

Abstract
Epithelial cells polarize their plasma membrane into biochemically and functionally distinct apical and basolateral domains where the apical domain faces the 'free' surfaces and the basolateral membrane is in contact with the substrate and neighboring cells. Both membrane domains are separated by tight junctions, which form a diffusion barrier. Apical-basolateral polarization can be recapitulated successfully in culture when epithelial cells such as Madin-Darby Canine Kidney (MDCK) cells are seeded at high density on polycarbonate filters and cultured for several days 1 2. Establishment and maintenance of cell polarity is regulated by an array of small GTPases of the Ras superfamily such as RalA, Cdc42, Rab8, Rab10 and Rab13 3 4 5 6 7. Like all GTPases these proteins cycle between an inactive GDP-bound state and an active GTP-bound state. Specific mutations in the nucleotide binding regions interfere with this cycling 8. For example, Rab13T22N is permanently locked in the GDP-form and thus dubbed 'dominant negative', whereas Rab13Q67L can no longer hydrolyze GTP and is thus locked in a 'dominant active' state 7. To analyze their function in cells both dominant negative and dominant active alleles of GTPases are typically expressed at high levels to interfere with the function of the endogenous proteins 9. An elegant way to achieve high levels of overexpression in a short amount of time is to introduce the plasmids encoding the relevant proteins directly into the nuclei of polarized cells grown on filter supports using microinjection technique. This is often combined with the co-injection of reporter plasmids that encode plasma membrane receptors that are specifically sorted to the apical or basolateral domain. A cargo frequently used to analyze cargo sorting to the basolateral domain is a temperature sensitive allele of the vesicular stomatitis virus glycoprotein (VSVGts045) 10. This protein cannot fold properly at 39°C and will thus be retained in the endoplasmic reticulum (ER) while the regulatory protein of interest is assembled in the cytosol. A shift to 31°C will then allow VSVGts045 to fold properly, leave the ER and travel to the plasma membrane 11. This chase is typically performed in the presence of cycloheximide to prevent further protein synthesis leading to cleaner results. Here we describe in detail the procedure of microinjecting plasmids into polarized cells and subsequent incubations including temperature shifts that allow a comprehensive analysis of regulatory proteins involved in basolateral sorting.
Keywords: Cellular Biology, Issue 51, Epithelial cells, cell polarity, microinjection, basolateral sorting, MDCK
Protocol
1. Isolation of Plasmid DNA
Use a Sigma-Aldrich endotoxin-free maxiprep kit to prepare endotoxin free DNA according to the manufacturer's protocol. This kit works for us, because it reliably removes any endotoxins from DNA preparations. Endotoxins that are injected with the DNA into cell nuclei will lead to cell death.
Add 100 μl phenol/chlorofom/isoamylalcohol (25:24:1) to the isolated DNA, vortex and spin for 1 min at 13,000 rpm in an Eppendorf microcentrifuge. Transfer the upper water phase into a new tube and add 100 μl chloroform/isoamylalcohol (24:1), vortex and spin as above. Transfer the upper water phase containing the DNA to a new tube. This step is needed to remove any protein from the DNA, which prevents clogging of the microinjection needle.
Precipitate the DNA by adding Na Acetate (pH 6.0) to a final concentration of 300 mM and 2 X vol 100% ethanol. Incubate at -20°C overnight. Spin DNA for 20 min at 13,000 rpm in an Eppendorf microcentrifuge, wash once with 70% ethanol, and resuspend in 300 μl endotoxin-free water (Sigma-Aldrich).
Determine DNA concentration. A typical DNA concentration ranges from 1 to 5 μg/μl.
2. Culture of MDCK Cells
Split MDCK cells. Count cells and seed 4 X 105 cells onto clear 12-mm Transwell filters (0.4 μm pore size, Corning Costar, 3460). For an experiment containing a mock-injection control and two different mutant Rab proteins you need to seed three filters.
Culture MDCK cells in MEM medium with 2 mM L-glutamine, 0.1 mg/ml penicillin/streptomycin and 7% (vol/vol) fetal bovine serum (= MEM growth media) at 37°C and 5% CO2.
Change media in the basolateral chamber every day. This aids the polarization process, because it mimics the situation of epithelial cells in animals.
2 days post-seeding check in a microscope whether the cells are growing in a closed monolayer. If you cannot detect any holes in the monolayer, perform your experiment on day 3. If there are still holes, perform your experiment on day 4.
