

Background: H2O2 is toxic to cells at micromolar levels.
Results: H2O2 inactivates a wide variety of mononuclear iron enzymes, but the hydroxyl radical is often quenched by metal-coordinating cysteine residue.
Conclusion: A surprising number of enzymes are acutely sensitive to inactivation by H2O2, but the injury is often reversible.
Significance: The use of iron to bind and activate enzymes creates a point of vulnerability to oxidants.
Keywords: Iron, Manganese, Metalloenzymes, Oxidative stress, Sulfhydryl, Fenton Reaction, Peptide Deformylase, Sulfhydryl Oxidation
Abstract
This study tested whether nonredox metalloenzymes are commonly charged with iron in vivo and are primary targets of oxidative stress because of it. Indeed, three sample mononuclear enzymes, peptide deformylase, threonine dehydrogenase, and cytosine deaminase, were rapidly damaged by micromolar hydrogen peroxide in vitro and in live Escherichia coli. The first two enzymes use a cysteine residue to coordinate the catalytic metal atom; it was quantitatively oxidized by the radical generated by the Fenton reaction. Because oxidized cysteine can be repaired by cellular reductants, the effect was to avoid irreversible damage to other active-site residues. Nevertheless, protracted H2O2 exposure gradually inactivated these enzymes, consistent with the overoxidation of the cysteine residue to sulfinic or sulfonic forms. During H2O2 stress, E. coli defended all three proteins by inducing MntH, a manganese importer, and Dps, an iron-sequestration protein. These proteins appeared to collaborate in replacing the iron atom with nonoxidizable manganese. The implication is that mononuclear metalloproteins are common targets of H2O2 and that both structural and metabolic arrangements exist to protect them.
Introduction
Reactive oxygen species, such as hydrogen peroxide and superoxide, arise continuously in aerobic organisms due to the oxidation of redox enzymes. The rate of reactive oxygen species formation is dependent upon the enzymatic cohort of a given organism and may be a primary determinant of which organisms can populate a given habitat. Studies of Escherichia coli demonstrate that the titer of scavenging enzymes is just sufficient to keep the intracellular concentrations of superoxide and hydrogen peroxide at tolerable levels (1, 2). As a consequence, any increment in stress from exogenous sources can quickly block growth. Both plants and animals exploit this situation by using NADPH oxidase to keep invading bacteria in check, and plants and microbes excrete redox-cycling compounds to suppress the growth of competitors.
Cells must turn up their defenses if they are to survive these challenges. Any influx of H2O2 into the cytoplasm activates the OxyR and PerR proteins of bacteria and the Yap1 protein of yeast (3–5). These transcription factors then induce peroxidases and catalases to drive back down the H2O2 level. They also induce an array of gene products whose contribution to stress resistance is less clear, in part because we do not understand the primary targets that H2O2 damages (6–8).
Even 1 μm intracellular H2O2 poisons individual pathways of E. coli (9–12). An early hypothesis was that H2O2 might disable metabolic enzymes by oxidizing key cysteine residues. This idea gained support from the discovery that the OxyR, PerR, and Yap1 systems all induce glutaredoxins and thioredoxins that can efficiently reduce sulfenic acid and disulfide derivatives (6–8). Yet kinetic analyses showed that H2O2 is actually a poor oxidant of typical cysteine residues, with rate constants that are too low to support enzyme inactivation by micromolar H2O2 (13, 14).
Work has shown that H2O2 can damage the solvent-exposed [4Fe-4S] clusters of dehydratases, resulting in their decomposition to a [3Fe-4S] state that is catalytically inactive (11). There are a handful of such enzymes in E. coli, and indeed the few pathways to which they belong fail during micromolar H2O2 stress. However, H2O2 is still acutely toxic even when culture conditions are chosen that do not rely upon the function of these enzymes. What then are the targets?
A recent study determined that H2O2 disables the pentose-phosphate pathway specifically by poisoning ribulose-5-phosphate 3-epimerase (RPE)3 (12). This enzyme requires a divalent metal for activity, and earlier in vitro studies had led investigators to believe that the metal was zinc. However, new work revealed that the intracellular enzyme is actually activated by ferrous iron; thus, through the Fenton reaction H2O2 oxidizes the metal and inactivates the enzyme.
Strikingly, manganese import, an essential element of the OxyR response and a universal protector of microbes against oxidative stress, protects this enzyme. When RPE is loaded with manganese in place of iron, it retains activity but does not react with H2O2. Manganese import thereby ensures the continued function of RPE during periods of oxidative stress.
The goal of this work was to determine whether RPE is a unique case or whether E. coli might contain many such iron-charged enzymes that are vulnerable to H2O2. Iron is not commonly believed to be an activator of nonredox mononuclear enzymes, which are typically annotated as being charged by zinc, manganese, or cobalt. However, the possibility that such enzymes actually use iron might be under-recognized because of the difficulty of conducting biochemical studies with ferrous iron in aerobic buffers. We examined three mononuclear enzymes that catalyze very distinct categories of reactions. All three were found to use ferrous iron in vivo, and all three were shown to be acutely sensitive to H2O2. Intriguingly, two of them use cysteine residues to absorb the hydroxyl radical that is made by the Fenton reaction, and the oxidized enzyme can therefore be reduced back to an active form. When manganese is available to be imported, these enzymes are protected from inactivation.
EXPERIMENTAL PROCEDURES
Reagents
Acid-hydrolyzed casamino acids (Hy-Case Amino), l-amino acids, ascorbic acid, antibiotics, o-nitrophenyl-β-d-galactopyranoside, catalase (from bovine liver), ferrous ammonium sulfate hexahydrate, cytosine, cobalt(II) chloride hexahydrate, manganese(II) chloride tetrahydrate, nickel(II) sulfate, zinc(II) chloride, diethylenetriaminepentaacetic acid (DTPA), EDTA, NADH, NAD+, monobromobimane, dl-dithiothreitol (DTT), and tris (2-carboxyethyl)phosphine (TCEP) were from Sigma. Guanidine hydrochloride was obtained from Fisher. Formate dehydrogenase (Candida boidinii) was from Roche Applied Science. Formyl-Met-Ala-Ser was from Bachem. OxyBlot protein oxidation detection kit (S7150) was from Chemical International.
Bacterial Growth
Luria broth (LB) contained (per liter) 10 g of tryptone, 5 g of yeast extract, and 10 g of NaCl (15). Minimal A casamino acids/glucose medium (MinCAAG) consisted of minimal A salts (15) supplemented with 0.2% casein hydrolysate, 0.5 mm l-tryptophan, and 0.2% glucose. When antibiotic selection was needed, media were supplemented with either 100 μg/ml ampicillin or 20 μg/ml chloramphenicol. Protein synthesis was stopped by adding 150 μg/ml chloramphenicol.
Anaerobic cultures were grown in an anaerobic chamber (Coy Laboratory Products, Inc.) under an atmosphere of 85% nitrogen, 10% hydrogen, and 5% carbon dioxide. Aerobic cultures were grown with vigorous shaking at 37 °C. To ensure that cells were growing exponentially before they were exposed to oxygen, anaerobic overnight cultures of oxygen-sensitive strains were diluted to A600 = 0.005 in fresh anaerobic medium and allowed to grow to A600 ∼0.15 at 37 °C. These cells were then subcultured into fresh aerobic medium to obtain an A600 of 0.005, with or without manganese chloride, and grown aerobically at 37 °C.
Strains and Plasmid Construction
Strains used in this study are listed in supplemental Table S1. Strain and plasmid constructions are also described in the supplemental material. All constructions in Hpx− (i.e. ΔkatE ΔkatG Δahp (9)) backgrounds were performed in an anaerobic chamber to ensure that suppressor mutations were not selected during outgrowth. Null mutations were created using the Red recombinase method (16). Mutations were then introduced into new strains by P1 transduction (15). The resultant mutations were confirmed by PCR analysis and, when possible, by enzyme assays.
