

Background: CARMIL inhibits the actin capping action of heterodimeric capping protein.
Results: Results argue against a steric blocking model and provide evidence for an allosteric mechanism.
Conclusion: The conformations of the actin- and CARMIL-binding sites on capping protein are linked.
Significance: Control of capping actin barbed ends is critical for cell motility.
Keywords: Actin, Allosteric Regulation, Cytoskeleton, Molecular Dynamics, Site-directed Mutagenesis, CARMIL, Capping Protein
Abstract
Capping protein (CP) controls the polymerization of actin filaments by capping their barbed ends. In lamellipodia, CP dissociates from the actin cytoskeleton rapidly, suggesting the possible existence of an uncapping factor, for which the protein CARMIL (capping protein, Arp2/3 and myosin-I linker) is a candidate. CARMIL binds to CP via two motifs. One, the CP interaction (CPI) motif, is found in a number of unrelated proteins; the other motif is unique to CARMILs, the CARMIL-specific interaction motif. A 115-aa CARMIL fragment of CARMIL with both motifs, termed the CP-binding region (CBR), binds to CP with high affinity, inhibits capping, and causes uncapping. We wanted to understand the structural basis for this function. We used a collection of mutants affecting the actin-binding surface of CP to test the possibility of a steric-blocking model, which remained open because a region of CBR was not resolved in the CBR/CP co-crystal structure. The CP actin-binding mutants bound CBR normally. In addition, a CBR mutant with all residues of the unresolved region changed showed nearly normal binding to CP. Having ruled out a steric blocking model, we tested an allosteric model with molecular dynamics. We found that CBR binding induces changes in the conformation of the actin-binding surface of CP. In addition, ∼30-aa truncations on the actin-binding surface of CP decreased the affinity of CBR for CP. Thus, CARMIL promotes uncapping by binding to a freely accessible site on CP bound to a filament barbed end and inducing a change in the conformation of the actin-binding surface of CP.
Introduction
The creation and regulation of free barbed ends of actin filaments is a critical determinant of where and when actin polymerizes in cells (1). Growth of free barbed ends creates force and drives the movement of various cell structures, notably membranes. Cells have specific mechanisms to nucleate the formation of new actin filaments, based on proteins such as the Arp2/3 complex, formins, and WH2 domain proteins, and these new filaments grow by adding subunits to their free barbed ends. In addition, barbed ends are the attachment point for actin filaments to certain cellular structures, such as Z lines of muscle.
Capping protein (CP)2 binds to and functionally caps free barbed ends both in vitro and in vivo (2). Across eukaryotes, CP sequences are conserved, and CP is important for actin assembly in cells. The concentration of CP in cells is in the micromolar range, above the concentration of actin filament barbed ends, and well above the sub-nanomolar binding affinity of CP for barbed ends (3, 4).
The biochemical properties of CP have physiological relevance. One molecule of CP is sufficient to attach a filament barbed end to an object, based on direct observations of single actin filaments (5, 6). CP is an essential element of the dendritic nucleation model describing how actin assembly nucleated by the Arp2/3 complex can produce force (7). In vertebrate cells, lamellipodia formation requires CP (8). In yeast, the ability of CP to cap barbed ends correlates with its ability to function in cells (9). In addition, a fundamental property of formins and VASP family proteins is to protect free barbed ends from the capping action of CP (10).
CP is an α/β heterodimer with subunits of ∼30 kDa. Single subunits are unstable, but the heterodimer is very stable. CP has the shape of a mushroom, with a cap and stalk (11). The two subunits have similar secondary structures, with a pseudo-2-fold axis of rotational symmetry down the center of the mushroom (11). The top surface of the mushroom includes the C-terminal regions of each subunit, and both are important for CP to bind barbed ends with high affinity (4, 12). The actin-binding surfaces of CP were defined by us in detail in a set of studies combining experimental analysis of mutations with computational analysis of binding interactions and dynamics (13).
CP is fully active in a wide range of solution conditions in vitro, with little variation resulting from physiological changes in pH, Ca2+, or other factors (2). A number of potential inhibitors of CP have been described, which suggest possible modes of CP regulation in cells. In lamellipodia, CP binds to the actin filament network very near the membrane, and it dissociates from the network after a short time (14, 15). This rate of dissociation is much higher than what would be expected from in vitro rate constants, suggesting that another factor plays a role, perhaps by removing CP from barbed ends or by severing filaments.
A protein sequence capable of binding and inhibiting CP, now called the CPI (capping protein interaction) motif, was first appreciated from comparative analysis of three otherwise unrelated proteins: CARMIL, CKIP-1, and CD2AP (16–19). For each protein, a fragment was found to bind directly to CP and inhibit capping activity. A small region within the fragment was defined as necessary and sufficient for binding to CP, and site-directed mutagenesis revealed the motif to be LXHXTXXRPK(6X)P (17). Crystal structures of co-complexes reveal this motif to be in close contact with CP (20, 21). The motif has also been found in other unrelated proteins, CapZIP and the WASH subunit FAM21 (also known as WASHCAP) (20).
CARMIL is a potent inhibitor and uncapper of CP. CARMIL binds tightly to CP, with a Kd in the nanomolar range (22), and CARMIL purified from Acanthamoeba contains CP in near-stoichiometric amounts (23). However, the large majority of Acanthamoeba CP in cell extracts is free and able to cap actin (3). CARMIL is important for actin-based motility in cells, based on gene knock-out and siRNA knockdown studies in Dictyostelium and cultured vertebrate cells (22, 24). Vertebrates have three conserved isoforms of CARMIL, encoded by three different genes, and human CARMIL1 appears to function in lamellipodial actin assembly (25).
CARMIL inhibits CP and uncaps barbed ends, based on actin polymerization assays in bulk solution (16, 22) and single-molecule total internal reflection fluorescence observations of GFP-CP on actin-filament barbed ends (26). CARMIL removes CP from capped barbed ends on a time scale of ∼10 s (16, 22); in contrast, the spontaneous dissociation of CP from a barbed end occurs on a time scale of ∼10 min (27).
Here, we investigated the molecular mechanism by which CARMIL inhibits CP and removes CP from barbed ends. Mutagenesis studies argue strongly against a steric blocking model, consistent with predictions from co-crystal structures (12, 20). All-atom molecular dynamics simulations provide evidence for an allosteric model in which the conformation of the binding sites on CP for CARMIL and actin are linked.
EXPERIMENTAL PROCEDURES
Plasmid Construction and Mutagenesis
His-tagged mouse CPα1β2 was prepared with a plasmid (pBJ 2041) provided by Drs. Ville Paavilainen and Pekka Lappalainen (University of Helsinki). CP mutations were generated in the plasmid by QuikChange site-directed mutagenesis (Stratagene). To prepare CPαCFPβ(CΔ29)YFP (pBJ 2037), we amplified the CPαβ(CΔ29) coding region, excluding the His tag, from a plasmid expressing His-tagged CPαβ(CΔ29) (pBJ 1891). We also PCR-amplified CFP and YFP from pECFP-N1 and pEYFP-N1 (Clontech), respectively. The CFP and YFP fragments were fused to the C-terminal ends of the coding regions for the α and β CP subunits, respectively, and then subcloned into pRSFDuet-1 vector (Novagen). Plasmids expressing CP mutations, CPα(E200R) (pBJ 1932), CPα(K256A) (pBJ 1878), CPα(R260A) (pBJ 1879), CPα(K268A) (pBJ 1881), CPβ(R195A) (pBJ 1944), CPβ(K223A) (pBJ 1949), CPβ(R225A) (pBJ 1884), CPβ(EN256/260SS) (pBJ 1954), and CPβ(LLL258/262/266SSS) (pBJ 1955) were generated from pBJ 2041 by QuikChange site-directed mutagenesis. A plasmid expressing GST-CBR (pBJ 1841) was produced by cloning the CBR fragment of human CARMIL1a (GenBankTM FJ009082), amplified from cDNA by PCR, into the pGEX-6P-3 vector (GE Healthcare) (20). Plasmids expressing GST fusions of the CBR fragment CBR71 (pBJ 2052) and the mutants CBR71-RL (pBJ 2055) and CBR115(R989A) (pBJ 2059) were generated from pBJ 1841 by QuikChange site-directed mutagenesis. The resulting plasmids were verified by DNA sequencing of the entire region derived from PCR reaction.
