

Background: β-Galactoside α2,6-sialyltransferase 1 (ST6Gal-1) action is essential for the anti-inflammatory activity in intravenous immunoglobulin (IVIG) therapy.
Results: Fc sialylation changes in accordance to the severity of inflammation. Inactivation of the P1 promoter abrogated IgG Fc sialylation.
Conclusion: Fc sialylation depends on ST6Gal-1 in the circulation. Defective Fc sialylation is a mechanism for the generally proinflammatory tendencies of the P1-ablated mutant mouse (Siat1ΔP1).
Significance: Anti-inflammatory bioactivity of IVIG requires sialylated Fc.
Keywords: Glycosylation, Immunochemistry, Immunotherapy, Inflammation, Sialyltransferase, IVIG, IgG, ST6Gal I, Sialylation
Abstract
The anti-inflammatory properties associated with intravenous immunoglobulin therapy require the sialic acid modification of the N-glycan of the Fc domain of IgG. Sialylation of the Fc fragment is mediated by β-galactoside α2,6-sialyltransferase 1 (ST6Gal-1), acting on the Gal(β4)GlcNAc terminal structure of the biantennary N-glycans on the Fc domain. However, little is known regarding the in vivo regulation of Fc sialylation and its role in the progression of inflammatory processes. Here, we report that decreased Fc sialylation of circulatory IgG accompanies the acute phase response elicited by turpentine exposure or upon acute exposure to either nontypeable Haemophilus influenzae or ovalbumin. However, Fc sialylation was increased 3-fold from the base line upon transition to chronic inflammation by repeated exposure to challenge. The P1 promoter of the ST6Gal-1 gene is critical for Fc sialylation, but P1 does not drive ST6Gal-1 expression in B cells. The Siat1ΔP1 mouse, with a dysfunctional P1 promoter, was unable to produce sialylated Fc in the systemic circulation, despite the presence of Gal(β4)GlcNAc termini on the Fc glycans. The major contribution of P1 action is to synthesize ST6Gal-1 enzymes that are deposited into the systemic circulation. The data strongly indicate that this pool of extracellular ST6Gal-1 in the blood impacts the sialylation of IgG Fc and that defective Fc sialylation is likely a major contributing mechanism for the proinflammatory tendencies previously noted in Siat1ΔP1 animals.
Introduction
Large-dose intravenous immunoglobulin (IVIG)2 therapy is widely used to treat autoimmune syndromes, including lupus erythematosus and rheumatoid arthritis, and to restore platelet counts in immune-related thrombocytopenia (1, 2). The effectiveness of IVIG therapy in significantly reducing mortality in a humanized xenogeneic model of graft-versus-host disease in NOD/SCID/γc− mice has also been demonstrated recently (3), although the effectiveness as a prophylactic therapy for hematopoietic stem cell transplantation remains controversial (4–6). The anti-inflammatory property associated with IVIG has an absolute requirement for the sialylation of the single biantennary N-glycan at Asn-297 on the Fc region of IgG, which induces the expression of inhibitory Fc receptors (FcγRIIB) (7, 8) via a complex signaling pathway starting with DC-SIGN on dendritic cells or SIGN-R1 on splenic macrophage in mice (7, 9). Sialylated Fc structures (Sia-Fc) are present in 2–4% of IgG in normal human circulation (8) and are generated by the addition of terminal α2,6-linked (but not α2,3-linked) sialic acids. β-Galactoside α2,6-sialyltransferase 1 (ST6Gal-1) mediates construction of α2,6-sialyl linkages on N-glycans and is directly responsible for generation of the anti-inflammatory effects of IVIG (10).
Our laboratory has demonstrated that ST6Gal-1 deficiency results in a generally proinflammatory state; mice with genetically induced ST6Gal-1 deficiency exhibit exaggerated acute peritonitis and Th2 pulmonary inflammation (11, 12). This phenotype was attributed specifically to the inactivation of the liver-specific promoter P1, one of six promoters regulating tissue- and cell-specific transcription of the ST6Gal-1 gene (13–15). Inactivation of P1 results in decreased ST6Gal-1 released into the systemic circulation but no appreciable alteration in the extent of sialylation of circulatory glycoproteins released into the blood by the liver (16). P1-mediated ST6Gal-1 expression in the liver is also regulated in inflammation (15, 17). Although P1 is not believed to be involved in ST6Gal-1 expression in B cells (14), here, we report the surprisingly absolute requirement for the P1 promoter in Sia-Fc production. Targeted ablation of the P1 region in Siat1ΔP1 mice that results in attenuated liver and circulatory ST6Gal-1 levels but normal expression elsewhere (17); these mice do not express Sia-Fc in their circulatory IgG. The data are highly suggestive of a role for the circulatory pool of ST6Gal-1 originating from P1-mediated transcription in the sialylation of the Fc region. This mechanism may explain the generally proinflammatory tendencies of animals with ST6Gal-1 deficiencies.