3. Microinjection Procedure and Post-injection Incubations
On the day of the experiment prepare 5 ml MEM growth media plus 50 mM HEPES in one 60 x 15 mm plate for each filter and place into a 39°C incubator. In addition, set up one well of a 12-well plate with 1 ml MEM growth media plus 50 mM HEPES and 0.1 mg/ml cycloheximide per filter, and place into a 31°C incubator.
Turn on your microinjection microscope and set its heated stage to 39°C. We use an inverted microscope (Axiovert 200, Carl Zeiss MicroImaging, Inc.) with a heated stage, 10X and 32X objectives, and an Eppendorf Femtojet (Injectman NI2). Lastly, open the nitrogen gas tank that supplies the air table with nitrogen.
Dilute DNA with filtered water (use 0.2 μm filter) to a final concentration of 0.2 mg/ml in a total volume of 10 - 100 μl. Subsequently, spin DNA in Eppendorf microcentrifuge at 13,000 rpm for 30 min. Remove top portion and place in a new tube. This step insures that when you load your diluted DNA into the microinjection needle you can accurately load "clean" DNA. Your DNA is now ready for microinjection.
Prepare your cells by taking the first filter out of its culture dish. With a surgical blade (Feather Surgical Blade, stainless steel, No 11), cut out the filter from the filter holder and place into 5 ml, 39°C warm MEM growth media plus 50 mM HEPES (60 x 15 mm plate). Weigh down the filter in the culture plate with the surgical blade you used for cutting it out. Place it on the filter such that the hole of the surgical blade surrounds the middle of the filter. Place your culture plate onto the heated stage of the microscope.
Load 2-3 μl of your diluted DNA (in this case plasmids encoding VSVGts045-GFP = mock injection control) into a microinjection needle (Femtotip II, Eppendorf, 930000043), twist the protective cover of the needle and let it fall to the floor. Now the needle is ready to be screwed into its holder. To do so, press the menu key of your injectman and make sure the valve is shut down. Screw the needle into its holder, beware not to screw in the needle too tightly as that may lead to breakage. Now, press the menu key again, the applied Pc will prevent the media from being sucked into the needle during the microinjeciton procedure. Finally, tap your joystick to erase stored homing.
To lower the needle onto the cells, use the 10X objective and bring the needle into the light beam above the liquid. Now focus on the cells, bring the focus up again by turning the focus wheel 180° up, and find the needle. Subsequently, slowly move needle into focus, then bring out of focus again (working towards bringing the cells into focus again), bring down needle into focus again. Repeat until needle touches the surface of the media, at which point all you will see is a halo. When you reach a point at which the cells are in focus, but the needle is still fuzzy (i.e. out of the focal plane) change to the 32X objective and fine coarse settings.
Set the z-limit by touching the apical membrane with the tip of the needle and subtracting about 10 μm as the nuclei are laying approximately 10 μm underneath the apical membrane.
After you set the z-limit, find the right injection pressure. Start at 95 PSI. If the pressure is too high, your cells will blow up. If your pressure is too low, you will see a white dot that stays, but nothing else happens. With a successful injection, you will see a phase change without a cell size change. For the injection, bring the needle a couple of microns out of the focal plane as fast as possible. Aim with the needle above the nucleus (i.e. the middle of an individual cell) and press the injection button of your joystick, but do not hold the button down.
Inject 100-500 cells within the hole of your surgical blade that lies on your cells. When you are done, place your cells with the culture dish and surgical blade into a 39°C incubator, and incubate for 2 h. During this time, VSVGts045-GFP is expressed and secreted into the ER. However, at 39°C, VSVGts045-GFP cannot fold correctly and thus cannot leave the ER. Meanwhile, the small GTPase of interest will accumulate in the cytosol in co-injection experiments.
After 2 h, place your cells into 1 ml MEM growth media plus 50 mM HEPES and 0.1 mg/ml cycloheximide in a 12-well plate and incubate for 2 h at 31°C. During this chase period, VSVGts045-GFP will fold correctly, leave the ER and is delivered to the cell surface.