Protein Expression and Purification
His-tagged mouse CP α1β2 was purified as described (20). GST-tagged CBR115 from human CARMIL1A was purified as described (20). Actin was purified from rabbit skeletal muscle acetone powder (Pel-Freez, Rogers, AK) and gel-filtered on Sephacryl S-200 (4). Spectrin actin seeds were purified as described (4).
His-tagged CPαCFPβ(CΔ29)YFP (pBJ 2037) was purified by the same method used for His-tagged WT CPαβ, with the following modification: BL21(DE3) cells were grown at 25 °C and induced at 13 °C for ∼12 h with 10 μm isopropyl β-d-1-thiogalactopyranoside. Protein eluted from the Talon column was dialyzed into 10 mm imidazole, pH 7.5, 50 mm KCl, 0.5 mm EDTA, and 1 mm DTT, and then purified by chromatography in the same buffer on a HiPrep 26/60 Sephacryl S-300 column (GE Healthcare). The protein was kept on ice and used within 2 weeks. The concentrations of CPαCFPβ(CΔ29)YFP (ϵ280 nm = 127,840 m−1 cm−1), GST-CBR115(ϵ280 nm = 48,610 m−1 cm−1), GST-CBR71 (ϵ280 nm = 43,110 m−1 cm−1), and GST-CBR71-RL (ϵ280 nm = 43,110 m−1 cm−1) were calculated from A280, based on predicted extinction coefficients (28) and confirmed by SDS-PAGE with Coomassie Blue staining.
Actin Polymerization Assays
Pyrene actin polymerization assays, including capping by CP and inhibition of CP by the CARMIL fragment CBR, were performed as described (20). Pyrene actin subunits (1.5 μm, 5% pyrene label) polymerized over time, by addition to barbed ends of filaments nucleated by spectrin actin seeds, the concentration of which was determined by fitting to Reaction 1 below.
To analyze the data from experiments in which CBR was added at time zero to inhibit the capping activity of CP, we determined binding constants for CP and CBR by least-squares fitting of the complete time courses of the reaction, using Berkeley Madonna (version 8.3), with the following three-step mechanism.
 |
 |
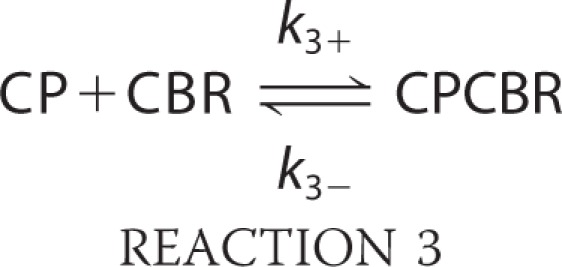 |
Here, A represents actin monomer, Nb indicates free barbed ends, CP indicates capping protein, and CBR is the CBR fragment of CARMIL. A capped barbed end, CPNb, can neither add nor lose actin subunits. This mechanism makes two simplifying assumptions: 1) CPCBR does not bind to a free barbed end, and 2) CBR does not bind to CPNb.
For the uncapping assay, the mechanism was more complex, with the addition of Reactions 4 and 5 below. Here, CBR can bind to a capped barbed end, CPNb, in the forward reaction of Reaction 5, the first step of uncapping. Uncapping is completed by the reverse reaction of Reaction 4, which generates a free barbed end. In this mechanism, the CPCBR complex can bind to a free barbed end, represented as the forward reaction of Reaction 4; thus, the CPCBR complex has a finite, non-zero, ability to cap.
 |
 |
For Reaction 1, the rate constants were fixed, from previous results, with k1+ = 11.6 μm−1 s−1 and k1− = 1.4 s−1 (29). The rate constants for capping in Reaction 2 were determined by fitting experimental data for seeded actin assembly in the presence of CP. A range of CP concentrations was used, and the full time courses of all of the experimental curves were fitted together. For experiments in which CBR was added at time zero to inhibit the capping activity of CP, the rate constants for CP binding to CBR in Reaction 3 were determined by fitting a set of curves produced by addition of CBR at various concentrations.
For uncapping experiments, in which CBR was added after CP had bound to and capped barbed ends, the rate constants for CP binding to CBR in Reaction 3 were fixed at values determined from surface plasmon resonance (SPR) analysis of CP binding to CBR, with k3+ = 1.6 ± 0.68 × 106 m−1 s−1 and k3− = 1.4 ± 0.49 × 10−3 s−1. The rate constants for CPCBR binding to the barbed end in Reaction 4 and for CBR binding to the capped barbed end, CPNb, in Reaction 5 were determined by fitting all the data from a set of curves produced by addition of CBR at various concentrations.
The fitting routine did not impose the requirement that the product of the equilibrium binding constants for Reactions 2 and 5 had to be equal to the product of the equilibrium binding constants for Reactions 3 and 4. The rate constants for Reactions 2 and 3 were set to fixed values determined from independent prior experiments, and the rate constants for Reactions 4 and 5 were allowed to vary. In the end, the two products of the equilibrium binding constants differed by a factor of four. More complex models, such as including a conformation change in CP induced by CBR, would give a better fit and bring the two products closer together, but we did not feel that the precision of the data were sufficient to justify the additional complexity.
Surface Plasmon Resonance Analysis
Measurements of binding by SPR were carried out using a Biacore T100 at 25 °C. Goat anti-GST antibody (GE Healthcare) was cross-linked to CM5 chips using amine coupling (GE Healthcare). As a negative control, goat anti-Dab12 (Santa Cruz Biotechnology) was cross-linked to CM5 chips using amine coupling. GST-CBR fusion proteins were loaded onto the chips containing the immobilized anti-GST antibody in 10 mm HEPES, pH 7.4, 150 mm NaCl, 4 mm EDTA, and 0.005% Tween 20. Next, CP was injected at 50 μl/min.
CP preparations were injected at various concentrations, and the kinetic rate constants, kon and koff were obtained with Biaevalution T100 software (Biacore GE Healthcare). The forms of CP tested were as follows: wild-type CP (pBJ 2041), CPα(CΔ28)β (pBJ 1883), CPαβ(CΔ29) (pBJ 1891), and CPα(CΔ28)β(CΔ29) (pBJ 2043). For the mutants, the whole curve was not fit well. For the single mutants, we were able to fit the dissociation phase to an exponential decay model as discussed in the text. The binding to the double mutant was too low to permit this analysis.