Protein Purification
Peptide deformylase (PDF) and threonine dehydrogenase (TDH) were each purified aerobically by standard methods, with modifications (17, 18); for full details, see the supplemental material. PDF was purified with 0.2 mm NiSO4 continuously present as a stabilizing cofactor; TDH was purified in the presence of 0.25 mm ZnCl2. The final purities of the enzymes were determined to be >95% by SDS-PAGE, with enzyme activities of ∼150 units/ml PDF and ∼100 units/ml TDH. The metallated enzymes were stable at both 0 °C and RT. In contrast, the apo-forms were each stable only when concentrated (200 μm); upon dilution, PDF and TDH converted to nonreactivatible states (t½ ∼3 and ∼5 min, respectively).
Enzyme Assays
All enzymes assays were conducted in anaerobic buffers within the anaerobic chamber. All enzyme assays were performed at 25 °C unless specified otherwise specified. Glyceraldehyde-3-phosphate dehydrogenase (19) and isocitrate lyase (20) enzyme activities were assayed as described. PDF (21), TDH (18), and cytosine deaminase (CDA) (22) assays were conducted by standard methods, with slight modifications. Peptide deformylase was assayed in 50 mm HEPES buffer, pH 7.5, with 25 mm NaCl. A typical assay (500 μl) consisted of enzyme (pure or extract), 10 mm NAD+, 1 unit of formate dehydrogenase, 1 mm formyl-Met-Ala-Ser, and 100 μm metal. Threonine dehydrogenase was assayed in 500 μl of 50 mm Tris-HCl buffer, pH 8.4, with enzyme (pure or extract), 1 mm NAD+, 30 mm threonine, and 100 μm metal. PDF and TDH activities were measured at 340 nm. Km and kcat values were determined using double-reciprocal plots, with variation of formyl-Met-Ala-Ser peptide for PDF and of both threonine and NAD+ for TDH. Metal dissociation rates were determined in the presence of substrates and 200 μm DTPA, a chelator that does not actively extract prosthetic metals from these enzymes but that traps dissociated metals. A typical assay of CDA (500 μl), included buffer, enzyme (extract), 100 μm metal, and 400 μm cytosine. CDA activities were measured by monitoring cytosine disappearance at 286 nm. Total protein content was determined using the Coomassie Blue dye binding assay (Pierce).
In Vitro Studies of Enzyme Oxidation
Pure PDF was stored metallated with 50 μm NiSO4. To load PDF with other metals, pure PDF was incubated without dilution for ∼45 min with 25 mm EDTA (50 mm HEPES, pH 7.5, with 25 mm NaCl) at RT to chelate the nickel. The apoenzyme was then diluted 1:50 into buffer (50 mm HEPES, pH 7.5, with 25 mm NaCl) containing 600 μm of the desired metal (enough to exceed the 500 μm carry-over EDTA). Pure TDH was stored metallated with 50 μm ZnCl2. To load TDH with other metals, TDH was incubated for ∼30 min with 10 mm EDTA to chelate the zinc, and the apoenzyme was then diluted 1:50 into buffer (50 mm Tris-HCl, pH 8.4) containing 300 μm of the desired metal (enough to exceed the 200 μm carry-over EDTA). CDA was chelated in extracts in the presence of 1 mm 1,10-phenanthroline for 10 min as described (22).
To study the inactivation of metal-loaded enzymes, the metallated enzymes (above) were further diluted 100-fold into buffer containing 100 μm metal, substrate, and (for PDF) formate dehydrogenase. Substrate was required to ensure manganese and iron binding; in the absence of substrate, chelators immediately inactivated both enzymes. Chelator (200 μm DTPA) was then added to suppress reactions between dissociated metals and H2O2, followed by the H2O2 after 30 s. The H2O2 was then scavenged by the addition of catalase (∼550 units/ml), and the assay was performed. In some cases, TCEP (500 μm) was included prior to the addition of H2O2; although TCEP can scavenge H2O2, we determined that the rate is too slow to be an issue in these experiments. Because H2O2 reacts so rapidly with iron-charged PDF and TDH, the inactivation rates were determined at 0 °C. The reversibility of damage was tested by incubating the H2O2-exposed enzyme with additional metal (nickel for PDF and zinc for TDH) and either TCEP or DTT (500 μm) for ∼5 min prior to assay. The added metals also displaced any iron/manganese from the active site of the undamaged enzyme, so that the activities of the oxidized and untreated enzymes could be directly compared.
The rate of inactivation of apo-PDF by H2O2 was also measured. At intervals an aliquot of the reaction mixture was then added to a tube containing catalase (550 units/ml) and NiSO4 (500 μm) ± TCEP (500 μm). Nickel immediately restored full activity to undamaged apoenzyme. The rate of apo-TDH inactivation was determined similarly, except ZnCl2 (500 μm) was used instead of NiSO4.
Calculations of second-order rate constants for the inactivation of iron-loaded enzymes and for the oxidation of apoenzymes were straightforward. These constants were then incorporated in the calculation of second-order rate constants for the over-oxidation of iron-loaded PDF enzymes from nominal sulfenic to sulfinic forms.
The PDF thiol content was determined by monobromobimane modification. Fluorescence was measured on a Thermo Scientific NanoDrop 3300 fluorometer under aerobic conditions. The excitation wavelength was 380 nm, and emission was measured at 475 nm. Measurement of sulfhydryl content required at least 5 μm enzyme. Apo-PDF was challenged with 20 μm H2O2 for 1 min, in 50 mm HEPES, pH 7.5, with 25 mm NaCl at RT, after which catalase was added (550 units/ml). The samples were then incubated with 500 μm monobromobimane for 30 min in the dark, and fluorescence was measured.
Mass spectrometric analysis (electrospray ionization) of purified PDF was performed on a micromass Q-TOF Ultima. Apo-PDF was oxidized with variable H2O2 concentrations and time intervals. These samples were then precipitated with 10% TCA in acetone, and the pellet was dissolved in acetonitrile for mass spectrometric analysis.
In Vivo Studies of Enzyme Oxidation
The inactivation of enzymes by intracellular H2O2 was studied in mutants lacking either catalase (Kat−, ΔkatE ΔkatG) or both catalase and peroxidase (Hpx−, ΔkatE ΔkatG Δahp). The absence of catalase from the former strain allows exogenous H2O2 to persist for the duration of exposure as long as the amount added exceeds 20 μm, as Ahp is ineffective at high doses. However, due to the presence of Ahp, the ΔkatE ΔkatG mutants do not experience endogenous H2O2 stress when grown aerobically and do not express the OxyR regulon. In contrast, the Hpx− mutants lack any significant scavenging activity and accumulate ∼1 μm endogenous H2O2 when cultured in aerobic medium. This stress is sufficient to activate the OxyR system, causing the induction of MntH and Dps.
For in vivo exposure with a bolus of H2O2, cells were grown at 37 °C ± MnCl2, as indicated, to A600 of ∼0.2–0.3. Protein synthesis was inhibited by adding 150 μg/ml chloramphenicol for 15 min. Cells were then stressed with 100 μm H2O2 for 10 min, followed by the addition of catalase (100 units/ml). Cells were then transferred to the anaerobic chamber, washed with 50 mm HEPES, 25 mm NaCl, resuspended in the same buffer, and quickly sonicated. PDF, TDH, and CDA were then immediately assayed as described. Where indicated, extracts were supplemented with 500 μm nickel (for PDF) or iron (TDH and CDA) ± 500 μm TCEP for 5 min prior to assay. Because these enzymes have high metal dissociation rates (Table 1), addition of exogenous metals is necessary to ensure full metallation of PDF, TDH, and CDA. The resultant activity represents the amount of undamaged metalloprotein polypeptide in the cell extract.
TABLE 1.
Kinetic values, PDF and TDH

1 Not determined.
Enzyme activities in Hpx− cultures were monitored after dilution from anaerobic log-phase cultures to 0.005 A600 in aerobic medium. Assays were conducted when cultures reached 0.2 A600, following the protocol above. Because Hpx− Δdps and Hpx− mntH strains only triple in biomass before growth ceases, this strain was inoculated into aerobic medium at an A600 of 0.050.