Repeat steps 3.1 - 3.10 for filters two (inject plasmids encoding VSVGts045-GFP and for example V5-tagged Rab13T22N) and three (inject plasmids encoding VSVGts045-GFP and for example V5-tagged Rab13Q67L). Each injection will take about 20 - 60 min. However, it is absolutely crucial to treat every filter the same way throughout the temperature shift incubations and surface staining until you fix the cells. After fixation, you may perform the rest of your staining protocol for all filters at the same time.
4. Surface Staining for Immunofluorescence Analysis
Note, in order to avoid bleaching of the GFP signal of VSVGts045-GFP, protect the specimens from light by covering with aluminum foil during all subsequent procedures.
If you wish to perform surface staining, place your cells in a culture dish onto a metal plate on ice and wash your cells 1X with ice cold PBS2+ (PBS [0.2 g/liter KCl, 0.2 g/liter KH2PO4, 8 g/liter NaCl, and 2.17 g/liter Na2HPO4 x 7 H2O] plus 0.1 g/liter CaCl2 and 0.1 g/liter MgCl2 x 6 H2O). Subsequently, place 30 μl of an antibody that recognizes the ectodomain of your protein, in this case the mouse monoclonal antibody TK1 (IgG1, obtained from the late Thomas Kreis), onto clean parafilm placed on the metal plate on ice. Place the filter with your cells upside down onto the drop and add a few drops of antibody onto the backside of the filter. Incubate for 1 h on ice.
Place cells into a 12-well plate, wash 3X with ice cold PBS2+ (or RT warm PBS2+ without prior surface staining), and fix with 3% paraformaldehyde for 15 min at RT.
Wash cells once with PBS2+ and leave in PBS2+ for 5 min.
Incubate cells in blocking/permeabilization buffer (BPB) (2% [wt/vol] BSA, 0.4% [wt/vol] saponin in PBS2+) with 10% [vol/vol] goat serum. Incubate 1 h at RT.
Dilute primary antibodies to detect the expressed Rab GTPase, in this example anti-V5 (mouse monoclonal antibody IgG2a, Invitrogen), 1:200 in BPB. Spin diluted antibodies in an Eppendorf microcentrifuge for 10 min at 13,000 rpm. Place 30 μl of the antibody solution on clean parafilm placed in a wet chamber. Place the cells on filter upside down onto the antibody drop and incubate for 1 h at RT.
Place cells back (right side up) into a 12-well plate and wash 5X over 30 min with BPB at RT.
Dilute appropriate secondary antibodies, in this case goat anti-mouse IgG1 Alexa 594-labeled (for VSVG detection on the surface, Invitrogen), and Cy5-labeled secondary antibodies to recognize your Rab GTPase, in this example goat anti-mouse IgG2a Cy5-labeled (Jackson ImmunoResearch), 1:200 into BPB and spin as above. Place 30 μl antibody solution onto clean parafilm in a wet chamber and place cells on filter upside down onto the antibody drop. Incubate 1 h at RT.
Repeat step 4.6.
Dip cells on filter 3X into deionized water and place right side up onto microslides. Add 10-15 μl mount (10% [wt/vol] DABCO, 50% [wt/vol] glycerol in distilled water) on top of the cells. Place 18X18 mm micro cover glass on top and using facial tissues gently press the micro cover glass onto the cells. Seal with nail polish.
Analyze specimens with a confocal microscope. We used a Microsystem LSM 510, Carl Zeiss MicroImaging, Inc. that was equipped with a 63X water immersion lens.
For the preparation of figures, adjust and combine images using programs such as Adobe Photoshop and Adobe Illustrator.
5. Representative Results
For examples on how the co-expression of small GTPases interferes with VSVG sorting kindly refer to published articles for either apical missorting 3, 4 or inhibition of surface delivery 7.
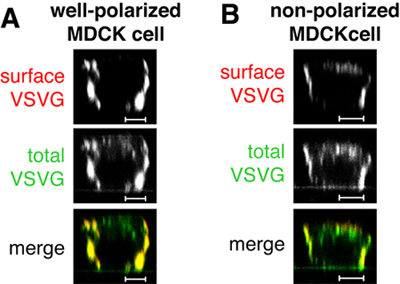 Figure 1. In the mock-injection, i.e. an injection of only the plasmid encoding VSVGts045-GFP, the protein is delivered to the basolateral surface of well-polarized MDCK cells as judged by surface staining (in red, Figure 1 A). Note that not all of the VSVGts045-GFP is delivered to the basolateral membrane during the chase at 31°C as evidenced by the extensive intracellular signal for the total protein (in green, Figures 1 A and B). In cells that are not well-polarized VSVGts045-GFP will be delivered also to the apical membrane (Figure 1 B). If your control specimens look like the cell in Figure 1 B, you cannot trust your data and have to repeat the experiment with better-polarized cells. Scale bars are 5 μm.