Isothermal Titration Calorimetry
Experiments were carried out using a MicroCal VP-isothermal calorimeter (ITC) (GE Healthcare) at 25 °C. CBR was prepared from GST-CBR by removing the GST tag as described (20). Wild-type CP and truncated forms of CP and CBR were dialyzed into 10 mm Tris-HCl, pH 8.0, 50 mm KCl, 0.5 mm EDTA, and 1 mm Tris (2-carboxyethyl)phosphine hydrochloride. Protein concentrations were determined after dialysis by absorbance at 280 nm using molar extinction coefficients determined by the method of Gill and Von Hippel (28). The proteins, extinction coefficients and expression plasmids were as follows: WT CPαβ, 78,310 m−1 cm−1, pBJ 2041; CPα(ΔC28)β, 71,320 m−1 cm−1, pBJ 1883; CPαβ(ΔC29), 78,310 m−1 cm−1, pBJ 1891; CPα(ΔC28)β(ΔC29), 71,320 m−1 cm−1, pBJ 2043; and GST-CBR115, 5500 m−1 cm−1, pBJ 1841. Samples were degassed at room temperature prior to use. In general, experiments were carried out by titrating solutions of 5 μm CP with CBR at 50 μm.
Data from the titrations were analyzed with the MicroCalTM OriginR software by iterative fitting to the following equations,
 |
 |
where Q(i) is total heat after the ith injection, V0 is the volume of the calorimetric cell, K is the binding constant, ΔH is the molar heat of ligand binding, and n is the number of binding sites. Mt is the concentration of CP as the macromolecule, and Xt is the concentration of CBR as the ligand.
Bead Pulldown Assays
Wild-type CPαβ (pBJ 2041), CPα(ΔC28)β(ΔC29) (pBJ 2043), and GST-CBR (pBJ 1841) were dialyzed into buffer A (40 mm KCl, 10 mm Tris, pH 8.0, 0.1 mm EDTA, 1 mm DTT). Glutathione-Sepharose beads were equilibrated with buffer A. In a 2-ml microcentrifuge tube, 500 nm of CP or CPα(ΔC28)β(ΔC29) was mixed with 100 μl of beads containing varying concentrations of GST-CBR (0–2 μm). Tubes were incubated for 1 h at 4 °C with shaking, and beads were pelleted by centrifuging at 15,000 rpm for 2 min in a microcentrifuge. Supernatants were collected and electrophoresed on a 12.5% SDS-polyacrylamide gel. The gel was stained with Coomassie Blue, and the intensities of the CP bands were compared.
FRET Analysis
CPαCFPβ(CΔ29)YFP was diluted to 100 nm in 300 mm NaCl, 50 mm sodium phosphate, pH 7.5, 0.5 mm EDTA, and 1 mm DTT. To measure the FRET efficiency of CPαCFPβ(CΔ29)YFP, the solution was excited at 430 nm, and an emission scan was performed at 1-nm intervals from 450 nm to 550 nm on a PTI Quantmaster spectrofluorometer (Photon Technology Intl., Santa Clara, CA). The prominent peak at 475 nm was used to calculate the FRET efficiency. A similar scan was performed after treating with 20 μg of trypsin at 25 °C for 3 min, which cleaves the link between the CP and two fluorescent proteins (30).
FRET efficiency was calculated according to the following equation (31, 32),
where CTRYPSIN is the intensity of CFP after trypsin treatment and CFRET is the intensity of CFP before trypsin treatment. To test whether CBR binding to CP causes a change in FRET efficiency, 500 nm of WT CBR or CBR(R989A) was mixed with CP.
Molecular Simulations and Analysis
Molecular dynamics simulations were performed on three separate structures: the CP heterodimer, CP bound to CBR, and CP bound at the barbed end of an actin filament. For the CP heterodimer, we used the Protein Data Bank structure 1IZN (11) where the chicken CPβ2 sequence was threaded onto the chicken CPβ1 structure using MODELLER (33), whereas the chicken CPα1 sequence was left unaltered. For the CP-CBR complex, we used the 3LK3 structure as a starting point (20). This structure is also chicken CPα1/β1, and the missing N and C termini were rebuilt for the α and β subunits again using MODELLER. For the bound CBR fragment, we found we could unambiguously build back the C-terminal portion of the missing peptide, but beyond Thr-1044, the models became highly disordered, as one might expect because this region could not be resolved in the crystal (20). The final CBR structure used corresponded to the 81-amino acid stretch from Glu-964 to Thr-1044. For the CP actin structure, we used our capped filament model, with CP bound to the barbed end of a four-protomer filament (13).
All molecular simulations were performed using NAMD (34) with the CHARMM27 force field and periodic boundary conditions in a cubic box. Each system was solvated in TIP3P waters with 15 Å padding, and counter ions were added to neutralize the system and set the ionic strength at 50 mm. Following minimization, the systems were heated in 50 K steps for 100 ps per step with Cα restraints. At 300 K, the restraints were removed, and the systems were equilibrated as detailed below. In all simulations, we used SHAKE for hydrogens and a 2-fs time step, a cut-off of 10 Å with a smooth switching function starting at 8.5 Å and a pair-list distance of 11.5 Å. Particle mesh Ewald was used for long-range electrostatics with grid spacing of >1/Å in each dimension. NpT dynamics were performed at a pressure of 1 atm, temperature of 300 K, sampling coefficient of 1/ps, and a Langevin piston period and decay of 200 fs and 100 fs, respectively.
Because the conformation of the free CP dimer was the reference point for our analysis, we performed more simulations of this structure. For the CP dimer, we ran four independent simulations (i.e. different starting velocities) of 110 ns. The first 10 ns of each trajectory were discarded, resulting in 400 ns of total simulation time. For the CP/F-actin structure, we used a single simulation of 150 ns. In this case, we discarded the first 50 ns to allow more extensive equilibration, and we used the final 100 ns for analysis. Similarly, for the CP-CBR complex, we had 10 ns of equilibration followed by 100 ns of production simulation.
We performed principal component analysis (PCA) on the simulations to determine the conformation changes induced by binding to the barbed end of the filament versus the CBR of CARMIL. We did not want the analysis to be influenced by the highly dynamic C termini of CP nor by the restricted dynamics of the stalk region when the CBR fragment was bound. As such, we limited our analysis to the cap region of CP, using only residues 61–258 for CPα and residues 48–243 for CPβ. Because the unbound CP dimer was the reference, we used the 400 ns of simulation to determine the eigenvectors of the covariance matrix. The first two eigenvectors captured the largest conformational modes (roughly bending and twisting of the cap), and they were used to create the plot in Fig. 5. Conformations from the CBR and F-actin bound simulations were projected onto this same conformational space to compare their structures. All of the available crystal and NMR structures were likewise projected onto this conformation space. These included Protein Data Bank codes 1IZN (two structures), 2KZ7, 3AA0, 3AA1, 3AA6, 3AA7, 3AAA, 3AAE (five structures), 3LK2, and 3LK3.
FIGURE 5.

PCA analysis of CP cap conformations. The top graph shows the conformational space spanned by the first two PCA eigenvectors calculated from the unbound CP simulation. The gray contours show the space covered by unbound CP, the red points/contours show the space spanned by CARMIL-bound CP, and the blue points/contours show the cap conformations for F-actin-bound CP. The green squares are projections of the available CP structures on to this same basis. The protein conformations corresponding to the peaks of the CARMIL and F-actin distributions are shown below, highlighting the conformational changes between the two structures. See supplemental Movie 1 for more information about the structures.