Protein carbonylation was measured in 100 ml of Hpx− Δdps cultures that were handled the same way. Cells were washed twice with ice-cold 50 mm potassium phosphate buffer, pH 7.0, after which they were suspended in 500 μl of the same buffer containing 5 mm DTPA. DTPA prevents further protein oxidation in extracts and thus allows the manipulations to be performed aerobically. Cells were sonicated, and then the protein carbonyl content of the extract (2 mg/ml total protein) was measured using the OxyBlot protein oxidation detection kit (S7150) (Chemicon International). Protein carbonyl groups were derivatized to their 2,4-dinitrophenylhydrazones by reaction with 2,4-dinitrophenylhydrazine for 15 min in 3% (w/v) SDS. β-Mercaptoethanol (1% v/v) was then added to these derivatized samples, after which the samples were subjected to polyacrylamide denaturing gel electrophoresis (30 μg/lane) using a pre-cast 4–15% gel (Bio-Rad). Proteins were then transferred to a PVDF membrane (HybondTM-ECLTM, GE Healthcare) for 120 min at 20 V. The membrane was then incubated with primary antibody specific to the dinitrophenylhydrazone moiety attached to the derivatized proteins. This step was followed by incubation with a horseradish peroxidase antibody conjugate directed against the primary antibody. The membranes were then treated with chemiluminescent substrate (ECLTM Western Blotting Analysis System, GE Healthcare) and imaged by exposure to light-sensitive films (Hyperfilm ECLTM chemiluminescence film, GE Healthcare).
To monitor the stability of PDF in stressed cells, strains containing pPDF-FLAG were grown in anaerobic medium containing 2.5 mm lactose to A600 ∼0.2 (100 ml of each culture). Chloramphenicol (150 μg/ml) was then added to inhibit further protein synthesis. After 15 min, the culture was aerated, and at specified time intervals samples were harvested. These cells were washed with ice-cold 50 mm HEPES containing 25 mm NaCl and then resuspended in the same buffer (500 μl). The samples were then Western-blotted as described above (30 μg/lane), using ProteoQwest anti-FLAG peroxidase conjugate antibody.
RESULTS
PDF, TDH, and CDA Are Very Sensitive to H2O2 in Vitro
To test whether mononuclear enzymes are commonly sensitive to H2O2, we selected PDF, TDH, and CDA for inspection. These enzymes catalyze diverse categories of reactions, but they have in common the use of a transition metal to bind substrate and stabilize an anionic reaction intermediate. They were each known to be capable of using a variety of metals in vitro, but the identities of their prosthetic metals in vivo were uncertain. PDF purifies with bound zinc and was originally believed to be a zinc enzyme, but the instability of the just-extracted enzyme in aerobic buffers matches the instability of the iron-constituted enzyme (23). CDA purified from cells is predominantly occupied by zinc with a minor amount of iron, and mechanistic studies have usually employed the zinc-loaded enzyme; however, kinetic efficiency is somewhat higher with iron (24, 25). TDH exhibits good activity when constituted with zinc and is tentatively regarded as using zinc as an active-site metal in vivo; it is less active with manganese, and activity with iron has not been reported (26, 27).
Two of the enzymes (PDF and TDH) were purified, stripped of their native metals by incubation with chelators, and then charged with various metals. Both enzymes exhibited some activity when constituted with Fe(II), Mn(II), and Co(II); PDF also was active when treated with Ni(II), whereas TDH exhibited activity with Zn(II) (Table 1). Neither enzyme was active with Mg(II), Ca(II), or Cd(II). Of the activating metals, only iron and zinc are customarily present inside E. coli. Manganese is apparently imported only under conditions of oxidative stress or iron starvation (28, 29), and nickel is imported only under anaerobic conditions when hydrogenase synthesis is activated (30). These observations provide circumstantial support for the idea that iron is likely to be the prosthetic metal for these mononuclear enzymes during routine growth.
Purified iron-metallated PDF and TDH were extremely sensitive to H2O2, whereas the manganese-, nickel-, cobalt-, and zinc-metallated enzymes were resistant (Fig. 1, A and B). Nonmetalloenzymes are also fully resistant to H2O2 (11, 12). The slight residual sensitivity of manganese-loaded TDH arose from a metal-independent reaction (see below). The rate constants with which H2O2 inactivated iron-loaded PDF (>2200 m−1 s−1) and TDH (7200 m−1 s−1) were very high even at 0 °C; rates at room temperature were too fast to quantify (>20,000 m−1 s−1 for PDF).
FIGURE 1.

PDF, TDH, and CDA are sensitive to H2O2 stress in vitro and in vivo. A, purified PDF was metallated with various transition metals (100 μm) and then challenged with H2O2 (10 μm for 5 min). B, purified TDH was metallated with various transition metals (100 μm) and then challenged with H2O2 (10 μm for 5 min). C, as-isolated PDF, TDH, and CDA activities of crude extracts were sensitive to H2O2. Catalase-deficient (ΔkatE ΔkatG) strains were grown in aerobic medium, and extracts were exposed to 10 μm H2O2 for 5 min. D, enzymes are sensitive to a bolus of H2O2 in vivo. Catalase-deficient (ΔkatE ΔkatG) strains were grown in aerobic medium and briefly exposed to H2O2 (100 μm for 10 min). Exogenous metal was added to the assay to ensure full activity of undamaged enzymes, as described under “Experimental Procedures.” E, enzyme activities are low in cells that lack scavenging enzymes. Wild-type (MG1655) and Hpx− (LC106) strains were grown in aerobic medium, and enzyme activities were measured.
Cell extracts were then prepared from catalase-deficient (ΔkatE ΔkatG) mutants so that added H2O2 would not be scavenged. To avoid incidental oxidation, anaerobic conditions were employed for cell lysis and for extract handling, and the inclusion of DTPA ensured that any loose metals in the extracts would not adventitiously activate apoenzyme. A brief challenge with low amounts of H2O2 (10 μm, 5 min) quickly eliminated the activity of all three enzymes (Fig. 1C). This result indicated that these enzymes are each charged with iron in vivo.
PDF, TDH, and CDA Are Also Sensitive to H2O2 in Vivo
We wished to confirm that the metal occupancy and H2O2 sensitivity of these enzymes in extracts were replicated inside live cells. When catalase-deficient (Kat−, ΔkatE ΔkatG) mutants were challenged with a bolus of H2O2 in vivo (100 μm, 10 min), the PDF, TDH, and CDA activities were lost (Fig. 1D).
The OxyR response of E. coli is calibrated so that it is activated when intracellular levels of H2O2 exceed 0.2 μm, which implies that such low doses are hazardous and that they represent the pertinent concentrations when E. coli encounters exogenous H2O2 in natural habitats (2, 31). Such protracted low grade stress cannot be replicated by adding a bolus of H2O2 to laboratory cultures of wild type, because dense cultures rapidly degrade it. However, E. coli strains that are deficient in peroxidase and catalase activities (Hpx−, Δahp ΔkatE ΔkatG) gradually accumulate up to 1 μm intracellular H2O2 when they are cultured in aerobic medium, due to the endogenous formation of H2O2 by the autoxidation of enzymes (9). Indeed, all three enzyme activities were low in this background (Fig. 1E). Thus, H2O2 sensitivity seems to be a general trait of nonredox mononuclear iron enzymes. This enzyme class joins [4Fe-4S] dehydratases as a second category of enzymes that are targeted by physiological concentrations of this oxidant.
Oxidation of Iron-loaded PDF and TDH Is Coupled to the Oxidation of Metal-binding Cysteine Residues
Such enzymes are vulnerable to Fenton chemistry because their catalytic metals must be solvent-exposed so that they can coordinate substrate (Fig. 2); this exposure also allows H2O2 to bind. We anticipated that the loss of enzyme activity might result from either of two consequences of the Fenton reaction as follows: the conversion of iron to its ferric form, prompting dissociation from the metal-binding site, or covalent polypeptide damage mediated by the ferryl or hydroxyl radical that the Fenton reaction would produce (Reaction 1),
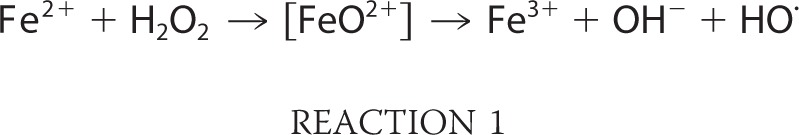 |
To test these alternatives, the iron-charged enzymes were inactivated with H2O2 in vitro, and residual H2O2 was then removed by catalase. When metal was added back to CDA and TDH, about one-third of the lost activity was regained, suggesting either that in these cases the hydroxyl radical did not react with the enzyme or that it did so in a way that did not prevent reactivation (Fig. 3). In contrast, metal addition did not restore any PDF activity at all. We inferred that active-site residues of PDF had been covalently modified.