EXPERIMENTAL PROCEDURES
Animals and Models of Inflammation
Mouse strains C57BL/6 (from JAX®), Siat1ΔP1, and Siat1-null were as described previously (17, 18). For the turpentine model of sterile acute phase response, 100 μl of turpentine was injected subcutaneously into the scruff of the neck. The mice were killed, and sera were collected via terminal heart puncture at the following points: before injection (base line) and 72 h after turpentine treatment. The nontypeable Haemophilus influenzae (NTHi) model of chronic pulmonary inflammation was performed as described earlier (19–21). Briefly, mice received biweekly oropharyngeal instillations of 1 × 106 colony-forming units of live NTHi strain 1479 for 16 consecutive weeks. Blood samples were taken weekly by retro-orbital bleed. For induction of acute allergic airway inflammation by ovalbumin (OVA), mice were sensitized by two intraperitoneal injections of 20 μg of OVA (grade IV, Sigma) adsorbed to 2.25 mg of Imject alum (Al(OH)3-Mg(OH)2, Pierce) in 100 μl of saline on days 0 and 14. Mice were challenged on days 24, 25, 26, and 27 with 20-min inhalations of an aerosol generated by nebulization of a 1% OVA solution prepared in saline. Mice were killed by intraperitoneal injection of 1 ml of Avertin (2.5 g of 2,2,2-tribromethanol and 5 ml of 2-methyl-2-butanol in 200 ml of sterile deionized water) on day 29. For induction of chronic allergic airway inflammation by OVA, mice were sensitized as per the acute model but challenged by OVA nebulization for 2 consecutive days every other week for 3 months (days 28 and 29, 42 and 43, 56 and 57, 70 and 71, 84 and 85, and 98 and 99) and killed on day 101. In both allergy models, bronchoalveolar lavage was performed. The thoracic cavity was opened to expose the trachea, which was cannulated with a 22-gauge intravenous catheter. PBS (750 μl) was injected and withdrawn from the lung two times using a tuberculin syringe. A white blood cell count of the bronchoalveolar lavage fluid was performed using a Z2 COULTER COUNTER (Beckman). The Roswell Park Cancer Institute Animal Care and Use Committee approved all animal studies presented here.
Isolation and Analysis of Fc Regions of Circulatory IgG
Protein A-agarose beads (Sigma) were washed three times with wash buffer (10 mm Tris and 0.1% Nonidet P-40, pH 7), resuspended in wash buffer at the initial volume, and added to an equal volume of serum (50 μl) pooled from five animals. Samples were then shaken vigorously for 90 min at room temperature before being washed three times with wash buffer. The remaining protein A beads were resuspended in elution buffer (0.1 m glycine, 0.1 m sodium acetate, and 5 mm MgCl2, pH 3.5) equal to the volume of the original serum sample and shaken at room temperature for 10 min. After incubation, samples were immediately spun down, and the supernatant was drawn off and adjusted to pH 7 with an equal volume of neutralization solution (0.1 m HEPES, 5 mm MgCl2, and 50 mm NaCl, pH 12) to reach pH 7. Immobilized papain-agarose beads (Pierce) were washed three times with digest buffer (20 mm cysteine, 20 mm sodium phosphate, and 10 mm EDTA, pH 7), resuspended in digest buffer at the initial volume, and added to IgG preparations at a 1:2 ratio. Samples were shaken at room temperature for 24 h and then briefly spun down, and the supernatant containing the Fc fragments was removed.
For Western blot analysis of Fc fragments, samples were separated by either 10 or 12% SDS-PAGE and transferred to a PVDF membrane (Millipore). Fc fragment gels were loaded to equalize the Fab signal (typically, 10 μl of digested IgG). Blots were blocked in TBS/Tween containing 5% BSA for 1 h at room temperature or overnight at 4 °C. Lectin probes used are as follows. Sambucus nigra agglutinin (SNA)-biotin (Vector Laboratories) at a working concentration of 0.08 μg/ml or Polyporus squamosus lectin (PSL; EY Laboratories) at 2.5 μg/ml was used for the detection of α2,6-sialic acids, and Erythrina crista-galli lectin (ECL)-biotin (Vector Laboratories) at 5 μg/ml was used for the detection of terminal galactose. Lectin blots were subsequently incubated with streptavidin-Cy5 (GE Healthcare) at 1:1000 or streptavidin-DyLight 649 (Jackson ImmunoResearch Laboratories) at 1:2000. After lectin binding, Fc fragment blots were incubated with goat anti-mouse Fc-Cy3 and goat anti-mouse Fab-Cy2 (Jackson ImmunoResearch Laboratories) at 1:1000. Fluorescent blots were visualized on a Typhoon Trio (GE Healthcare) and quantified with ImageQuant.