RESULTS
Kinetic Modeling of Actin Polymerization
The CBR of CARMIL inhibits capping by CP and uncaps barbed ends capped by CP, as illustrated by pyrene actin polymerization assays in Fig. 1. For experiments with inhibition of capping (Fig. 1A), kinetic modeling of the full time courses of the polymerization curves for a range of CBR concentrations revealed that a simple model (represented by Reactions 1 to 3 under “Experimental Procedures”) in which CBR binds to and inhibits CP and the CBR-CP complex does not bind barbed ends provides a good fit for the data. The affinity of CBR for CP was high, with a Kd of ∼1 nm (Reaction 3), consistent with previous measurements. Note that this value is ∼10-fold higher than the Kd of CP for the barbed end (Reaction 2).
FIGURE 1.

Inhibition and uncapping of CP by CBR. Concentrations of CP and the CBR fragment of CARMIL were as indicated on the graphs. A, inhibition of capping, with CBR added to CP at time zero. B, reversal of capping, i.e. uncapping. CP was present at time zero, and CBR was added at 200 s. In both panels, experimental data are black, and fitted curves are red, based on simultaneous fitting of all the data points in all the curves to polymerization rate equations (see “Experimental Procedures”). In the inhibition assay, the Kd for the interaction of CP and CBR was 0.94 ± 0.6 nm. In the uncapping assay, the Kd for the interaction of CP and CBR (Reaction 3) was fixed at 0.88 nm, based on SPR binding data. The Kd for the interaction of CPCBR with the free barbed end (Reaction 4) was fit to 25 nm, and the Kd for the interaction of CPN with CBR (Reaction 5) was 60 nm. a.u., arbitrary units.
For the uncapping experiments (Fig. 1B), in contrast, achieving a good fit for the data required adding steps in which CBR can associate with CP bound to the barbed end and the CBR-CP complex can associate with and dissociate from the barbed end (represented by Reactions 4 and 5 under “Experimental Procedures”). In this analysis, the Kd for binding of the CBR-CP complex to the barbed end was 25 nm (Reaction 4), ∼250-fold higher than the Kd of CP alone for the barbed end. Also, the Kd for binding of CBR to CP bound to the barbed end was 60 nm (Reaction 5), ∼60-fold higher than the Kd for the binding of CBR to free CP. Thus, the binding of CP to CBR and the binding of CP to the barbed end are mutually inhibitory, consistent with the principle of linked functions (35).
As discussed below, we found evidence that CBR induces a conformation change in CP. One might therefore add additional steps to the kinetic scheme, including two states for CP. Including more steps and parameters will always improve a fit, but in this case, the fit was sufficiently good, within the error of the data, that the inclusion of these states was not required.
Interaction of CBR with CP Actin-binding Mutants: Actin Polymerization Assays
To investigate the molecular mechanism by which CBR inhibits CP binding to actin, we first asked whether any of the surface residues of CP involved in binding to CBR were also involved in the binding of CP to the barbed end. Crystal structures of complexes of CP with the CBR of CARMIL and CD2AP resolved two regions of the CARMIL CBR in close contact with CP (20). The regions are labeled in a sequence alignment of the CBR of vertebrate CARMILs (Fig. 2). The structure of the complex of CP with CBR is shown in Fig. 3. The first resolved region of 34 aa included the CP-binding motif defined previously and called the CPI motif. The second resolved region of 15 aa, conserved among CARMILs, is called the CARMIL-specific interaction motif. The two regions correspond to amino acid residues Ile-971–Cys-1004 and Arg-1021–Thr-1035 of human CARMIL1A, the sum of which is roughly equivalent to a 71-aa fragment that potently inhibits capping and causes uncapping (20). These two regions were resolved in the structure, but they were separated by a 16-aa region that has not been resolved in this or any structure. The sequence of this 16-aa unresolved region, corresponding to residues Ala-1005–Gly-1020, also shows sequence similarity among CARMILs, especially among the isoforms 1, 2, and 3, suggesting that it may have functional importance (Fig. 2).
FIGURE 2.

Sequence alignment of vertebrate CARMILs, illustrating the CPI and CARMIL-specific interaction (CSI) regions.
FIGURE 3.

Interaction of CP mutants with GST-CBR in actin polymerization assays. A, structure of the CP-CBR co-complex indicating the positions of the CP residues tested by mutagenesis. The CPα and CPβ subunit are colored yellow and red, respectively, and the CBR fragment is green. The residues tested by mutagenesis are blue. B, a representative experiment with uncapping of a CP mutant (CPα(R260A)) by CBR. The mutant CP was present at time zero, and CBR was added at 200 s. Concentrations of CP and CBR were as indicated. Black curves are controls with either no CP or WT CP, and red curves are mutant CPα(R260A). All CP mutants were tested in this manner, including CPα(E200R), CPα(K256A), CPα(K268A), CPβ(R195A), CPβ(K223A), CPβ(R225A), CPβ(LLL258/262/266SSS), and CPβ(EN256/260SS). For each mutant, the results were similar (see supplemental Fig. 1). a.u., arbitrary units.
The biochemical significance of the close contact residues was tested in a previous study (20). For the CPI of CARMIL CBR, residues Glu-978, Arg-989, Arg-992, and Lys-994 were in close contact with CP β-subunit residues Arg-15, Asp-44, Tyr-79, and Asp-63, respectively. Each residue was changed to Ala, one by one. All eight single mutants showed a loss of interaction affinity, from severalfold to several hundredfold. Here, we found that these CP mutants bound to actin with at most a small decrease of affinity (data not shown), showing that the CARMIL-binding residues of CP are not important for binding actin.
We asked whether CBR binding to CP requires any of the surface residues of CP involved in binding to the barbed end of the actin filament, as would be predicted by a simple steric blocking model. Our previous study defined the actin-binding surfaces of CP by a combination of experimental and computational approaches, which included a collection of CP mutants tested for actin-binding ability (13). Atomic structures of co-complexes of CARMIL fragments with CP showed the interaction to be located on the underside of the mushroom-shaped CP (20, 21), not on the top surface where actin binds (13). However, the fact that a portion of the CBR region was not resolved in the crystal structure left open the possibility that the unresolved regions of CBR might come into direct contact with the actin-binding surfaces of CP.
First, we tested the ability of CBR to interact with a set of CP actin-binding mutants. The locations of the CP residues tested are illustrated in Fig. 3A, and a typical uncapping assay with a mutant CP is shown in Fig. 3B. For each CP mutant, our previous results revealed the capping activity of the mutant (13). For example, in Fig. 3B, the CPα(R260A) mutant exhibited approximately half of the capping activity of WT CP. Therefore, in designing the uncapping assay, we chose a concentration of the CP mutant twice that of WT CP, to achieve similar levels of actin capping. Here, we observed that two concentrations of CBR inhibited the mutant CPα(R260A) as well as they inhibited WT CP (Fig. 3B). We conclude that this residue has essentially no role in the interaction of CP with CBR. For all the other mutants tested, including CPα(E200R), CPα(K256A), CPα(R260A), CPα(K268A), CPβ(R195A), CPβ(K223A), CPβ(R225A), CPβ(LLL258/262/266SSS), and CPβ(EN256/260SS), the results were similar, with CBR inhibiting the CP mutant as well as it inhibited WT CP (supplemental Fig. 1.).