FIGURE 2.

Metal-substrate interactions within the actives sites of PDF (35), TDH (60), and CDA (61).
FIGURE 3.

H2O2 stress reversibly oxidizes the cysteine residues of iron-charged PDF and TDH. Iron-charged enzymes were exposed to 20 μm H2O2 for 5 min. After the addition of catalase, enzyme activity was measured before and after the addition of iron (CDA and TDH) or nickel (PDF) and of TCEP.
PDF and TDH differ from CDA in that they each employ a cysteine residue to help coordinate the catalytic iron atom (Fig. 2) (32, 33). Cysteine reacts at diffusion-limited rates with hydroxyl radicals, raising the prospect that the nascent ferryl/hydroxyl radical might immediately oxidize the coordinating cysteine. Cysteine radicals can decompose to sulfenic acids (see under “Discussion”), which thiol compounds can reduce back to the original thiol form (Fig. 4, reactions 1 and 2). Indeed, treatment of the oxidized PDF and TDH with either TCEP or DTT plus metal fully restored activity (Fig. 3). No further activity was recovered from CDA. Therefore, it appears that the radical that is formed by the Fenton reaction quantitatively oxidizes the coordinating sulfhydryl ligand in PDF; in TDH the sulfhydryl is oxidized about half the time. In the remaining cases, the ferryl/hydroxyl radical of TDH may have oxidized the bound substrate, threonine, although this was not shown. It was not possible to run the same experiment without threonine, because in its absence the metal quickly dissociates from the enzyme.
FIGURE 4.

Proposed pathways for the damage and reactivation of PDF and TDH.
Sulfenic acids can further react with nearby sulfhydryl and amine moieties to generate disulfide and sulfenamide species, and reactivation by TCEP or DTT would not distinguish between these products and a sulfenic acid per se (34). Disulfide formation can occur on a time scale that is physiologically relevant, and so this possibility was examined. The sole other cysteine residue of PDF (Cys-130) appears to be too far from the active site Cys-91 to form a disulfide bond (35), and indeed analysis of a C130S mutant protein did not show any change in inactivation or reactivation behavior (data not shown). Attempts to demonstrate the presence of a sulfenic acid by reaction with 7-chloro-4-nitrobenz-2-oxa-1,3-diazole or by mass spectrometry were unsuccessful (data not shown). This failure is not interpretable, however, as 7-chloro-4-nitrobenz-2-oxa-1,3-diazole also failed to produce a diagnostic signal with the reduced (thiolate) form of the enzyme, possibly indicating difficulty in accessing the active site, whereas mass spectrometric analysis of thiolate and thiol derivatives can fail due to further oxidation of the sulfur species during the analytical process (see below). Sulfenamide formation is a slower process that is less likely to be biologically important but that may also have confounded the protein analysis.
H2O2 Also Slowly Oxidizes the Active-site Cysteine Residues of the Apoproteins in a Metal-independent Reaction
We also tested whether H2O2 could oxidize the cysteine residues of PDF and TDH directly, without the mediation of iron. Reduced apoenzymes would be present in vivo when the PDF and TDH proteins are first synthesized and whenever the oxidized proteins are repaired by reduction. The direct oxidation of cysteine residues by H2O2 is a divalent process that is chemically distinct from univalent oxidation by hydroxyl radicals, and vulnerability to one does not connote vulnerability to the other. Nevertheless, the oxidation of sulfhydryl residues has long been suspected to underlie aspects of H2O2 toxicity.
The apoprotein forms of PDF and TDH enzymes were prepared by treatment with metal chelators. When the apoproteins were then exposed to H2O2, they were rapidly converted to a form that could no longer be activated by the simple addition of metal (Fig. 5A). CDA, in contrast, was unaffected; it still regained full activity once metals were supplied. Monobromobimane tagging experiments confirmed that the sole solvent-exposed thiol of apo-PDF was rapidly oxidized (Fig. 5B). The same result was obtained with the PDF C130S mutant that lacks the noncoordinating cysteine (data not shown). We infer that the active-site cysteine was oxidized to a sulfenic acid form through a metal-independent reaction (Fig. 4, reaction 4).
FIGURE 5.

Apo-PDF and apo-TDH can be directly oxidized by H2O2. A, purified apo-PDF and apo-TDH and apo-CDA in cell extracts were exposed to 0 or 10 μm H2O2 for 5 min. After addition of catalase, enzymes were reconstituted with iron (CDA), zinc (TDH), or nickel (PDF) and assayed. B, direct measurement of sulfhydryl oxidation in PDF. Purified apo-PDF was challenged with zero or 20 μm H2O2 for 1 min. The enzyme was then treated with monobromobimane, and fluorescence was measured. C, comparison of the sensitivities of apo-PDF, apo-TDH, and GAPDH to H2O2. Pure apo-PDF and pure apo-TDH were challenged with 1 and 6 μm H2O2, respectively, at RT prior to remetallation and assay. The sensitivity of GAPDH to 10 μm H2O2 was measured in cell extracts (JI367, ΔkatE ΔkatG). Extracts did not protect PDF or TDH (data not shown).
The rates at which H2O2 oxidized apo-PDF and apo-TDH were remarkable; the measured rate constants at room temperature were 13,000 and 1000 m−1 s−1, respectively (Fig. 5C). These values greatly exceed those for the divalent oxidation of free cysteine at neutral pH (2 m−1 s−1) (13). Local environments can favor cysteine deprotonation, but even so typical cysteinate oxidation is only moderately faster (20 m−1 s−1). For the sake of comparison, we also measured the rate constants with which H2O2 oxidizes the active-site cysteine residues of glyceraldehyde-3-phosphate dehydrogenase and isocitrate lyase; these cysteines serve as nucleophiles and therefore might be predisposed to react with H2O2. Those rates were 50 and 20 m−1 s−1, respectively. The reactivity of the PDF and TDH active-site cysteine residues is unprecedented. This reaction may be biologically relevant in H2O2-stressed cells, as newly synthesized enzyme exists in the apoprotein form while awaiting metallation.
It should be noted that the oxidation of cysteine in iron-loaded enzyme still occurred faster than the oxidation of cysteine in apoenzyme. Thus, in the former case the oxidation of iron and of cysteine (Fig. 4, reaction 1) is a concerted rather than stepwise process.
H2O2 Can Further Oxidize Damaged PDF and TDH to a Nonreactivatible Form
The glutaredoxin and thioredoxin systems rapidly reduce intracellular sulfenic acid residues and disulfide bonds to their original thiol state (36); therefore, relatively few oxidized cysteine residues are found in extracts prepared from H2O2-treated cells (37). Yet although 5 min was required to centrifuge and prepare anaerobic extracts from H2O2-stressed Hpx− cells, the recovered PDF and TDH activities were both low, suggesting that they had incurred an injury that could not be reversed. Indeed, subsequent treatment with TCEP did not restore any activity (Fig. 6A). Furthermore, when the stressed cells were moved to anaerobic medium in the presence of chloramphenicol, the enzyme activities did not rebound at all. Western blots of PDF established that the inactive enzyme had not been degraded (Fig. 6B). Thus, the damaged enzymes were not in the same form as the enzymes that had been inactivated in vitro. The same result was observed when catalase− mutants were exposed to a bolus of 100 μm H2O2 for 10 min; 90% of the PDF and 70% of the TDH activity were lost (Fig. 1D), but the activity could not be restored by subsequent TCEP treatment in vitro. Implicitly, the irreversible loss of activity that occurred in vivo seemed likely to arise from an additional type of injury that was secondary to the initial Fenton reaction.
FIGURE 6.