Sample Preparation and Release of N-Linked Glycans for Glycomics
Fc fragments were recovered by preparative SDS-PAGE. All bands between 26 and 34 kDa in each sample gel were extracted by slicing carefully with a scalpel blade, cut into ∼1-mm squares, and transferred into prerinsed screw-cap glass tubes. The gel pieces were destained by alternate addition/exchange of 50 mm ammonium bicarbonate and acetonitrile, which was repeated at least five times until the gels turned clear. After destaining, the gel slices were reswelled with 50 mm ammonium bicarbonate (previously chilled at 4 °C) and allowed to set at 4 °C for 45 min before adding peptide:N-glycosidase F. The enzyme-treated gels were placed in a heating block (37 °C) overnight. The released N-linked glycans were extracted in series with 20% acetonitrile in 5% formic acid, 50% acetonitrile in 5% formic acid, and 80% acetonitrile in 5% formic acid. Each extraction step was accomplished by allowing the gel in extractant solution to set for at least 25 min. The acetonitrile was evaporated under a stream of nitrogen gas, and the extracts were eventually lyophilized. Prior to per-O-methylation, the eluates were passed through a Sep-Pak C18 cartridge (Waters) for purification. The N-linked glycans were permethylated for structural characterization by mass spectrometry. Briefly, the dried eluates were dissolved with dimethyl sulfoxide and methylated with NaOH and methyl iodide. The reaction was quenched with water, and per-O-methylated carbohydrates were extracted with methylene chloride and dried under N2. For profiling by MALDI-TOF-MS, the permethylated glycans were dissolved with methanol and crystallized with α-dihydroxybenzoic acid matrix (20 mg/ml in 50% methanol/water). Analysis of glycans present in the samples was performed in the positive ion mode by MALDI-TOF/TOF-MS using an AB SCIEX TOF/TOF 5800 system (Applied Biosystems/MDS Analytical Technologies). For analysis by linear ion trap-MS, the remainder of each of the permethylated N-linked glycans was diluted with 1 mm NaOH in 50% methanol. Each of the prepared samples was infused directly into an LTQ Orbitrap Discovery mass spectrometer (Thermo Scientific) at a flow rate of 0.5 μl/min to obtain nanospray ionization. The full mass spectrum of each sample was obtained in the positive ion mode. MS/MS analysis of selected parent ions and total ion mapping were performed at 2-mass unit interval.
RESULTS
Sia-Fc Is Decreased in Acute Phase But Increased during Chronic Phases in Mouse Models of Pulmonary Inflammation
To quantify Fc sialylation, we developed a multichannel immunofluorescence Western blot method that permits simultaneous detection of signals from three different fluorescent probes using a tri-laser scanner (Typhoon Trio). We quantified Fc and Fab regions using anti-mouse Fc and anti-mouse Fab antibodies, respectively, and glycan structures were probed with SNA or ECL to visualize the presence of α2,6-sialic acids or the exposed underlying disaccharide structure (Gal(β4)GlcNAc), respectively. In combination, these three channels allowed quantification of the relative sialylation levels of Fc and Fab.
Lungs of smokers with chronic obstructive pulmonary disease are frequently colonized with NTHi strains during exacerbations (22). This colonization (with constant bacterial turnover) is akin to a low-grade smoldering infection that induces chronic airway inflammation and augments the inflammatory effects of tobacco smoke. We utilized a murine model of chronic obstructive pulmonary disease-like pulmonary inflammation to examine the extent of Fc sialylation. Repeated exposure of C57BL/6 mice to NTHi elicited chronic pulmonary inflammation highlighted by the accumulation of immune cells and proinflammatory cytokines in the airways. In this study, we measured the levels of serum Sia-Fc in NTHi-challenged mice. IgG was isolated from base-line and post-challenge serum samples. Proteolyzed Fc and Fab fragments were assayed on multichannel Western blots (Fig. 1). The overall IgG levels, as well as SNA reactivity against the Fc fragment (Fig. 1, inset), increased steadily over the 16-week course of repeated NTHi exposure. When expressed as SNA/Fc signal ratios, the degree of α2,6-sialylation of the Fc fragments also increased steadily, with a maximum signal at 12 weeks of chronic NTHi exposure. Interestingly, compared with the basal SNA/Fc ratio (Fig. 1, dashed line denoted Resting), the early phases of NTHi exposure showed dramatically suppressed α2,6-Fc sialylation. This was followed by a gradual restoration of Sia-Fc, such that circulatory Sia-Fc peaked at 12 weeks with close to a 3-fold elevation compared with the base line.
FIGURE 1.

Circulatory Fc sialylation in NTHi-mediated pulmonary inflammation. C57BL/6 mice (WT) were subjected to biweekly intra tracked instillations of NTHi for 16 consecutive weeks as detailed under “Experimental Procedures,” and serum (n = 5) was harvested weekly and pooled (where D1 represents a post-challenge blood draw on the first day of treatment). Fc fragments of IgG were isolated from each of the weekly pools, subjected to SDS-PAGE, and probed with SNA (green), anti-Fc antibody (blue), and anti-Fab antibody (red) as displayed in the inset. The SNA reactivity of the Fc fragments is expressed as a ratio of the relative SNA to anti-Fc signals, where untreated denotes the ratio found in the serum IgG of animals without treatment.