The C-terminal region of the CP β subunit, termed the β tentacle, is an amphipathic helix that extends away from the body of the protein and is highly mobile. We considered whether the mobility of the tentacle might allow it move to a position where it contacted CARMIL. A previous study found no evidence that CARMIL interacts with the β tentacle, based on several types of experiments (20). Here, point mutations of the β tentacle, CPβ(LLL258/262/266SSS) and CPβ(EN256/260SS), had no effect on CARMIL binding (supplemental Fig. 1). Therefore, the actin-binding residues of CP are not critical for its functional interaction with CBR.
Interaction of CBR with CP Actin-binding Mutants: SPR Assays
To complement these results from actin polymerization functional assays, we measured the interaction between CBR and the set of CP mutants using a physical method, SPR. CBR was immobilized on the SPR chip, and CP was the analyte. Results from a typical experiment with CBR and WT CP are shown in supplemental Fig. 2. For this single experiment, a simple 1:1 binding mechanism provided a good fit, with fitted rate constants kon = 1.6 ± 0.7 × 106 m−1 s−1 and koff = 1.4 ± 0.5 × 10−3 s−1, which yielded a calculated Kd of 0.88 nm. This Kd value is similar to the result from pyrene actin polymerization assays, in this and previous studies (22). We measured binding affinities between CBR and all of the CP mutants discussed above. All of the CP mutants had binding affinities similar to that of WT CP (Table 1).
TABLE 1.
Binding constants for the interaction of CP and CP mutants with CBR based on SPR
Values are mean ± S.D. for three to five experiments.
| CP species | Kd |
|---|---|
| nm | |
| αβ WT | 0.75 ± 0.12 |
| α(E200R)β | 0.52 ± 0.01 |
| α(K256A)β | 1.38 ± 0.10 |
| α(R260A)β | 0.63 ± 0.10 |
| α(K268A)β | 0.94 ± 0.13 |
| αβ(R195A) | 0.50 ± 0.18 |
| αβ(K223A) | 0.57 ± 0.04 |
| αβ(R225A) | 0.67 ± 0.13 |
| αβ(LLL258/252/266SSS) | 0.72 ± 0.11 |
| αβ(EN256/260SS) | 0.63 ± 0.10 |
Potential Role for Unresolved Region of CBR in Binding CP
X-ray crystallography of the complex of CP with the 115-aa CBR (CBR115) resolved the structure of two parts of CBR115 (Glu-964 to Ser-1078), the CP-interaction (CPI) motif (Ile-971 to Cys-1004) and the CARMIL-specific interaction motif (Arg-1021 to Thr-1035) (20). However, a middle region of CBR115 from Ala-1005 to Gly-1020 was not resolved, presumably due to having a disordered structure in the crystal. We noted that the sequence of the middle region is conserved among CARMIL vertebrate isoforms (Fig. 2). Therefore, we considered the possibility that the unresolved middle region of CBR might be involved in inhibiting or uncapping CP.
To test the functional importance of the unresolved region, we made a mutant where all the residues in this region were changed to either glycine or alanine. We refer to this construct as the CBR random linker (CBR-RL) (Fig. 4A). We previously found that CBR71 (Glu-964–Val-1034) and CBR115 have similar activity toward CP, so we used CBR71 for this analysis (20). We tested the activities of WT CBR71 and CBR71-RL using pyrene actin polymerization assays. In capping inhibition assays, there was no difference between CBR71 and CBR71-RL (Fig. 4B). Also, in uncapping assays, CBR71-RL was able to uncap CP with the same ability as CBR71 (Fig. 4C). Finally, we used SPR to measure the binding affinity between CP and CBR71 or CBR71-RL. The Kd values were essentially identical, with 1.26 ± 0.12 nm for CBR71 and 1.38 ± 0.34 nm for CBR71-RL. Therefore, the middle region of the CBR does not appear to have an important role in the interaction of CBR with CP, additional evidence against a steric-blocking model.
FIGURE 4.

Ability of CBR71 random linker mutant to inhibit CP in actin polymerization assays. A, sequences of WT CBR71 and the CBR71 random linker mutant. The mutated region is shaded pink. B, inhibition of capping activity of CP by WT and mutant CBR71. C, uncapping of CP by WT and mutant CBR71. Concentrations of CP and CBR are as indicated. Red curves are WT CBR71, and green curves are CBR71 random linker mutant. a.u., arbitrary units.
In sum, none of the results supported a steric-blocking model. We next considered whether an allosteric model could account for the effects of CBR on CP.
Molecular Simulation of CP/CBR
We performed a series of all-atom molecular dynamics simulations to determine how the binding of CP to actin or to CARMIL CBR may affect the conformation and/or dynamics of CP. Using 400 ns of simulation for unbound CP, we performed PCA to determine the major conformational changes in CP. Because we were concerned about the influence of the C termini as well as how CBR may affect the stalk region of CP, we limited the PCA to the cap of CP, namely residues 61–258 for CPα and residues 48–243 for CPβ. The first two PCA modes correspond roughly to bending and twisting of the cap, and these modes were able to account for the majority of the conformational variance, and these formed the basis for the plot shown in Fig. 5, where the gray density contours show the range of conformations for unbound CP.
Next, we performed a simulation for the CBR-CP complex. Projecting the cap conformations from this simulation onto the same conformational space (red points and contour lines in Fig. 5), we see that CBR binding only marginally perturbs the cap conformation, shifting the peak of the distribution slightly from the unbound case. This result is consistent with our FRET results below, which found no significant change in the distance between the CFP and YFP connection points.
Finally, we simulated the CP-F-actin complex (13) and projected the results onto the conformation space (blue points and contour lines in Fig. 5). We see that binding to F-actin significantly changes the bend and twist of the cap by a relatively large amount and in a direction opposite to that induced by CBR.
Examining the structures at the peaks of the CBR and F-actin bound simulations, one sees significant differences in the helices and β sheets that make up the cap and F-actin binding region. These differences are illustrated in supplemental Movie 1. The F-actin cap conformation has a more curved conformation, whereas the CBR cap is slightly flatter. Based on these structures and the PCA analysis, we suggest that CARMIL CBR keeps CP in a conformation close to the unbound form of CP, thus inhibiting F-actin binding by inducing the cap to have an unfavorable actin-binding configuration. Furthermore, CBR may uncap a filament by inducing a conformational change in the cap of CP, leading to an increased dissociation rate for CP from the barbed end.
FRET Analysis
To test the predictions from the molecular dynamics simulations regarding the magnitude of distance changes associated with the proposed conformational change, we engineered a recombinant protein to measure intramolecular FRET efficiency for CP, alone and complexed with CBR. We placed the probes on the actin binding-surface, with CFP fused to the C terminus of the CPα subunit, and YFP fused to the C terminus of the CPβ subunit. Because the CPβ C-terminal tentacle is highly flexible, we removed the CPβ tentacle in the construct, placing YFP at the site on the body of CP where the tentacle would normally attach. We refer to this construct as CPαCFPβ(CΔ29)YFP. From the CP structure, the distance between CFP and YFP in this construct was ∼55 Å, which is within the range of FRET observance, considering that the R0 for GFP-based FRET pairs is ∼40 Å (Fig. 6A) (30).
FIGURE 6.

FRET analysis of binding of CP and CBR. A, structure of CPαCFPβ(CΔ29)YFP, indicating the positions of CFP and YFP on CP. The CPα and -β subunits are yellow and red, respectively. B, SDS-PAGE analysis of purified CPαCFPβ(CΔ29)YFP. C, emission scans of CPαCFPβ(CΔ29)YFP (excitation at 430 nm) before and after trypsin digestion (black and blue, respectively) without CBR. D, FRET efficiency was expressed as the difference in fluorescence of CFP after trypsin digestion (excitation, 430 nm; emission, 475 nm) minus before trypsin, as a percentage of the value after trypsin. The samples are 100 nm CP (CPαCFPβ(CΔ29)YFP), alone or in the presence of 500 nm WT CBR or CBR(R989A). a.u., arbitrary units.