Extended exposure to H2O2 converts PDF and TDH to nonreactivatible forms in vivo and in vitro. A, TCEP did not restore PDF and TDH activities in the extracts of Hpx− cells that had been grown in aerobic medium. Both enzymes were assayed in the presence of metal (500 μm Ni2+ and 500 μm Fe2+ respectively) to ensure full activity of undamaged enzymes. B, PDF polypeptide was not degraded in vivo. Anaerobic cells expressing PDF-FLAG were treated with chloramphenicol and aerated starting at time 0. Polypeptide content was monitored by Western blot. The strains used were MG1655/pPDF-FLAG, LC106/pPDF-FLAG (Hpx−) and AA30/pPDF-FLAG (Hpx− ΔmntH). C, iron-PDF and apo-PDF can be overoxidized in vitro. Iron-charged PDF, apo-PDF, and nickel-charged PDF were exposed to H2O2 (133, 100, and 500 μm, respectively) for the indicated times. They were then treated ± TCEP and reconstituted with nickel prior to assay.
Because H2O2 can further oxidize sulfenic acids to sulfinic and sulfonic forms (38, 39), which are not reducible by DTT or TCEP, we considered that such over-oxidation might occur to PDF and TDH (Fig. 4, reaction 5). Indeed, when the iron-loaded enzyme was incubated in vitro for an extended time with H2O2, the lost activity could no longer be restored by TCEP treatment (Fig. 6C). Enzyme loaded with nickel was completely resistant to this treatment. The measured rate constant for conversion to a nonreactivatible form at room temperature was 220 ± 30 m−1 s−1.
The same irreversible outcome occurred when apo-PDF and apo-TDH were exposed to H2O2 for an extended time, they were ultimately converted to a form that could not be revived by TCEP/metal treatment (Fig. 6C and supplemental Fig. S1). Nevertheless, the conversion to a nonreactivatible form was inhibited when TCEP was present during the H2O2 exposure, demonstrating that the irreversible oxidation (Fig. 4, reaction 5) competed with reduction of the nominal sulfenate species (reaction 2). Mass spectrometric analysis of the nonreactivatible apo-PDF demonstrated an increase of 48 mass units for much of the enzyme population, suggesting that the critical cysteine had ultimately been oxidized to the sulfonic acid form. The intermediate sulfinic form is presumably an obligatory intermediate, but it was not detected, suggesting that it is unstable to the mass spectrometric analysis and impeding a determination of the cysteine status in enzyme recovered from H2O2-stressed cells. Other workers have made the same observation (23). Again, the rate constant for conversion of the inactive-but-reactivatible (presumptive sulfenic) form of PDF to the nonreactivatible form was 260 ± 60 m−1 s−1, in agreement with the value that had been determined starting with holoenzyme. This rate constant would support a half-time of 40 min inside cells containing 1 μm H2O2 and so could explain the recovery of nonreactivatible enzyme from the Hpx− mutants.
In sum, both the iron-charged and apoprotein forms of PDF and TDH are vulnerable to oxidation by H2O2. In both cases, the oxidized product is consistent with a sulfenic acid form that lacks activity but can be easily reactivated. However, continued exposure to H2O2 further oxidizes the cysteine to a form that cannot be reactivated. The inactive enzyme that gradually accumulates in H2O2-stressed cells is in a form consistent with this outcome. CDA, in contrast, does not use a cysteine residue to coordinate its iron atom, and Fenton chemistry immediately and irreversibly inactivates many of the protein molecules.
Manganese Import and Iron Sequestration Protect Mononuclear Enzymes from H2O2
During H2O2 stress, the OxyR system strongly induces both a manganese importer (MntH) and an iron sequestration protein (Dps) (28, 40). Although Hpx− mutants can grow in aerobic media, growth soon stops if they are defective in either of these proteins (Fig. 7) (10, 29). We hypothesized that the essential role of MntH and Dps is to substitute manganese for iron in nonredox mononuclear proteins, thereby charging them with a metal that is not vulnerable to Fenton chemistry (12, 29).
FIGURE 7.

Manganese import (by MntH) and iron sequestration (by Dps) are key elements in protecting PDF, TDH, and CDA from H2O2. A–C, PDF, TDH, and CDA activities in wild-type (MG1655, WT), Hpx− (LC106), Hpx− ΔmntH (AA30), and Hpx− Δdps (SP66) strains. Where indicated, extra manganese was included in the growth medium. Assays were conducted after anaerobic reconstitution with nickel (PDF) or iron (TDH and CDA). Time courses of PDF and TDH inactivation are shown as supplemental Fig. S2. D, overexpression of PDF relieves the Hpx− ΔmntH growth defect. At time 0, anaerobic cultures were diluted into aerobic medium. The strains used were LC106 (Hpx−, circles), AA30 (Hpx− ΔmntH, squares), and SP66 (Hpx− Δdps, diamonds). Closed symbols, plasmid overexpressing PDF; open symbols, vector control.
Fig. 7, A–C supports this idea, as PDF, TDH, and CDA activities were extremely low in Hpx− ΔmntH and Hpx− Δdps mutants. Activities remained high in both strains if the cells were supplemented with manganese.
Although many mononuclear enzymes are annotated in the EcoCyc E. coli data base, few of these enzymes are essential for growth under laboratory conditions. PDF is an exception, and we wondered whether the loss of its activity was specifically responsible for the aerobic inviability of Hpx− ΔmntH and Hpx− Δdps mutants. To test this idea, we overproduced PDF from a multicopy plasmid. The growth defects of those strains were largely suppressed (Fig. 7D). We considered the possibility that the overproduced protein might protect enzymes indirectly, by sequestering iron; however, measurements showed that the activity of TDH was not enhanced (7% activity in Hpx− mntH cells containing overexpression plasmid, compared with 10% with vector only). We infer that the inability of these strains to grow in aerobic medium does indeed stem from a lack of PDF activity.
Fig. 1 showed that when PDF, TDH, and CDA were charged with manganese in vitro, they resisted inactivation by a large bolus of H2O2. This phenomenon was tested in vivo; a bolus of 100 μm H2O2 was added to aerobically grown strains that lacked catalase, both catalase and peroxidase, or iron-import systems (Fig. 8). The latter two strains contain substantial manganese due to the induction of MntH by OxyR and by derepression of Fur, respectively (28, 29, 41). MntH is not induced in the catalase mutant, and in this strain the three enzymes were rapidly inactivated by the H2O2. In the iron-import mutant, the enzymes were more resistant, suggesting that the imported manganese protected a subset of the enzymes (Fig. 8). In the catalase/peroxidase mutants the initial PDF, TDH, and CDA activities were lower, as shown previously, but the residual activities were completely unaffected by the H2O2 bolus. Thus, the data are consistent with the notion that imported manganese protects these enzymes by blocking the initial Fenton reaction.
FIGURE 8.

Intracellular manganese protects PDF (A), TDH (B), and CDA (C) from a bolus of H2O2. Cells were grown in aerobic medium that was supplemented with 5 μm MnCl2 where indicated. Chloramphenicol was added, and cells were exposed to 100 μm H2O2 for 10 min. Enzyme activities were measured in extracts after metallation with nickel (PDF) or iron (TDH and CDA). Strains used are as follows: JI367 (Kat−), AA301 (Kat− import−), and LC106 (Hpx−).
Finally, if the protective effect of manganese stems from its ability to occupy the active sites of these enzymes, then cobalt might be expected to exert the same effect, as it can similarly metallate these enzymes in vitro. E. coli does not normally import cobalt, but when high levels are added to growth medium, some can slip into the cell through nonspecific cation-import processes. We observed that 30 μm exogenous cobalt(II) suppressed the growth defect of Hpx− ΔmntH mutants (Fig. 9A). At the same time, PDF activity, which was undetectable (<2%) in the unsupplemented cultures, rose to 55% of the wild-type level. Cobalt supplements were also able to suppress the growth defect of the Hpx− Δdps mutants. Finally, like manganese, cobalt blocked the general carbonylation of cellular proteins that otherwise occurs in the Hpx− Δdps strain (Fig. 9B). Thus the protective effects of manganese can be replicated by cobalt. Although manganese has some low level ability to scavenge reactive oxygen species, cobalt does not. We infer that the protective effects of both metals arise primarily from the displacement of iron from metal-binding sites.
FIGURE 9.