Regulation of circulatory Sia-Fc in a pulmonary Th2 inflammation model mimicking allergic asthma was also examined using OVA-sensitized (primed) mice subjected to short-term or long-term exposure to aerosolized OVA. This is summarized in Fig. 2 as either the raw tri-probe data (A) or ratios of lectin reactivity to Fc (B). As with the NTHi-induced pulmonary response, the OVA-induced pulmonary response elicited strikingly higher Fc sialylation (expressed as SNA/Fc ratios) in WT mice after long-term repeated allergen exposure (chronic) compared with short-term allergen exposure (acute) or no exposure (primed but not challenged). Interestingly, Sia-Fc increased slightly despite allergen removal for 2 weeks after acute allergen challenge (resolution). ECL was used to assess the availability of exposed galactose termini on Fc that serve as acceptor substrate for α2,6-sialylation. In WT animals, exposed galactose termini on Fc (expressed as ECL/Fc ratios) increased strikingly during the resolution phase after acute challenge, and the availability of exposed galactose was not further increased significantly upon chronic allergen exposure. The data indicate a two-step process in Fc sialylation during exposure of WT animals to inflammatory challenge: acute challenge led to increase production of galactose termini, whereas continued conversion of these termini to sialylated structures was less pronounced, as observed in the acute resolution phase. Then, as the inflammation progressed into the chronic stages, increased ST6Gal-1-mediated conversion into sialylated structures was observed.
FIGURE 2.

Circulatory Fc sialylation in OVA model of allergic pulmonary inflammation. C57BL/6 (WT) and Siat1ΔP1 mice were sensitized to OVA and subjected to acute and chronic protocols of pulmonary inflammation as described under “Experimental Procedures.” Sera from five animals representing each of the conditions were pooled, and Fc fragments of IgG were isolated from pooled sera from animals sensitized but not challenged by OVA (Primed), challenged with the acute protocol (Acute), and challenged with the chronic protocol (Chron). A, sera were subjected to SDS-PAGE analysis, probed with SNA or ECL (green), anti-Fc antibody (blue), and anti-Fab antibody (red). Resol, resolution. B, quantitation of the data from A expressed as SNA/anti-Fc or ECL/anti-Fc ratios. Ratio values <30 were regarded as random noise.
In a separate model of systemic inflammation elicited by the subcutaneous instillation of the sterile irritant turpentine, WT mice responded within 72 h not only with depressed circulatory IgG but also with a 8.3-fold decreased sialylation of the Fc glycans (Fig. 3). Decreased sialylation is consistent with the decreased Sia-Fc noted in the acute stages of NTHi-elicited pulmonary inflammation models (Fig. 1). Decreased Sia-Fc was not noted in the acute OVA-induced Th2 response (Fig. 2), but that might have been due the initial priming or sensitizing step, which altered the Sia-Fc response profile.
FIGURE 3.

Acute phase response is accompanied by reduced circulatory Sia-Fc. Fc fragments were isolated from the pooled sera of C57BL/6 (WT), Siat1ΔP1 (ΔP1), and Siat1-null (KO) mice at base line (Rest) or 72 h after elicitation of the acute phase response by subcutaneous injection of turpentine (T72). The samples were separated by SDS-PAGE and probed with SNA (Cy5), anti-Fc antibody (Cy3), and anti-Fab antibody (Cy2). In each lane, serum pools from five animals were used.
Siat1ΔP1 Animals Are Unable to Express Sia-Fc
When tested in parallel with their WT cohorts in the OVA model of pulmonary allergic inflammation, Siat1ΔP1 mice, with an inactivated P1 promoter, were completely unable to express Sia-Fc in their circulatory IgGs, either at base line or after acute or chronic inflammatory challenges (Fig. 2). This is reflected in the SNA/Fc ratios, where values <30 were below the limits of detection (Fig. 2B). However, the inability to express Sia-Fc is not due to limitation in acceptor substrate for ST6Gal-1, as the ECL/Fc ratios indicate an abundance of free galactose termini. Fig. 3 clearly shows the absence of all SNA reactivity in Siat1ΔP1 circulatory Fc. Circulatory Fc from Siat1-null animals also had the expected lack of Sia-Fc due to a genetic inactivation that rendered impossible all ST6Gal-1-mediated α2,6-sialylation (18, 23).