To measure the intramolecular FRET of CPαCFPβ(CΔ29)YFP, emission scans were performed with excitation at 430 nm for 100 nm of CPαCFPβ(CΔ29)YFP. To calculate FRET efficiency, CPαCFPβ(CΔ29)YFP was treated with trypsin to cleave the link between the CP and the GFPs, and similar emission scans were performed (Fig. 6C). The results showed that CPαCFPβ(CΔ29)YFP had an intramolecular FRET efficiency of 6.0 ± 1.1% (mean ± S.D., n = 5) (Fig. 6D). Then, we added 500 nm of CBR to 100 nm CP and found that the FRET efficiency increased to 7.1 ± 1.6% (mean ± S.D., n = 5). This small difference was not statistically significant (p = 0.26). As a negative control, we measured the FRET efficiency using the CBR mutant R989A, which fails to bind CP. For CBR(R989A), the FRET efficiency was 6.0 ± 1.7% (mean ± s.d., n = 6). Thus, CBR binding to CP causes at most a small conformational change on the actin-binding surface of CP, consistent with the molecular dynamics analysis above.
Effects of CP C-terminal Truncations on CBR Binding
The allosteric model predicts that CBR binding influences the conformation of the actin-binding surface of CP. Conversely, one might expect that mutations of the actin-binding surface would influence the conformation of the CBR-binding site of CP. As described above, we found that numerous point mutations of the actin-binding surface, which involved changes of one to several surface amino acids, had essentially no effect on the binding of CBR. These changes may not have had much effect on the structure of the protein, because the residues were on the surface.
To test this prediction more stringently, we made much larger changes in the actin-binding surfaces of CP by truncating the C terminus of each subunit; these truncations produced large independent and additive effects on the capping activity in our previous work (4). We tested whether these mutations would affect the affinity of CARMIL for CP using physical binding assays. Pyrene actin polymerization assays, similar to what we used above with the CP point mutants, were not possible in this case because the CP double truncation mutant has essentially no capping activity that might be inhibited by CARMIL CBR (4). We used ITC, SPR, and bead pulldown experiments (Fig. 7).
FIGURE 7.

Binding of CBR to C-terminal truncation mutants of CP. A, isothermal calorimetry. Upper panels are raw titration data plotted as heat (μcal/s) versus time (seconds). Each experiment consisted of 28 injections of 10 μl of 50 μm CP into a solution of 5 μm CBR at 25 °C. Titrations with CPα(ΔC28)β, CPαβ(ΔC29), and CPα(ΔC28)β(ΔC29) were performed with varied injection volumes of 10 and 5 μl. The lower panels are integrated heat responses plotted as normalized heat per mole of injectant. Smooth curves represent best fits of the data to the equation as described under “Experimental Procedures” using software provided by the instrument manufacturer (MicroCal). B, surface plasmon resonance binding. GST-CBR was immobilized by anti-GST antibody, and CP was injected onto the chip, at 25 °C, at the following concentrations: wild-type (0.25, 0.5, 1.0, 2.0, 4.0, and 10 nm), CPα(ΔC28)β (20, 40, 60, 80, and 100 nm), CPαβ(ΔC29) (2.5, 5.0, 10, 20, 30, and 40 nm), and CPα(ΔC28)β(ΔC29) (300, 400, 500, and 1000 nm). C, bead pulldown analysis of CBR binding to the CPα(ΔC28)β(ΔC29) double truncation mutant, labeled CPΔ/Δ on the figure. Loss of CP from the supernatant was assessed by SDS-PAGE. The CP concentration was constant at 500 nm, and the concentration of GST-CBR was varied as indicated, up to 2 μm. For WT CP, the intensities of the α (upper) and β (lower) subunit bands are observed to decrease with increasing amounts of GST-CBR beads. In contrast, no decrease is observed for the CPΔ/Δ mutant, which migrate to lower positions in the gel. The first lane, M, indicates the molecular weight standards.
For ITC, we titrated a solution of CP (5–20 μm) with CARMIL CBR (50–150 μm). Typical titration curves are shown in Fig. 7A, with fitting of the binding isotherm to a one-site binding model. No titrations required correction for heat of dilution because no significant signal was observed when buffer alone was titrated with CBR. For WT CP and the single truncation mutants, the affinity was too high to allow for a reliable measurement. Relatively high concentrations of CP and CBR were required to overcome the sensitivity limit of the ITC instrument (0.1 μcal). Thus, the Kd values obtained, from 1 to 10 nm, represent upper limits. The Kd values for the single mutants were each somewhat higher than that of the wild-type; however, the error in the values was too high to allow one to draw firm conclusions. For the double truncation mutant, the affinity could be measured, and the Kd values were 30–100 nm, substantially less than the values for the WT and single mutants. The stoichiometry values were reproducible at 1:1, for WT CP and all the mutants.
For SPR analysis, 50 nm GST-CBR was flowed onto the chip and immobilized via anti-GST antibody, as the ligand. WT and mutant CP were injected at a range of concentrations, as the analyte. Representative tracings in Fig. 7B illustrate that the value of the resonance units of bound material was less for the mutants, despite the fact that the [CP] in the analyte was greater for the mutants. Both single truncation mutants showed decreased binding compared with WT, with the α truncation mutant showing worse binding than the β truncation mutant. The double mutant showed very little binding, far less than either single mutant alone. We were able to fit the dissociation portions of the curves, and the dissociation rate constants (koff) were 2.4 ± 0.4 × 10−3 s−1 for WT CPαβ, 68 ± 6 × 10−3 s−1 for the α truncate CPα(ΔC28)β, and 5.3 ± 0.2 × 10−3 s−1 for the β truncate CPαβ(ΔC29). Representative single tracings for each sample are shown in supplemental Fig. 3. The amount of material bound for the double mutant CPα(ΔC28)β(ΔC29) was too low to allow for fitting the dissociation curves.
Finally, we performed a bead pulldown assay to assess binding (Fig. 7C). CP in solution was added to GST-CBR attached to glutathione-Sepharose beads. The beads were centrifuged, and CP in the supernatant was analyzed by SDS-PAGE. The CP concentration was held constant at 500 nm, and the amount of beads was increased, with the maximum amount equivalent to a GST-CBR concentration of 2 μm. WT CP was depleted from the supernatant, in amounts consistent with the high affinity observed in other assays. In contrast, the CPα(ΔC28)β(ΔC29) double truncation mutant showed no detectable interaction, also consistent with the other assays. Single truncation mutants were not tested in this assay.
DISCUSSION
Regulation of capping barbed ends is a critical issue for the control of actin polymerization in cells. CP is the major factor, present in essentially all eukaryotic cells, which caps barbed ends. CARMIL is a prominent potential regulator of CP in cells. CARMIL is a member of a set of otherwise unrelated proteins that bind and inhibit CP via a common motif. Therefore, we were interested to investigate the molecular mechanism by which CARMIL inhibits CP and causes uncapping of CP-capped actin filaments.