Cobalt can substitute for manganese in protecting H2O2-stressed cells. A, cobalt supplements restore the growth of both Hpx− ΔmntH and Hpx− Δdps cells. At time 0, anaerobic cultures were diluted into aerobic medium. Where indicated, 30 μm CoCl2 was included (closed symbols). Strains used are as follows: LC106 (Hpx−, circles), AA30 (Hpx− ΔmntH, squares), and SP66 (Hpx− Δdps, diamonds). B, cobalt suppresses protein carbonylation in the Hpx− Δdps cells.
DISCUSSION
Mononuclear Enzymes Are Key Targets of H2O2
E. coli employs over 100 nonredox enzymes that can be activated in vitro by various transition metals. All three of the enzymes from that category that were examined in this study turned out to use iron as their native cofactor. The implication, of course, is that iron might also cofactor many or most of these other enzymes, too. If so, one upshot is that low doses of H2O2 will incapacitate many more branches of metabolism than had been appreciated. Previously, H2O2 was known to efficiently damage only a half-dozen iron-sulfur-dependent dehydratases (11).
The ability of ferrous iron to activate mononuclear enzymes is frequently overlooked because in aerobic buffers it is quickly oxidized to its insoluble ferric form. However, intracellular thiols and other reductants are abundant inside cells, and EPR analysis of E. coli confirms that the pool of 20–50 μm unincorporated iron exists almost entirely in the ferrous form (42). Such a pool is likely to be sufficient to activate any enzyme with a high affinity mononuclear site. In contrast, manganese is typically scarce (<10 μm) (29), E. coli lacks a dedicated cobalt transport system, and nickel is imported only under anaerobic conditions when the nickel-dependent hydrogenase is induced (30).
The mononuclear enzymes examined thus far catalyze diverse reactions types, including amide cleavage, dehydrogenation, deamination, and epimerization. What they have in common is the use of iron to stabilize an oxyanion intermediate. In PDF and RPE, a reaction intermediate forms a bidentate complex with the metal atom, and in PDF and CDA, the metal activates a nucleophilic water molecule (Fig. 2); iron is particularly good at both behaviors. Neither magnesium nor zinc, the other bioavailable metals, matches iron in these attributes. Magnesium is generally unsuitable as a surface catalyst because it has an especially high barrier to ligand-exchange reactions; none of the tested enzymes exhibit any activity at all with it. Interestingly, magnesium also requires tightly constrained bond angles, a fact that is apparently exploited by these metal-binding sites, which exclude it. The full d-orbital configuration of zinc disfavors ligand rearrangements (43, 44), and accordingly, zinc confers upon PDF, CDA, and RPE <10% the activity than does iron. However, in accordance with the Irving-Williams series, zinc binds more tightly to these active sites than does iron (Table 1), which creates a problem for the cell. We expect that the devices that limit zinc accumulation inside E. coli must be important in guarding against the deactivation of these enzymes by mis-metallation.
More work will be needed to establish which mononuclear enzymes actually employ iron in vivo. Sensitivity to H2O2 should provide a straightforward method by which to do so.
Sacrificial Cysteine Residues at the Metal-binding Site
The use of cysteine to coordinate iron is common in mononuclear enzymes, and the work with PDF and TDH indicates that this residue absorbs the force of the oxidant that is generated by the Fenton reaction. This arrangement avoids the release of a hydroxyl radical that might otherwise damage residues in the active site. The sulfenic acid that is presumably formed can be reduced by TCEP or DTT in vitro, and in vivo a similar reduction is presumably mediated by the glutathione/glutaredoxin or thioredoxin systems. Both systems are induced as part of the OxyR response (6). They are very efficient, and by the point of extract formation, no detectable fraction of the enzyme was in the inactive-but-reactivatible form.
Interestingly, when H2O2 oxidizes the substrate-binding iron atom of [4Fe-4S] dehydratases, the ferryl radical that is formed also pulls a second electron from coordinating ligands, in that case from the iron-sulfur cluster itself (11). The effect is the same as was observed with PDF and TDH; the polypeptide is spared damage, and the enzyme activity is quickly restored by cellular reactivation systems. In contrast, when H2O2 oxidizes the prosthetic iron atom of PerR, the regulator that governs the H2O2 resistance regulon in B. subtilis, a coordinating histidine residue can be oxidized in a way that inactivates the protein (5). CDA and RPE may be in the same category, as 25–50% of activity is irreversibly lost per each cycle of Fenton events (Fig. 3) (12). This observation raises the question, which is currently unanswerable, as to why all iron-charged enzymes do not employ a sacrificial cysteine ligand.
The fact that the cysteine of the apoprotein is so easily oxidized is striking. Hydrogen peroxide can oxidize sulfhydryls on H2O2 sensors such as OxyR and H2O2 peroxidases such as Ahp with rate constants in excess of 107 m−1 s−1 (31, 45); however, the oxidation rate constants of other protein sulfhydryls have not been reported to exceed 60 m−1 s−1, including those of glyceraldehyde-3-phosphate dehydrogenase (GAPDH), 40–60 m−1 s−1 (14), and protein-tyrosine phosphatases, 10–20 m−1 s−1 (46, 47). The latter constants seem too low to have physiological relevance; for example, 1 μm H2O2, a toxic dose for E. coli, would require 10 h to oxidize half the isocitrate lyase proteins. Yet this dose would need only 1–10 min to do the same to PDF and TDH (104 and 103 m−1 s−1). The reason for the high reactivity of the cysteine is unclear. The physiological impact may be that if metallation of apoprotein is a slow step, then a dynamic equilibrium between sulfhydryl and sulfenate forms may be established in H2O2-stressed cells.
Further oxidation of the sulfenate to more oxidized states (250 m−1 s−1) occurs in vitro and is one plausible explanation for the gradual conversion of these enzymes to nonreactivatible forms inside oxidatively stressed cells. Other possibilities include the mis-metallation of enzyme by errant zinc and the progressive denaturation of the apoprotein, which is notably unstable in vitro. Occupancy of the active site by manganese would block any of these three outcomes.
Protection of Mononuclear Enzymes by Imported Manganese
Manganese can defend both bacteria and eukarya against oxidative stress (48–54). Manganese is imported into E. coli under two conditions, H2O2 stress and iron starvation (28). Distinct regulators, OxyR and Fur, drive these effects by controlling MntH synthesis. In both situations, the import of manganese is needed to avert stasis, and it apparently does so by preserving the activities of mononuclear iron enzymes.
We believe that during H2O2 stress the sequestration of iron by Dps and the import of manganese by MntH comprise a concerted strategy that ensures that manganese will preferentially occupy metal-binding sites. The data of Figs. 7 and 9B confirm that excess manganese can compensate for the absence of Dps, as this model would predict. The activities of PDF, TDH, and CDA are all protected. However, the presence of manganese in the active sites of these enzymes has not yet been physically demonstrated, and we observed that the active enzymes that were recovered from the manganese-loaded cells exhibited H2O2 sensitivity in extracts, indicating that by that point they had been reloaded with ferrous iron. We suspect that manganese, which dissociates easily from these enzymes, was replaced by iron during the 5+ min required to centrifuge, wash, and harvest the cells.
An an alternative model, by which imported manganese might chemically degrade oxidants, does not apply to E. coli, as shown by direct measurements of H2O2 scavenging (29). Furthermore, the ability of cobalt to substitute for manganese and the benefit of manganese import during iron starvation (41, 55, 56) fit the notion that manganese directly binds the enzymes. We note that the manganese-loaded TDH has a substantial kcat but binds threonine poorly. Implicitly either threonine must accumulate to restore pathway flux or else manganese is simply a placeholder during the period of stress, protecting the protein from overoxidation or denaturation without providing continuous activity.