However, despite the absence of sialylation in the Fc region, IgGs from Siat1ΔP1 animals were not completely deficient in α2,6-sialylation. Fig. 4A shows the α2,6-sialylation of intact IgG that contains both Fab and Fc regions. Intact circulatory IgGs from Siat1ΔP1 animals had significant albeit reduced SNA signals compared with wild-type IgG (Fig. 4A). However, α2,6-sialylation was notably absent in the Fc fragments, as shown in Fig. 4B, where the SNA/Fc ratio was below the limits of detection. Another α2,6-sialic acid lectin, PSL, was used to confirm the findings with SNA for the lack of α2,6-sialylation in the Fc fragment of Siat1ΔP1 circulatory IgG, and the results are shown in Fig. 5. Together, these data strongly imply that the Siat1ΔP1 animals are specifically unable to sialylate the Fc glycan while retaining the capability to sialylate the Fab glycans. Moreover, the inability to sialylate Fc is not due to the absence of terminal galactose acceptor structures, as visualized by ECL.
FIGURE 4.

Siat1ΔP1 Fc region is undersialylated. The serum pools of three C57BL/6 (WT), three Siat1ΔP1 (ΔP1), and three Siat1-null (KO) mice at rest were probed as described in the legends to Figs. 1–3, and the results are expressed as SNA/anti-Fc ratios. A, signal ratio of intact IgG; B, signal ratio of an isolated Fc fragment.
FIGURE 5.
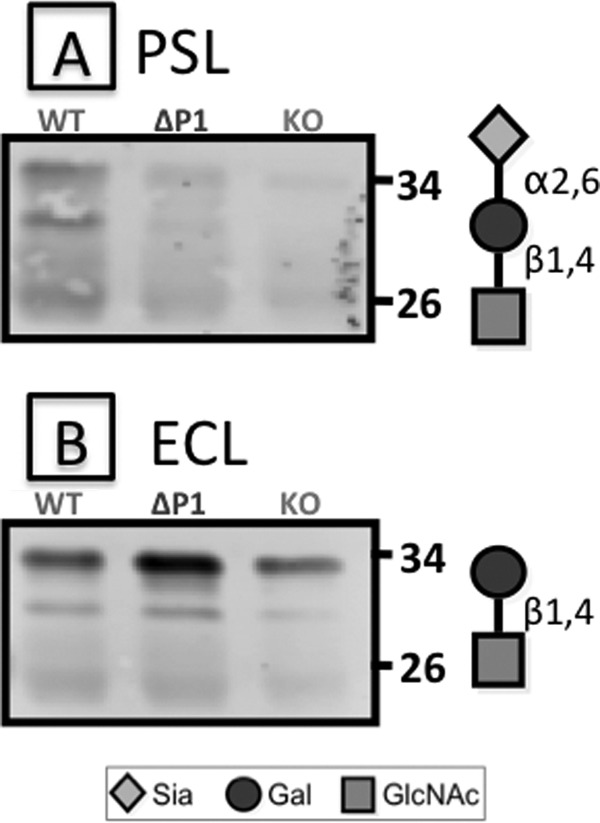
PSL and ECL reactivity of Fc fragments. Fc fragments from serum pools of five C57BL/6 (WT), Siat1ΔP1 (ΔP1), and Siat1-null (KO) mice were subjected to SDS-PAGE and blotted for reactivity against PSL (A) and ECL (B), recognizing the α2,6-sialyl type 2 lactosamine and unsialylated type 2 lactosamine structures, respectively, as diagrammed.
Mass Spectrometry Confirmation for Absence of α2,6-Sia-Fc in Siat1ΔP1 IgG
Glycans released from gel-purified Fc fragments of WT and Siat1ΔP1 mice were profiled by MALDI-TOF-MS and confirmed by linear ion trap-MS. The summary of this analysis is given in Fig. 6, with the MALDI-TOF spectra presented in supplemental Fig. 1. At base line, Fc glycans exist as biantennary structures; tri- and tetra-antennary structures were not detected. Consistent with published reports (24), 9% of the wild-type Fc glycans terminated with sialic acid, and all Fc glycans were modified by core fucosylation. All sialylated structures were singly sialylated, indicating that only 4.5% of the available nonreducing termini are capped by a sialic acid. 41% of the wild-type Fc glycan termini at base line had an exposed Gal that was not further modified by sialylation. Moreover, the majority of the termini (54%) had an exposed GlcNAc that was not occupied by Gal. The abundance of these precursor termini supports further the idea that Sia-Fc formation has at least two rate-limiting steps: the formation of Gal-GlcNAc from GlcNAc and the sialyl modification by ST6Gal-1 to form Sia-Gal-GlcNAc. MS revealed no sialylated structures in the Siat1ΔP1 Fc glycans. Importantly, the distribution between Gal and GlcNAc termini was similar to that in wild-type Fc (42 and 57%, respectively), indicating that the defect in Siat1ΔP1 relates only to the sialylation event and not the ability to generate the precursor Gal-GlcNAc structure.
FIGURE 6.