We first considered a simple steric blocking model, in which CARMIL binds to some of the same surface locations on CP that contact the barbed end of the actin filament. We have considered such a model to be unlikely on the basis of two observations: 1) CARMIL causes CP to dissociate from barbed ends very rapidly (26), much faster than the spontaneous time for dissociation of CP from the barbed end (27). 2) The complex of CARMIL with CP appears to have a finite, albeit greatly decreased, ability to cap barbed ends (22). These two facts suggest that CARMIL binds to CP at a site distinct from the actin-binding site and that a ternary complex of CARMIL, CP, and the barbed end of actin can exist.
This view was supported by structural studies of the CARMIL-CP complex in which CARMIL was observed to be in contact with a surface of CP distinct from the actin-binding surface (20, 21). However, substantial portions of the CARMIL fragment were not resolved in the structure, raising the possibility that these unresolved regions might be in contact with one of the several actin-binding surfaces of CP. In addition, a separate structural study, utilizing NMR, concluded that certain residues of CARMIL were in close contact with residues of CP known to contact actin (36).
Therefore, we tested the steric-blocking model. First, we tested a set of point mutations of CP that we previously showed impair the ability of CP to bind actin (13). Those mutations involve CP residues that are in close contact with actin in a structural model that we developed (13). We found that none of those point mutations had any significant effect on the ability of CARMIL to bind CP, based on functional assays involving pyrene actin polymerization and physical binding assays with SPR. Of note, we tested point mutations involving the highly mobile C terminus of the CP β subunit which is involved in binding actin; we reasoned that the mobility of this region might allow it to move into direct contact with a region of CARMIL not resolved in the structure of the CP-CARMIL complex. This particular notion was tested previously with a distinct set of biochemical studies; no evidence for such an interaction was observed (20).
We also tested the steric blocking model by extensively mutating the unresolved region of CARMIL that lies between the two CP-binding motifs that were resolved in the crystal structure. In one mutant form of CARMIL, we changed all of these residues to glycine or alanine. This mutant CARMIL interacted with CP with essentially normal affinity. We conclude that together, these results exclude a steric-blocking model as thoroughly as reasonably practical.
We then sought evidence for an allosteric model in which the binding of CARMIL to one surface of CP affected the structure of CP at a distance, on its actin-binding surface. A previous NMR study indicated that the structure of CP might undergo at most subtle rearrangements upon binding to CARMIL (36). We used molecular dynamics to examine how the structure of CP was affected by binding to CARMIL or to actin. Concentrating on the actin-binding surface of CP, we found that CARMIL binding had only a small effect on the range of structures, comparing free CP with CARMIL-bound CP. In contrast, the structure of CP bound to actin was quite distinct from those of free CP or CARMIL-bound CP. These results support the hypothesis that the binding of CARMIL, to a site on CP distant from the actin-binding site, has a substantial influence on the structure of the actin-binding surface of CP, one that might account for the decrease in affinity of CP for actin when CARMIL is bound.
It is interesting to compare our findings here with elastic network model results of Takeda and colleagues (12). In this report, the authors performed normal mode analysis using an elastic network model, and they concluded that CARMIL binding alters a twisting motion within CP that could hypothetically decrease the affinity for the barbed end. At a qualitative level, our simulations provide similar observations; however, the all-atom molecular dynamics simulations that we used here are more detailed than an elastic network model, and the observed dynamics are not restricted by artificial harmonic constraints. The mean root mean square deviation between the various crystal and NMR structures of CP plotted in Fig. 5 is only ∼1 Å, and the range of conformations exhibited by these structures is obviously less than what we see in our molecular dynamics simulations. Furthermore, the root mean square deviation between the peak structures shown in Fig. 5 is >4 Å, and the conformation of F-actin bound CP is completely outside the range of either CARMIL-bound or F-actin-bound CP. The fact that the F-actin- and CARMIL-bound conformations do not overlap strongly supports the allosteric mechanism that we propose.
In addition, we sought experimental evidence for this allosteric mechanism with two additional approaches. First, we used intramolecular FRET, with CFP and YFP probes attached to different locations on the actin-binding surface of CP. Addition of CARMIL resulted in little change in distance between the probes, which is consistent with the differences in distances expected from the molecular dynamics simulations. Second, we mutated, rather harshly, the actin-binding surface of CP, truncating the α and β subunits at their C termini by ∼30 aa residues. These mutations on the actin-binding surface of CP had measurable effects on the affinity of CP for CARMIL, confirming the proposed linkage between the conformations of the CARMIL-binding and actin-binding sites. Truncating each subunit alone had an effect, and those effects were additive in the double mutant. We reached this conclusion with three independent physical binding approaches: ITC, SPR, and bead pulldown analyses. We took extra care to be thorough in this analysis because a previous study from our laboratory did not reach this conclusion (22). That study (22) employed only the GST bead pulldown analysis. The shapes of the binding curves suggested that the concentrations of the reactants were rather high compared with the equilibrium dissociation constant. Here, we attempted to replicate the conditions used in the previous study, but the results were distinctly different. We do not have a ready explanation for this difference. However, the three independent approaches used in this current study all lead to the same conclusion, and we are confident that this conclusion is the correct one.
In summary, we conclude that CARMIL binds to CP at a site distinct from the actin-binding site and that this binding influences the conformation of the actin-binding site, which lowers the affinity of CP for the barbed end. We anticipate that this mechanism will be relevant to other CPI motif proteins, such as CD2AP, CKIP-1, and WASHCAP, which remains to be investigated. On the physiological role of CARMIL and other CPI motif proteins, we note that the CARMIL-CP complex does have capping activity, albeit much less than that of CP alone, which raises the possibility that, in cells, CARMIL serves to target CP to a location, which promotes barbed end capping in that region. This idea is an interesting alternative to the simpler notion that CARMIL serves to inhibit or reverse the action of CP, which promotes the formation and presence of free barbed ends.
Supplementary Material
Acknowledgments
We are grateful to Dr. Robert Robinson for continued interactions in general and, in particular, for the idea of extensively mutating the unresolved middle region of CARMIL CBR. We thank Drs. Ville Paavilainen and Pekka Lappalainen for the His-tagged CP expression plasmid. For advice and assistance with SPR, we thank Kyle Austin, Dr. Daved Fremont, and Dr. Paul Schlesinger. Dr. Schlesinger analyzed the SPR data. We thank Drs. Tim Lohman and Roberto Galletto for continued discussions of this work, especially the ITC and FRET analyses.
This work was supported, in whole or in part, by National Institutes of Health Grants R01GM38542 (to J. A. C.) and R01GM67246 (to D. S.).

This article contains supplemental Table 1, Figs. 1–3, and Movie 1.
- CP
- capping protein
- CBR
- capping protein-binding region of CARMIL
- CD2AP
- CD2-associated protein
- CKIP-1
- casein kinase interaction protein
- CPI
- capping-protein-interaction motif
- ITC
- isothermal calorimetry
- SPR
- surface plasmon resonance
- aa
- amino acid(s)
- PCA
- principal component analysis.