E. coli is an iron-centric organism, acquiring it from its usually anoxic habitat and employing it to activate myriad iron-sulfur and heme proteins, in addition to the mononuclear enzymes. This dependence is not universal, however, as organisms that routinely contend with oxidative stress or iron scarcity may constitutively express the manganese strategy that E. coli only turns to in extremes. Many lactic acid bacteria generate endogenous H2O2 at very high rates through the actions of pyruvate and lactate oxidases (57). It appears that their tolerance of the resultant stress depends upon the import of manganese, which they accumulate at intracellular levels of up to 20 mm, orders of magnitude higher than the 0.2 mm concentration inside H2O2-stressed E. coli (29, 58). It therefore seems probable that these bacteria use manganese in place of iron inside mononuclear enzymes. Data from the Daly laboratory suggest that manganese may additionally protect nonmetallated enzymes (59); perhaps the abundant manganese outcompetes iron even for adventitious binding sites on the protein surfaces and thereby circumvents incidental Fenton reactions. Whether less extreme aerobic organisms, from yeast to humans, employ manganese in place of iron remains to be determined.
Supplementary Material
Acknowledgments
We thank Peter Yau and Brian S. Imai (University of Illinois Biotechnology Center) and Haijun Yao (University of Illinois SCS Mass Spectrometry Laboratory) for assistance with protein mass spectrometry.
This work was supported, in whole or in part, by National Institutes of Health Grant GM049640.

This article contains supplemental “Experimental Procedures,” Figs. S1 and S2, Table S1, and additional references.
- RPE
- ribulose-5-phosphate 3-epimerase
- peptide deformylase
- TDH
- threonine dehydrogenase
- TCEP
- tris (2-carboxyethyl)phosphine
- CDA
- cytosine deaminase
- DTPA
- diethylenetriaminepentaacetic acid.
REFERENCES
- 1. Gort A. S., Imlay J. A. (1998) Balance between endogenous superoxide stress and antioxidant defenses. J. Bacteriol. 180, 1402–1410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Seaver L. C., Imlay J. A. (2001) Hydrogen peroxide fluxes and compartmentalization inside growing Escherichia coli. J. Bacteriol. 183, 7182–7189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Delaunay A., Pflieger D., Barrault M. B., Vinh J., Toledano M. B. (2002) A thiol peroxidase is an H2O2 receptor and redox-transducer in gene activation. Cell 111, 471–481 [DOI] [PubMed] [Google Scholar]
- 4. Choi H., Kim S., Mukhopadhyay P., Cho S., Woo J., Storz G., Ryu S. (2001) Structural basis of the redox switch in the OxyR transcription factor. Cell 105, 103–113 [DOI] [PubMed] [Google Scholar]
- 5. Lee J. W., Helmann J. D. (2006) The PerR transcription factor senses H2O2 by metal-catalyzed histidine oxidation. Nature 440, 363–367 [DOI] [PubMed] [Google Scholar]
- 6. Zheng M., Wang X., Templeton L. J., Smulski D. R., LaRossa R. A., Storz G. (2001) DNA microarray-mediated transcriptional profiling of the Escherichia coli response to hydrogen peroxide. J. Bacteriol. 183, 4562–4570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Helmann J. D., Wu M. F., Gaballa A., Kobel P. A., Morshedi M. M., Fawcett P., Paddon C. (2003) The global transcriptional response of Bacillus subtilis to peroxide stress is coordinated by three transcription factors. J. Bacteriol. 185, 243–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jamieson D. J. (1998) Oxidative stress responses of the yeast Saccharomyces cerevisiae. Yeast 14, 1511–1527 [DOI] [PubMed] [Google Scholar]
- 9. Seaver L. C., Imlay J. A. (2001) Alkyl hydroperoxide reductase is the primary scavenger of endogenous hydrogen peroxide in Escherichia coli. J. Bacteriol. 183, 7173–7181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Park S., You X., Imlay J. A. (2005) Substantial DNA damage from submicromolar intracellular hydrogen peroxide detected in Hpx− mutants of Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 102, 9317–9322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jang S., Imlay J. A. (2007) Micromolar intracellular hydrogen peroxide disrupts metabolism by damaging iron-sulfur enzymes. J. Biol. Chem. 282, 929–937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sobota J. M., Imlay J. A. (2011) Iron enzyme ribulose-5-phosphate 3-epimerase in Escherichia coli is rapidly damaged by hydrogen peroxide but can be protected by manganese. Proc. Natl. Acad. Sci. U.S.A. 108, 5402–5407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Winterbourn C. C., Metodiewa D. (1999) Reactivity of biologically important thiol compounds with superoxide and hydrogen peroxide. Free Radic. Biol. Med. 27, 322–328 [DOI] [PubMed] [Google Scholar]
- 14. Zaffagnini M., Michelet L., Marchand C., Sparla F., Decottignies P., Le Maréchal P., Miginiac-Maslow M., Noctor G., Trost P., Lemaire S. D. (2007) The thioredoxin-independent isoform of chloroplastic glyceraldehyde-3-phosphate dehydrogenase is selectively regulated by glutathionylation. FEBS J. 274, 212–226 [DOI] [PubMed] [Google Scholar]
- 15. Miller J. H. (1972) Experiments in Molecular Genetics, pp. 431–435, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 16. Datsenko K. A., Wanner B. L. (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 97, 6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Smith K. J., Petit C. M., Aubart K., Smyth M., McManus E., Jones J., Fosberry A., Lewis C., Lonetto M., Christensen S. B. (2003) Structural variation and inhibitor binding in polypeptide deformylase from four different bacterial species. Protein Sci. 12, 349–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Boylan S. A., Dekker E. E. (1981) l-Threonine dehydrogenase. Purification and properties of the homogeneous enzyme from Escherichia coli K-12. J. Biol. Chem. 256, 1809–1815 [PubMed] [Google Scholar]
- 19. Duggleby R. G., Dennis D. T. (1974) Nicotinamide adenine dinucleotide-specific glyceraldehyde 3-phosphate dehydrogenase from Pisum sativum. Assay and steady state kinetics. J. Biol. Chem. 249, 167–174 [PubMed] [Google Scholar]
- 20. Giachetti E., Pinzauti G., Vanni P. (1984) A new continuous optical assay for isocitrate lyase. Cell. Mol. Life Sci. 40, 227–228 [Google Scholar]
- 21. Lazennec C., Meinnel T. (1997) Formate dehydrogenase-coupled spectrophotometric assay of peptide deformylase. Anal. Biochem. 244, 180–182 [DOI] [PubMed] [Google Scholar]
- 22. Porter D. J., Austin E. A. (1993) Cytosine deaminase. The roles of divalent metal ions in catalysis. J. Biol. Chem. 268, 24005–24011 [PubMed] [Google Scholar]
- 23. Rajagopalan P. T., Pei D. (1998) Oxygen-mediated inactivation of peptide deformylase. J. Biol. Chem. 273, 22305–22310 [DOI] [PubMed] [Google Scholar]
- 24. Porter D. J. (2000) Escherichia coli cytosine deaminase. The kinetics and thermodynamics for binding of cytosine to the apoenzyme and the Zn2+ holoenzyme are similar. Biochim. Biophys. Acta 1476, 239–252 [DOI] [PubMed] [Google Scholar]
- 25. Hall R. S., Fedorov A. A., Xu C., Fedorov E. V., Almo S. C., Raushel F. M. (2011) Three-dimensional structure and catalytic mechanism of cytosine deaminase. Biochemistry 50, 5077–5085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Johnson A. R., Chen Y. W., Dekker E. E. (1998) Investigation of a catalytic zinc binding site in Escherichia coli l-threonine dehydrogenase by site-directed mutagenesis of cysteine 38. Arch. Biochem. Biophys. 358, 211–221 [DOI] [PubMed] [Google Scholar]
- 27. Higashi N., Tanimoto K., Nishioka M., Ishikawa K., Taya M. (2008) Investigating a catalytic mechanism of hyperthermophilic l-threonine dehydrogenase from Pyrococcus horikoshii. J. Biochem. 144, 77–85 [DOI] [PubMed] [Google Scholar]
