences for all genotypes (P < 0.0001), suggesting an additive model of inheritance for these QTL.
The genotype means on chromosomes 12 show evidence of dominant inheritance with significant difference (P < 0.0001) observed between the N/N and C/C and also N/N and N/C genotypes, but not between C/C and N/C. The region of linkage spans over 20 cM, which includes many potential candidate genes such as Prpl3 (prion protein ligand 3) (33). Prpl3 was identified by its ability to bind PrPc and was mapped to chromosome 12 by restriction fragment length polymorphism analysis in a Jackson Laboratory interspecific backcross (33). Prpl3 shows 81% sequence identity to the human EST H06169, which on a blast search against the GenBank htgs database gives a 97% sequence identity match to a sequence derived from bacterial artificial chromosome (BAC) RP11-422L23 (GenBank accession no. AL356322), which has been mapped to the human X chromosome. This region of mouse chromosome 12 is reported to have homology to human chromosome 14; therefore, to clarify the location of Prpl3, primers were designed to the mouse cDNA sequence and used to screen the T31 radiation hybrid panel. Our data place Prpl3 13.93 cR from DXMit38, confirming its location on mouse chromosome X and excluding it as a candidate QTL for prion disease incubation time. However, it remains a possibility that the X chromosome locus represents a pseudogene and that the real gene is on mouse chromosome 12 and human chromosome 14.
A preliminary survey for genetic interaction between the linked loci was conducted by using two-way ANOVA. Only marginal significance was detected between D11Mit179 and D12Mit28 (P = 0.016), suggesting that the three regions generally act independently (additively) to influence the incubation time phenotype.
We have demonstrated unequivocally that genetic loci other than the ORF of Prnp can have a major effect on prion disease incubation time in mice and, furthermore, have identified eight QTL on three chromosomes that explain 82% of the variance. Although refining the regions of linkage identified in this study and characterizing relevant polymorphisms will still require considerable work, this study provides hope that we may also be able to identify human alleles that predispose to a shorter incubation time for BSE in humans.
Previous reports suggested a QTL for incubation time on chromosome 17 within the H2 locus of the major histocompatibility complex, although this was not replicated by others or identified in our cross (18, 34). However, challenge with different prion strains by different routes of inoculation and in other strains of mice may result in the segregation of alternative susceptibility genes. Recently, a much smaller study, which looked at only 153 F2 animals from an SJL/J and CAST/Ei intercross, identified loci on chromosomes 9 and 11 (35). The chromosome 9 locus was not identified in our cross, suggesting that this may be an SJL/J-specific effect. The chromosome 11 locus falls within the broad region of linkage described here, which is consistent with the suggestion that mouse chromosome 11 contains multiple QTL.
The identification of QTL for prion disease incubation time cast doubt on the validity of the genetic models used in current epidemiological studies, which may result in overly optimistic predictions of the size of the vCJD epidemic (36). These models assume that only methionine homozygous individuals are susceptible to vCJD. This in itself appears unlikely because the other acquired human prion diseases, iatrogenic CJD and kuru, occur in all codon 129 genotypes as the epidemic evolves, with codon 129 heterozygotes having the longest mean incubation periods (37–39). By definition, the patients identified to date with vCJD are those with the shortest incubation periods for BSE. These in turn, given that no unusual history of dietary, occupational, or other exposure to BSE has been identified, would be expected to be predominantly those individuals with short incubation time alleles at these multiple genetic loci in addition to having the codon 129 methionine homozygous PRNP genotype (38). The vCJD cases reported to date may, therefore, represent a distinct genetic subpopulation with unusually short incubation periods to BSE prions. Collectively, the effect on susceptibility and incubation time of the loci mapped could be more significant than that exerted by the PRNP locus itself. It should also be considered that variation of prion incubation periods between inbred mouse lines is even greater when considering transmission from one mammalian species to another (for instance with BSE transmission to mice). Here, additional genes involved in the species barrier itself would also be relevant. Because the frequencies of such genetic polymorphisms, alone or in combination, are unknown, this severely limits the utility of epidemiological predictions based only on these early vCJD patients.
Acknowledgments
We thank R. J. Bond and D. Moore for technical assistance. This work was funded by the United Kingdom Medical Research Council. P.T. was supported through the Mayo Foundation Scholarship scheme.