Structural composition of Fc glycans. Shown are the results from linear ion trap-MS analysis for the glycan structural composition (expressed as percent of total glycans) present on the Fc fragments in untreated WT and Siat1ΔP1 pooled sera. MALDI-TOF-MS data from which this information was derived are shown in supplemental Fig. 1.
DISCUSSION
When present in vivo, sialic acids on Fc glycans exist only in α2,6-linkages requiring ST6Gal-1. Fc fragments that are artificially forced to acquire α2,3-sialyl linkages do not have anti-inflammatory activity (10). Here, we confirmed the absolute requirement for functional ST6Gal-1 in the creation of sialic acid linkages on the Fc glycan of circulatory IgG. The Siat1-null mouse, with an inactivated ST6Gal-1 gene, does not express Sia-Fc.
Hepatic expression and systemic circulatory levels of ST6Gal-1 are tightly regulated during inflammation (25). Therefore, we hypothesized that the degree of Fc sialylation might likewise be altered during the inflammatory process. To date, the most characterized human conditions that alter circulatory Sia-Fc content are rheumatoid arthritis and pregnancy, which suppress and increase Sia-Fc, respectively (26, 27). Reduction of Fc sialylation upon induction of an antigen-specific immune response has been previously reported (8). We have recapitulated this observation in mouse models upon challenge with the sterile irritant turpentine (Fig. 4) and also in the earliest stages of pulmonary response to acute NThi challenge (Fig. 1). The rationale for decreased Sia-Fc in acute inflammation is not understood, but altered biosynthesis is unlikely to be the sole mechanism due to the rapidity of Sia-Fc disappearance from the circulation, e.g. within hours. Another explanation is that existing Sia-Fcs are preferentially bound and removed from the circulation by Sia-Fc-binding cells (e.g. splenic macrophages in mice and dendritic cells in humans) responding to inflammatory cues. Concomitantly, Sia-Fc in the circulation may be further diluted by the de novo synthesis of antigen-specific Ig with undersialylated Fc to generate a protective response (8). An unexpected observation was that progression from acute to chronic inflammation in our animal models was paralleled by a paradoxical 3-fold increase in Sia-Fc. A possible explanation is that our chronic inflammation models were achieved by forced and repeated exposures to challenge, and the dramatic increase in Sia-Fc might represent a futile attempt to limit destructive inflammation in the face of an unrelenting challenge. Our data suggest that increased Fc sialylation accompanying chronic inflammatory conditions is a two-stage sequential process. A near-term response to an initial challenge is increased production of Gal-terminated Fc glycans. Increased conversation of these Gal termini by ST6Gal-1, e.g. to produce Sia-Fc, occurs subsequently upon prolonged and repeated exposure to the challenge. At present, there is no information on whether the increased Gal termini are due to increased branching of core N-glycans or to increased conversion of GlcNAc to Gal-GlcNAc or both. Whatever the case, our data show there is an even greater surplus of Gal termini in the Siat1ΔP1 Fc, further supporting the idea that ST6Gal-1-mediated sialylation is the abrogated step in the Siat1ΔP1 animals.
Our data also unexpected show that inactivation of the P1 promoter of the ST6Gal-1 gene in mice abrogated the ability to express Sia-Fc in circulatory IgG. Because ablation of P1 results only in decreased ST6Gal-1 in the liver and systemic circulation (16, 17), our observation strongly infers a role for circulatory ST6Gal-1, previously considered a functionless byproduct of metabolic inefficiency, in the construction of the sialic acid linkage on the Fc glycans. Reduced circulatory ST6Gal-1 resulting in the lack of Sia-Fc may explain the generally proinflammatory tendencies noted in the Siat1ΔP1 mouse (11).
IgG is made in B cells that do not utilize the P1 promoter to express ST6Gal-1 (14), and the precise reason for the requirement of a functional P1 for Sia-Fc formation is unclear. Because P1 ablation dramatically decreases ST6Gal-1 levels in the systemic circulation (16, 17), a tantalizing possibility is that the Fc glycans are sialylated directly by the pool of extracellular or extrinsic ST6Gal-1 in the circulation. An alternative explanation is the existence of clonal populations of B cells with a differential ability to express Sia-Fc, and circulatory ST6Gal-1 influences the clonal selection process for these populations. Thus, Siat1ΔP1 mice might lack the particular subset of B cells that produce sialylated Fc glycans. Indeed, circulatory ST6Gal-1 deficiency has been attributed to altered hematopoietic activity in bone marrow (11, 12, 16). IgG acquires anti-inflammatory properties upon Fc sialylation. Differential Fc sialylation may serve as a mechanism in the normal switching between pro- and anti-inflammatory environments. Although IgGs containing Sia-Fc are the effective portions of large-dose IVIG therapy, they typically comprise only 5–10% of the IgGs in human IVIG pools. Enriching for Sia-Fc for a more effective clinical outcome is an issue of current intense interest (28, 29). Elucidating the mechanism by which Sia-Fc is naturally generated will have significant therapeutic value and, at the very least, illuminate an approach to enhance the proportion of Sia-Fc in IVIG therapy.