Figure 1. In the mock-injection, i.e. an injection of only the plasmid encoding VSVGts045-GFP, the protein is delivered to the basolateral surface of well-polarized MDCK cells as judged by surface staining (in red, Figure 1 A). Note that not all of the VSVGts045-GFP is delivered to the basolateral membrane during the chase at 31°C as evidenced by the extensive intracellular signal for the total protein (in green, Figures 1 A and B). In cells that are not well-polarized VSVGts045-GFP will be delivered also to the apical membrane (Figure 1 B). If your control specimens look like the cell in Figure 1 B, you cannot trust your data and have to repeat the experiment with better-polarized cells. Scale bars are 5 μm.
Discussion
The most critical steps for a successful microinjection experiment are the quality and purity of the DNA and the polarity of your cells. Without polarized cells, your injection control will already have mistargeted VSVG and the experiment cannot be used. If the DNA is of poor quality, the DNA may clog the injection needle leading to poor or no expression of the desired protein at all. Also, it is advisable to use expression plasmids that are known to lead to high expression levels such as pRKV.
If you desire to test for sorting of other cargo proteins, the temperature shift protocols can be modified. For example, to express low-density lipoprotein receptors, we perform the microinjection at 37°C followed by a 1 h incubation at 37°C, 4 h at 20°C (to arrest the cargo in the TGN) and 2 h at 37°C in the presence of 0.1 mg/ml cycloheximide to chase the protein to the surface. For some proteins such as Fc receptors, the incubation time at 20°C may be lowered to 2 h dependent on how fast your cargo protein is synthesized and travels through the secretory pathway. For a detailed description of incubations that we used for different cargo proteins see Nokeset al., 2008 7.
Finally, this protocol can not only be used for the analysis of small GTPases, but in theory for all proteins or protein fragments that you suspect may have a function in (polarized) surface delivery.
Disclosures
No conflicts of interest declared.
Acknowledgments
This work was funded by a grant from the National Institutes of Health (GM070736) to H. Fölsch. S.F. Ang was supported by an A*STAR Graduate Scholarship award, and R.S. Kang was supported by the Cellular and Molecular Basis of Disease Training Program (GM8061)
References
- Mellman I, Nelson WJ. Nature reviews. 2008;9(11):833–833. doi: 10.1038/nrm2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Boulan E, Kreitzer G, Musch A. Nature reviews. 2005;6(3):233–233. doi: 10.1038/nrm1593. [DOI] [PubMed] [Google Scholar]
- Ang AL, Fölsch H, Koivisto UM. The Journal of cell biology. 2003;163(2):339–339. doi: 10.1083/jcb.200307046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroschewski R, Hall A, Mellman I. Nature cell biology. 1999;1(1):8–8. doi: 10.1038/8977. [DOI] [PubMed] [Google Scholar]
- Moskalenko S, Henry DO, Rosse C. Nature cell biology. 2002;4(1):66–66. doi: 10.1038/ncb728. [DOI] [PubMed] [Google Scholar]
- Schuck S, Gerl MJ, Ang A. Traffic (Copenhagen, Denmark) 2007;8(1):47–47. doi: 10.1111/j.1600-0854.2006.00506.x. [DOI] [PubMed] [Google Scholar]
- Nokes RL, Fields IC, Collins RN. The Journal of cell biology. 2008;182(5):845–845. doi: 10.1083/jcb.200802176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins RN. Molecular cell. 2003;12:1064–1064. doi: 10.1016/s1097-2765(03)00445-3. [DOI] [PubMed] [Google Scholar]
- Hall A. Science (New York, N.Y. 1998;279(5350):509–509. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- Scales SJ, Pepperkok R, Kreis TE. Cell. 1997;90(6):1137–1137. doi: 10.1016/s0092-8674(00)80379-7. [DOI] [PubMed] [Google Scholar]
- Keller P, Toomre D, Diaz E. Nature cell biology. 2001;3(2):140–140. doi: 10.1038/35055042. [DOI] [PubMed] [Google Scholar]