REFERENCES
- 1. Pollard T. D., Cooper J. A. (2009) Actin, a central player in cell shape and movement. Science 326, 1208–1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cooper J. A., Sept D. (2008) New insights into mechanism and regulation of actin capping protein. Int. Rev. Cell Mol. Biol. 267, 183–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cooper J. A., Blum J. D., Pollard T. D. (1984) Acanthamoeba castellanii capping protein: Properties, mechanism of action, immunologic cross-reactivity, and localization. J. Cell Biol. 99, 217–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wear M. A., Yamashita A., Kim K., Maéda Y., Cooper J. A. (2003) How capping protein binds the barbed end of the actin filament. Curr. Biol. 13, 1531–1537 [DOI] [PubMed] [Google Scholar]
- 5. Bearer E. L. (1991) Direct observation of actin filament severing by gelsolin and binding by gCap39 and CapZ. J. Cell Biol. 115, 1629–1638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pavlov D., Muhlrad A., Cooper J., Wear M., Reisler E. (2007) Actin filament severing by cofilin. J. Mol. Biol. 365, 1350–1358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Le Clainche C., Carlier M. F. (2008) Regulation of actin assembly associated with protrusion and adhesion in cell migration. Physiol. Rev. 88, 489–513 [DOI] [PubMed] [Google Scholar]
- 8. Mejillano M. R., Kojima S., Applewhite D. A., Gertler F. B., Svitkina T. M., Borisy G. G. (2004) Lamellipodial versus filopodial mode of the actin nanomachinery: Pivotal role of the filament barbed end. Cell 118, 363–373 [DOI] [PubMed] [Google Scholar]
- 9. Kim K., Yamashita A., Wear M. A., Maéda Y., Cooper J. A. (2004) Capping protein binding to actin in yeast: Biochemical mechanism and physiological relevance. J. Cell Biol. 164, 567–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chesarone M. A., DuPage A. G., Goode B. L. (2010) Unleashing formins to remodel the actin and microtubule cytoskeletons. Nat. Rev. Mol. Cell Biol. 11, 62–74 [DOI] [PubMed] [Google Scholar]
- 11. Yamashita A., Maeda K., Maéda Y. (2003) Crystal structure of CapZ: Structural basis for actin filament barbed end capping. EMBO J. 22, 1529–1538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Takeda S., Koike R., Nitanai Y., Minakata S., Maéda Y., Ota M. (2011) Actin capping protein and its inhibitor CARMIL: how intrinsically disordered regions function. Phys. Biol. 8, 035005 [DOI] [PubMed] [Google Scholar]
- 13. Kim T., Cooper J. A., Sept D. (2010) The interaction of capping protein with the barbed end of the actin filament. J. Mol. Biol. 404, 794–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Iwasa J. H., Mullins R. D. (2007) Spatial and temporal relationships between actin filament nucleation, capping, and disassembly. Curr. Biol. 17, 395–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Miyoshi T., Tsuji T., Higashida C., Hertzog M., Fujita A., Narumiya S., Scita G., Watanabe N. (2006) Actin turnover-dependent fast dissociation of capping protein in the dendritic nucleation actin network: Evidence of frequent filament severing. J. Cell Biol. 175, 947–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Uruno T., Remmert K., Hammer J. A. (2006) CARMIL is a potent capping protein antagonist: Identification of a conserved CARMIL domain that inhibits the activity of capping protein and uncaps capped actin filaments. J. Biol. Chem. 281, 10635–10650 [DOI] [PubMed] [Google Scholar]
- 17. Bruck S., Huber T. B., Ingham R. J., Kim K., Niederstrasser H., Allen P. M., Pawson T., Cooper J. A., Shaw A. S. (2006) Identification of a novel inhibitory actin-capping protein binding motif in CD2-associated protein. J. Biol. Chem. 281, 19196–19203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Canton D. A., Olsten M. E., Niederstrasser H., Cooper J. A., Litchfield D. W. (2006) The role of CKIP-1 in cell morphology depends on its interaction with actin-capping protein. J. Biol. Chem. 281, 36347–36359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Canton D. A., Olsten M. E., Kim K., Doherty-Kirby A., Lajoie G., Cooper J. A., Litchfield D. W. (2005) The pleckstrin homology domain-containing protein CKIP-1 is involved in regulation of cell morphology and the actin cytoskeleton and interaction with actin capping protein. Mol. Cell Biol. 25, 3519–3534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hernandez-Valladares M., Kim T., Kannan B., Tung A., Aguda A. H., Larsson M., Cooper J. A., Robinson R. C. (2010) Structural characterization of a capping protein interaction motif defines a family of actin filament regulators. Nat. Struct. Mol. Biol. 17, 497–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Takeda S., Minakata S., Koike R., Kawahata I., Narita A., Kitazawa M., Ota M., Yamakuni T., Maéda Y., Nitanai Y. (2010) Two distinct mechanisms for actin capping protein regulation–steric and allosteric inhibition. PLoS Biol. 8, e1000416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yang C., Pring M., Wear M. A., Huang M., Cooper J. A., Svitkina T. M., Zigmond S. H. (2005) Mammalian CARMIL inhibits actin filament capping by capping protein. Dev. Cell 9, 209–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Remmert K., Olszewski T. E., Bowers M. B., Dimitrova M., Ginsburg A., Hammer J. A., 3rd (2004) CARMIL is a bona fide capping protein interactant. J. Biol. Chem. 279, 3068–3077 [DOI] [PubMed] [Google Scholar]
- 24. Jung G., Remmert K., Wu X., Volosky J. M., Hammer J. A. (2001) The Dictyostelium CARMIL protein links capping protein and the Arp2/3 complex to type I myosins through their SH3 domains. J. Cell Biol. 153, 1479–1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liang Y., Niederstrasser H., Edwards M., Jackson C. E., Cooper J. A. (2009) Distinct roles for CARMIL isoforms in cell migration. Mol. Biol. Cell 20, 5290–5305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fujiwara I., Remmert K., Hammer J. A., 3rd (2010) Direct observation of the uncapping of capping protein-capped actin filaments by CARMIL homology domain 3. J. Biol. Chem. 285, 2707–2720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schafer D. A., Jennings P. B., Cooper J. A. (1996) Dynamics of capping protein and actin assembly in vitro: Uncapping barbed ends by polyphosphoinositides. J. Cell Biol. 135, 169–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gill S. C., von Hippel P. H. (1989) Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 182, 319–326 [DOI] [PubMed] [Google Scholar]
- 29. Pollard T. D. (1986) Rate constants for the reactions of ATP- and ADP-actin with the ends of actin filaments. J. Cell Biol. 103, 2747–2754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Makhina E. N., Nichols C. G. (2001) in Ion Channel Localization Methods and Protocols (Lopatin A., Nichols C. G., eds.) pp. 261–274, Humana Press, Totowa, NJ [Google Scholar]
- 31. Lakowicz J. R. (1999) Principles of Fluorescence Spectroscopy, p. 370, Kluwer Academic/Plenum, New York [Google Scholar]
- 32. Ohashi T., Galiacy S. D., Briscoe G., Erickson H. P. (2007) An experimental study of GFP-based FRET, with application to intrinsically unstructured proteins. Protein Sci. 16, 1429–1438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Eswar N., Eramian D., Webb B., Shen M. Y., Sali A. (2008) Protein structure modeling with MODELLER. Methods Mol. Biol. 426, 145–159 [DOI] [PubMed] [Google Scholar]
- 34. Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kalé L., Schulten K. (2005) Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wyman J., Jr. (1964) Linked functions and reciprocal effects in hemoglobin: A second look. Adv. Protein Chem. 19, 223–286 [DOI] [PubMed] [Google Scholar]
- 36. Zwolak A., Uruno T., Piszczek G., Hammer J. A., 3rd, Tjandra N. (2010) Molecular basis for barbed end uncapping by CARMIL homology domain 3 of mouse CARMIL-1. J. Biol. Chem. 285, 29014–29026 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.