Supplementary Material
Acknowledgments
We thank Christine Collins for excellent technical assistance and Dr. Parastoo Azadi (Complex Carbohydrate Research Center, University of Georgia) for providing glycan analytical services.
This work was supported, in whole or in part, by National Institutes of Health Grants R01AI38193 and P01HL107146 (to J. T. Y. L.) and R01 AI069379 (to Y. T). The core facilities used in this work were supported in part by National Cancer Institute Cancer Center Support Grant CA16056 to the Roswell Park Cancer Institute.

This article contains supplemental Fig. 1.
- IVIG
- intravenous immunoglobulin
- Sia-Fc
- sialylated Fc
- ST6Gal-1
- β-galactoside α2,6-sialyltransferase 1
- NTHi
- nontypeable H. influenzae
- OVA
- ovalbumin
- SNA
- S. nigra agglutinin
- PSL
- P. squamosus lectin
- ECL
- E. crista-galli lectin.
REFERENCES
- 1. Bruhns P., Samuelsson A., Pollard J. W., Ravetch J. V. (2003) Colony-stimulating factor-1-dependent macrophages are responsible for IVIG protection in antibody-induced autoimmune disease. Immunity 18, 573–581 [DOI] [PubMed] [Google Scholar]
- 2. Samuelson P. (2001) Anticircumvention rules: threat to science. Science 293, 2028–2031 [DOI] [PubMed] [Google Scholar]
- 3. Gregoire-Gauthier J., Durrieu L., Duval A., Fontaine F., Dieng M. M., Bourgey M., Patey-Mariaud de Serre N., Louis I., Haddad E. (2012) Use of immunoglobulins in the prevention of GvHD in a xenogeneic NOD/SCID/γc− mouse model. Bone Marrow Transplantation 47, 439–450 [DOI] [PubMed] [Google Scholar]
- 4. Sokos D. R., Berger M., Lazarus H. M. (2002) Intravenous immunoglobulin: appropriate indications and uses in hematopoietic stem cell transplantation. Biol. Blood Marrow Transplant. 8, 117–130 [DOI] [PubMed] [Google Scholar]
- 5. Sullivan K. M., Kopecky K. J., Jocom J., Fisher L., Buckner C. D., Meyers J. D., Counts G. W., Bowden R. A., Peterson F. B., Witherspoon R. P. (1990) Immunomodulatory and antimicrobial efficacy of intravenous immunoglobulin in bone marrow transplantation. N. Engl. J. Med. 323, 705–712 [DOI] [PubMed] [Google Scholar]
- 6. Sullivan K. M., Storek J., Kopecky K. J., Jocom J., Longton G., Flowers M., Siadak M., Nims J., Witherspoon R. P., Anasetti C., Appelbaum F. R., Bowden R. A., Buckner C. D., Crawford S. W., Deeg H. J., Hansen J. A., McDonald G. B., Sanders J. E., Storb R. (1996) A controlled trial of long-term administration of intravenous immunoglobulin to prevent late infection and chronic graft-vs.-host disease after marrow transplantation: clinical outcome and effect on subsequent immune recovery. Biol. Blood Marrow Transplant. 2, 44–53 [PubMed] [Google Scholar]
- 7. Anthony R. M., Kobayashi T., Wermeling F., Ravetch J. V. (2011) Intravenous γ-globulin suppresses inflammation through a novel TH2 pathway. Nature 475, 110–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kaneko Y., Nimmerjahn F., Ravetch J. V. (2006) Anti-inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science 313, 670–673 [DOI] [PubMed] [Google Scholar]
- 9. Anthony R. M., Wermeling F., Karlsson M. C., Ravetch J. V. (2008) Identification of a receptor required for the anti-inflammatory activity of IVIG. Proc. Natl. Acad. Sci. U.S.A. 105, 19571–19578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Anthony R. M., Nimmerjahn F., Ashline D. J., Reinhold V. N., Paulson J. C., Ravetch J. V. (2008) Recapitulation of IVIG anti-inflammatory activity with a recombinant IgG Fc. Science 320, 373–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nasirikenari M., Chandrasekaran E. V., Matta K. L., Segal B. H., Bogner P. N., Lugade A. A., Thanavala Y., Lee J. J., Lau J. T. (2010) Altered eosinophil profile in mice with ST6Gal-1 deficiency: an additional role for ST6Gal-1 generated by the P1 promoter in regulating allergic inflammation. J. Leukoc. Biol. 87, 457–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nasirikenari M., Segal B. H., Ostberg J. R., Urbasic A., Lau J. T. (2006) Altered granulopoietic profile and exaggerated acute neutrophilic inflammation in mice with targeted deficiency in the sialyltransferase ST6Gal-1. Blood 108, 3397–3405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dalziel M., Huang R. Y., Dall'Olio F., Morris J. R., Taylor-Papadimitriou J., Lau J. T. (2001) Mouse ST6Gal sialyltransferase gene expression during mammary gland lactation. Glycobiology 11, 407–412 [DOI] [PubMed] [Google Scholar]