Abbreviations
- BSE
bovine spongiform encephalopathy
- CJD
Creutzfeldt-Jakob disease
- vCJD
variant CJD
- QTL
quantitative trait loci
- lod
logarithm of odds
- PrP
prion protein
- Dpl
doppel protein
References
- 1.Collinge J, Sidle K C L, Meads J, Ironside J, Hill A F. Nature (London) 1996;383:685–690. doi: 10.1038/383685a0. [DOI] [PubMed] [Google Scholar]
- 2.Hill A F, Desbruslais M, Joiner S, Sidle K C L, Gowland I, Collinge J. Nature (London) 1997;389:448–450. doi: 10.1038/38925. [DOI] [PubMed] [Google Scholar]
- 3.Bruce M E, Will R G, Ironside J W, McConnell I, Drummond D, Suttie A, McCardle L, Chree A, Hope J, Birkett C, et al. Nature (London) 1997;389:498–501. doi: 10.1038/39057. [DOI] [PubMed] [Google Scholar]
- 4.Collinge J, Palmer M S, Dryden A J. Lancet. 1991;337:1441–1442. doi: 10.1016/0140-6736(91)93128-v. [DOI] [PubMed] [Google Scholar]
- 5.Palmer M S, Dryden A J, Hughes J T, Collinge J. Nature (London) 1991;352:340–342. doi: 10.1038/352340a0. [DOI] [PubMed] [Google Scholar]
- 6.Carlson G A, Kingsbury D T, Goodman P A, Coleman S, Marshall S T, DeArmond S J, Westaway D, Prusiner S B. Cell. 1986;46:503–511. doi: 10.1016/0092-8674(86)90875-5. [DOI] [PubMed] [Google Scholar]
- 7.Hunter N, Goldmann W, Smith G, Hope J. Arch Virol. 1994;137:171–177. doi: 10.1007/BF01311184. [DOI] [PubMed] [Google Scholar]
- 8.Westaway D, Zuliani V, Cooper C M, Da Costa M, Neuman S, Jenny A L, Detwiler L, Prusiner S B. Genes Dev. 1994;8:959–969. doi: 10.1101/gad.8.8.959. [DOI] [PubMed] [Google Scholar]
- 9.Baker H E, Poulter M, Crow T J, Frith C D, Lofthouse R, Ridley R M, Collinge J. Lancet. 1991;337:1286–1286. doi: 10.1016/0140-6736(91)92953-y. [DOI] [PubMed] [Google Scholar]
- 10.Collinge J, Beck J, Campbell T, Estibeiro K, Will R G. Lancet. 1996;348:56–56. doi: 10.1016/s0140-6736(05)64378-4. [DOI] [PubMed] [Google Scholar]
- 11.Westaway D, Goodman P A, Mirenda C A, McKinley M P, Carlson G A, Prusiner S B. Cell. 1987;51:651–662. doi: 10.1016/0092-8674(87)90134-6. [DOI] [PubMed] [Google Scholar]
- 12.Carlson G A, Goodman P A, Lovett M, Taylor B A, Marshall S T, Peterson-Torchia M, Westaway D, Prusiner S B. Mol Cell Biol. 1988;8:5528–5540. doi: 10.1128/mcb.8.12.5528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moore R C, Hope J, McBride P A, McConnell I, Selfridge J, Melton D W, Manson J C. Nat Genet. 1998;18:118–125. doi: 10.1038/ng0298-118. [DOI] [PubMed] [Google Scholar]
- 14.Hill A F, Butterworth R J, Joiner S, Jackson G, Rossor M N, Thomas D J, Frosh A, Tolley N, Bell J E, Spencer M, et al. Lancet. 1999;353:183–189. doi: 10.1016/s0140-6736(98)12075-5. [DOI] [PubMed] [Google Scholar]
- 15.Owen F, Poulter M, Collinge J, Crow T J. Am J Hum Genet. 1990;46:1215–1216. [PMC free article] [PubMed] [Google Scholar]
- 16.Will R G, Zeidler M, Stewart G E, Macleod M A, Ironside J W, Cousens S N, Mackenzie J, Estibeiro K, Green A J E, Knight R S. Ann Neurol. 2000;47:575–582. [PubMed] [Google Scholar]
- 17.Dickinson A G. Genetics. 1975;79,Suppl:387–395. [PubMed] [Google Scholar]
- 18.Kingsbury D T, Kasper K C, Stites D P, Watson J D, Hogan R N, Prusiner S B. J Immunol. 1983;131:491–496. [PubMed] [Google Scholar]
- 19.Lander E S, Green P, Abrahamson J, Barlow A, Daly M J, Lincoln S E, Newburg L. Genomics. 1987;1:174–181. doi: 10.1016/0888-7543(87)90010-3. [DOI] [PubMed] [Google Scholar]
- 20.Zeng Z. Proc Natl Acad Sci USA. 1993;90:10972–10976. doi: 10.1073/pnas.90.23.10972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zeng Z. Genetics. 1994;136:1457–1468. doi: 10.1093/genetics/136.4.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith C, Gavora J S, Benkel B, Chesnais J, Fairfull W, Gibson J P, Kennedy B W, Burnside E B. Proc 5th World Congr Genet Appl Livestock Prod. 1994;22:65–66. [Google Scholar]
- 23.Basten, C. J., Weir, B. S. & Zeng, Z.-B. (2000) QTL Cartographer 1.14.