- 28. Kehres D. G., Janakiraman A., Slauch J. M., Maguire M. E. (2002) Regulation of Salmonella enterica serovar Typhimurium mntH transcription by H2O2, Fe2+, and Mn2+. J. Bacteriol. 184, 3151–3158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Anjem A., Varghese S., Imlay J. A. (2009) Manganese import is a key element of the OxyR response to hydrogen peroxide in Escherichia coli. Mol. Microbiol. 72, 844–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rowe J. L., Starnes G. L., Chivers P. T. (2005) Complex transcriptional control links NikABCDE-dependent nickel transport with hydrogenase expression in Escherichia coli. J. Bacteriol. 187, 6317–6323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Aslund F., Zheng M., Beckwith J., Storz G. (1999) Regulation of the OxyR transcriptional factor by hydrogen peroxide and the cellular thiol-disulfide status. Proc. Natl. Acad. Sci. U.S.A. 96, 6161–6165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chan M. K., Gong W., Rajagopalan P. T., Hao B., Tsai C. M., Pei D. (1997) Crystal structure of the Escherichia coli peptide deformylase. Biochemistry 36, 13904–13909 [DOI] [PubMed] [Google Scholar]
- 33. Epperly B. R., Dekker E. E. (1991) l-Threonine dehydrogenase from Escherichia coli. Identification of an active site cysteine residue and metal ion studies. J. Biol. Chem. 266, 6086–6092 [PubMed] [Google Scholar]
- 34. Lee J. W., Soonsanga S., Helmann J. D. (2007) A complex thiolate switch regulates the Bacillus subtilis organic peroxide sensor OhrR. Proc. Natl. Acad. Sci. U.S.A. 104, 8743–8748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Becker A., Schlichting I., Kabsch W., Groche D., Schultz S., Wagner A. F. (1998) Iron center, substrate recognition, and mechanism of peptide deformylase. Nat. Struct. Biol. 5, 1053–1058 [DOI] [PubMed] [Google Scholar]
- 36. Ritz D., Beckwith J. (2001) Roles of thiol-redox pathways in bacteria. Annu. Rev. Biochem. 55, 21–48 [DOI] [PubMed] [Google Scholar]
- 37. Takanishi C. L., Ma L. H., Wood M. J. (2007) A genetically encoded probe for cysteine sulfenic acid protein modification in vivo. Biochemistry 46, 14725–14732 [DOI] [PubMed] [Google Scholar]
- 38. Wood Z. A., Poole L. B., Karplus P. A. (2003) Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science 300, 650–653 [DOI] [PubMed] [Google Scholar]
- 39. Tanner J. J., Parsons Z. D., Cummings A. H., Zhou H., Gates K. S. (2011) Redox regulation of protein-tyrosine phosphatases. Structural and chemical aspects. Antioxid. Redox. Signal. 15, 77–97 [DOI] [PubMed] [Google Scholar]
- 40. Altuvia S., Almirón M., Huisman G., Kolter R., Storz G. (1994) The dps promoter is activated by OxyR during growth and by IHF and σS in stationary phase. Mol. Microbiol. 13, 265–272 [DOI] [PubMed] [Google Scholar]
- 41. Martin J. E., Imlay J. A. (2011) The alternative aerobic ribonucleotide reductase of Escherichia coli, NrdEF, is a manganese-dependent enzyme that enables cell replication during periods of iron starvation. Mol. Microbiol. 80, 319–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Woodmansee A. N., Imlay J. A. (2002) Quantitation of intracellular free iron by electron paramagnetic resonance spectroscopy. Methods Enzymol. 349, 3–9 [DOI] [PubMed] [Google Scholar]
- 43. Lipscomb W. N., Sträter N. (1996) Recent advances in zinc enzymology. Chem. Rev. 96, 2375–2434 [DOI] [PubMed] [Google Scholar]
- 44. Jain R., Hao B., Liu R. P., Chan M. K. (2005) Structures of E. coli peptide deformylase bound to formate. Insight into the preference for Fe2+ over Zn2+ as the active site metal. J. Am. Chem. Soc. 127, 4558–4559 [DOI] [PubMed] [Google Scholar]
- 45. Peskin A. V., Low F. M., Paton L. N., Maghzal G. J., Hampton M. B., Winterbourn C. C. (2007) The high reactivity of peroxiredoxin 2 with H2O2 is not reflected in its reaction with other oxidants and thiol reagents. J. Biol. Chem. 282, 11885–11892 [DOI] [PubMed] [Google Scholar]
- 46. Denu J. M., Tanner K. G. (1998) Specific and reversible inactivation of protein-tyrosine phosphatases by hydrogen peroxide. Evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry 37, 5633–5642 [DOI] [PubMed] [Google Scholar]
- 47. Forman H. J., Fukuto J. M., Torres M. (2004) Redox signaling. Thiol chemistry defines which reactive oxygen and nitrogen species can act as second messengers. Am. J. Physiol. Cell Physiol. 287, C246–256 [DOI] [PubMed] [Google Scholar]
- 48. Archibald F. S., Fridovich I. (1981) Manganese and defenses against oxygen toxicity in Lactobacillus plantarum. J. Bacteriol. 145, 442–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chang E. C., Kosman D. J. (1989) Intracellular Mn(II)-associated superoxide scavenging activity protects Cu,Zn superoxide dismutase-deficient Saccharomyces cerevisiae against dioxygen stress. J. Biol. Chem. 264, 12172–12178 [PubMed] [Google Scholar]
- 50. Daly M. J., Gaidamakova E. K., Matrosova V. Y., Vasilenko A., Zhai M., Venkateswaran A., Hess M., Omelchenko M. V., Kostandarithes H. M., Makarova K. S., Wackett L. P., Fredrickson J. K., Ghosal D. (2004) Accumulation of Mn(II) in Deinococcus radiodurans facilitates γ-radiation resistance. Science 306, 1025–1028 [DOI] [PubMed] [Google Scholar]
- 51. Tseng H. J., Srikhanta Y., McEwan A. G., Jennings M. P. (2001) Accumulation of manganese in Neisseria gonorrhoeae correlates with resistance to oxidative killing by superoxide anion and is independent of superoxide dismutase activity. Mol. Microbiol. 40, 1175–1186 [DOI] [PubMed] [Google Scholar]
- 52. Tseng H. J., McEwan A. G., Paton J. C., Jennings M. P. (2002) Virulence of Streptococcus pneumoniae. PsaA mutants are hypersensitive to oxidative stress. Infect. Immun. 70, 1635–1639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sanchez R. J., Srinivasan C., Munroe W. H., Wallace M. A., Martins J., Kao T. Y., Le K., Gralla E. B., Valentine J. S. (2005) Exogenous manganous ion at millimolar levels rescues all known dioxygen-sensitive phenotypes of yeast lacking CuZn-SOD. J. Biol. Inorg. Chem. 10, 913–923 [DOI] [PubMed] [Google Scholar]
- 54. Inaoka T., Matsumura Y., Tsuchido T. (1999) SodA and manganese are essential for resistance to oxidative stress in growing and sporulating cells of Bacillus subtilis. J. Bacteriol. 181, 1939–1943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Grass G., Franke S., Taudte N., Nies D. H., Kucharski L. M., Maguire M. E., Rensing C. (2005) The metal permease ZupT from Escherichia coli is a transporter with a broad substrate spectrum. J. Bacteriol. 187, 1604–1611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cotruvo J. A., Stubbe J. (2011) Escherichia coli class Ib ribonucleotide reductase contains a dimanganese(III)-tyrosyl radical cofactor in vivo. Biochemistry 50, 1672–1681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Berthier F. (1993) On the screening of hydrogen peroxide-generating lactic acid bacteria. Lett. Appl. Microbiol. 16, 150–153 [Google Scholar]
- 58. Archibald F. (1986) Manganese. Its acquisition by and function in the lactic acid bacteria. Crit. Rev. Microbiol. 13, 63–109 [DOI] [PubMed] [Google Scholar]
- 59. Daly M. J., Gaidamakova E. K., Matrosova V. Y., Kiang J. G., Fukumoto R., Lee D. Y., Wehr N. B., Viteri G. A., Berlett B. S., Levine R. L. (2010) Small molecule antioxidant proteome-shields in Deinococcus radiodurans. PLoS ONE 5, e12570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ishikawa K., Higashi N., Nakamura T., Matsuura T., Nakagawa A. (2007) The first crystal structure of l-threonine dehydrogenase. J. Mol. Biol. 366, 857–867 [DOI] [PubMed] [Google Scholar]
- 61. Ireton G. C., McDermott G., Black M. E., Stoddard B. L. (2002) The structure of Escherichia coli cytosine deaminase. J. Mol. Biol. 315, 687–697 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.