- 14. Wuensch S. A., Huang R. Y., Ewing J., Liang X., Lau J. T. (2000) Murine B cell differentiation is accompanied by programmed expression of multiple novel β-galactoside α2,6-sialyltransferase mRNA forms. Glycobiology 10, 67–75 [DOI] [PubMed] [Google Scholar]
- 15. Hu Y. P., Dalziel M., Lau J. T. (1997) Murine hepatic β-galactoside α2,6-sialyltransferase gene expression involves usage of a novel upstream exon region. Glycoconj. J. 14, 407–411 [DOI] [PubMed] [Google Scholar]
- 16. Jones M. B., Nasirikenari M., Feng L., Migliore M. T., Choi K. S., Kazim L., Lau J. T. (2010) Role for hepatic and circulatory ST6Gal-1 sialyltransferase in regulating myelopoiesis. J. Biol. Chem. 285, 25009–25017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Appenheimer M. M., Huang R. Y., Chandrasekaran E. V., Dalziel M., Hu Y. P., Soloway P. D., Wuensch S. A., Matta K. L., Lau J. T. (2003) Biologic contribution of P1 promoter-mediated expression of ST6Gal-1 sialyltransferase. Glycobiology 13, 591–600 [DOI] [PubMed] [Google Scholar]
- 18. Hennet T., Chui D., Paulson J. C., Marth J. D. (1998) Immune regulation by the ST6Gal sialyltransferase. Proc. Natl. Acad. Sci. U.S.A. 95, 4504–4509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lugade A. A., Vethanayagam R. R., Nasirikenari M., Bogner P. N., Segal B. H., Thanavala Y. (2011) Nrf2 regulates chronic lung inflammation and B cell responses to nontypeable Haemophilus influenzae. Am. J. Respir. Cell Mol. Biol. 45, 557–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lugade A. A., Bogner P. N., Murphy T. F., Thanavala Y. (2011) The role of TLR2 and bacterial lipoprotein in enhancing airway inflammation and immunity. Front. Immun. 2, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lugade A. A., Bogner P. N., Thanavala Y. (2011) Murine model of chronic respiratory inflammation. Adv. Exp. Med. Biol. 780, 125–141 [DOI] [PubMed] [Google Scholar]
- 22. Murphy T. F. (2003) Respiratory infections caused by nontypeable Haemophilus influenzae. Curr. Opin. Infect. Dis. 16, 129–134 [DOI] [PubMed] [Google Scholar]
- 23. Martin L. T., Marth J. D., Varki A., Varki N. M. (2002) Genetically altered mice with different sialyltransferase deficiencies show tissue-specific alterations in sialylation and sialic acid 9-O-acetylation. J. Biol. Chem. 277, 32930–32938 [DOI] [PubMed] [Google Scholar]
- 24. Blomme B., Van Steenkiste C., Grassi P., Haslam S. M., Dell A., Callewaert N., Van Vlierberghe H. (2011) Alterations of serum protein N-glycosylation in two mouse models of chronic liver disease are hepatocyte and not B cell-driven. Am. J. Physiol. Gastrointest. Liver Physiol. 300, G833–F842 [DOI] [PubMed] [Google Scholar]
- 25. Baumann H., Gauldie J. (1994) The acute phase response. Immunol. Today 15, 74–80 [DOI] [PubMed] [Google Scholar]
- 26. Kötz K., Hänsler M., Sauer H., Kaltenhäuser S., Häntzschel H. (1996) Immunoglobulin G galactosylation deficiency determined by isoelectric focusing and lectin affinoblotting in differential diagnosis of rheumatoid arthritis. Electrophoresis 17, 533–534 [DOI] [PubMed] [Google Scholar]
- 27. van de Geijn F. E., Wuhrer M., Selman M. H., Willemsen S. P., de Man Y. A., Deelder A. M., Hazes J. M., Dolhain R. J. (2009) Immunoglobulin G galactosylation and sialylation are associated with pregnancy-induced improvement of rheumatoid arthritis and the postpartum flare: results from a large prospective cohort study. Arthritis Res. Ther. 11, R193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stadlmann J., Weber A., Pabst M., Anderle H., Kunert R., Ehrlich H. J., Peter Schwarz H., Altmann F. (2009) A close look at human IgG sialylation and subclass distribution after lectin fractionation. Proteomics 9, 4143–4153 [DOI] [PubMed] [Google Scholar]
- 29. Stadlmann J., Pabst M., Altmann F. (2010) Analytical and functional aspects of antibody sialylation. J. Clin. Immunol. 30, Suppl. 1, 15–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.