- 24.Meng X, Rubin D B. Biometrika. 2001;80:267–268. [Google Scholar]
- 25.Hunter N, Dann J C, Bennett A D, Somerville R A, McConnell I, Hope J. J Gen Virol. 1992;73:2751–2755. doi: 10.1099/0022-1317-73-10-2751. [DOI] [PubMed] [Google Scholar]
- 26.Lander E, Kruglyak L. Nat Genet. 1995;11:241–247. doi: 10.1038/ng1195-241. [DOI] [PubMed] [Google Scholar]
- 27.Moore R C, Lee I Y, Silverman G L, Harrison P M, Strome R, Heinrich C, Karunaratne A, Pasternak S H, Chishti M A, Liang Y, et al. J Mol Biol. 1999;292:797–817. doi: 10.1006/jmbi.1999.3108. [DOI] [PubMed] [Google Scholar]
- 28.Beck J A, Lloyd S, Hafezparast M, Lennon-Pierce M, Eppig J T, Festing M F, Fisher E M. Nat Genet. 2000;24:23–25. doi: 10.1038/71641. [DOI] [PubMed] [Google Scholar]
- 29.Westaway D, Cooper C, Turner S, Da Costa M, Carlson G A, Prusiner S B. Proc Natl Acad Sci USA. 1994;91:6418–6422. doi: 10.1073/pnas.91.14.6418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baybutt H, Manson J. Gene. 1997;184:125–131. doi: 10.1016/s0378-1119(96)00600-2. [DOI] [PubMed] [Google Scholar]
- 31.Prusiner S B, Scott M, Foster D, Pan K M, Groth D, Mirenda C, Torchia M, Yang S L, Serban D, Carlson G A, et al. Cell. 1990;63:673–686. doi: 10.1016/0092-8674(90)90134-z. [DOI] [PubMed] [Google Scholar]
- 32.Bueler H, Raeber A, Sailer A, Fischer M, Aguzzi A, Weissmann C. Mol Med. 1994;1:19–30. [PMC free article] [PubMed] [Google Scholar]
- 33.Yehiely F, Bamborough P, Da Costa M, Perry B J, Thinakaran G, Cohen F E, Carlson G A, Prusiner S B. Neurobiology. 1997;3:339–355. doi: 10.1006/nbdi.1997.0130. [DOI] [PubMed] [Google Scholar]
- 34.Mohri S, Tateishi J. J Gen Virol. 1989;70:1391–1400. doi: 10.1099/0022-1317-70-6-1391. [DOI] [PubMed] [Google Scholar]
- 35.Stephenson D A, Chiotti K, Ebeling C, Groth D, DeArmond S J, Prusiner S B, Carlson G A. Genomics. 2000;69:47–53. doi: 10.1006/geno.2000.6320. [DOI] [PubMed] [Google Scholar]
- 36.Ghani A C, Ferguson N M, Donnelly C A, Anderson R M. Nature (London) 2000;406:583–584. doi: 10.1038/35020688. [DOI] [PubMed] [Google Scholar]
- 37.D'Aignaux J H, Costagliola D, Maccario J, de Villemeur T B, Brandel J P, Deslys J P, Hauw J J, Chaussain J L, Agid Y, Dormont D, et al. Neurology. 1999;53:1197–1201. doi: 10.1212/wnl.53.6.1197. [DOI] [PubMed] [Google Scholar]
- 38.Collinge J. Lancet. 1999;354:317–323. doi: 10.1016/S0140-6736(99)05128-4. [DOI] [PubMed] [Google Scholar]
- 39.Cervenáková L, Goldfarb L G, Garruto R, Lee H S, Gajdusek D C, Brown P. Proc Natl Acad Sci USA. 1998;95:13239–13241. doi: 10.1073/pnas.95.22.13239. [DOI] [PMC free article] [PubMed] [Google Scholar]