

Background: Synaptic signaling is modulated by protein-protein interactions involving receptor ion channels, cytoskeletal proteins, and protein kinases.
Results: α-Actinin enhances CaMKII signaling to GluN2B-NMDARs, but interferes with CaMKII signaling to GluA1-AMPARs.
Conclusion: CaMKII actions on key synaptic targets can be differentially modulated by a Ca2+-independent interaction with α-actinin.
Significance: These findings provide new molecular insights into the synaptic mechanisms underlying learning and memory.
Keywords: Actin, CaMKII, Cytoskeleton, Glutamate Receptors, Synaptic Plasticity
Abstract
Protein-protein interactions are thought to modulate the efficiency and specificity of Ca2+/calmodulin (CaM)-dependent protein kinase II (CaMKII) signaling in specific subcellular compartments. Here we show that the F-actin-binding protein α-actinin targets CaMKIIα to F-actin in cells by binding to the CaMKII regulatory domain, mimicking CaM. The interaction with α-actinin is blocked by CaMKII autophosphorylation at Thr-306, but not by autophosphorylation at Thr-305, whereas autophosphorylation at either site blocks Ca2+/CaM binding. The binding of α-actinin to CaMKII is Ca2+-independent and activates the phosphorylation of a subset of substrates in vitro. In intact cells, α-actinin selectively stabilizes CaMKII association with GluN2B-containing glutamate receptors and enhances phosphorylation of Ser-1303 in GluN2B, but inhibits CaMKII phosphorylation of Ser-831 in glutamate receptor GluA1 subunits by competing for activation by Ca2+/CaM. These data show that Ca2+-independent binding of α-actinin to CaMKII differentially modulates the phosphorylation of physiological targets that play key roles in long-term synaptic plasticity.
Introduction
Transient changes in intracellular Ca2+ concentrations can be decoded by the ubiquitous Ca2+/calmodulin (CaM)3-dependent protein kinase II (CaMKII) to elicit diverse physiological responses. Binding of Ca2+/CaM activates CaMKII and Thr-286 autophosphorylation, creating an autonomously active form of CaMKII that is a molecular memory of transient Ca2+ signals. Dissociation of Ca2+/CaM allows for rapid autophosphorylation at Thr-305 or Thr-306 (Thr-305/6), blocking re-association of Ca2+/CaM such that CaMKII is desensitized to subsequent Ca2+ signals (reviewed in Refs. 1–4). These autophosphorylation reactions are key for CaMKII-dependent modulation of synaptic glutamate receptors and other neuronal functions related to long-term synaptic plasticity, learning, and memory (5–8).
As for many signaling proteins, precise targeting of CaMKII is crucial for its actions (9, 10). Neuronal Ca2+ signals can be tightly compartmentalized in dendritic spines (11), F-actin-rich synaptic structures that contain postsynaptic densities (PSDs) enriched in glutamate receptors and other signaling proteins, including CaMKIIα. CaMKIIα appears to be initially cytosolic, but Ca2+/CaM binding initiates translocation to dendritic spines; a recent study suggests that only a small fraction of CaMKII in spines is tightly associated with PSDs (12). Retention of CaMKII in the spines is modulated by autophosphorylation; Thr-286 autophosphorylation stabilizes spine localization, whereas Thr-305/6 autophosphorylation promotes CaMKII dissociation from spines (13). N-Methyl-d-Aspartate (NMDA)-type glutamate receptor (NMDAR) GluN2B subunits are the most widely recognized binding partner for CaMKII; this interaction modulates CaMKII activity and is important for synaptic plasticity and memory (14–19). GluN2B selectively binds activated CaMKII, perhaps contributing to CaMKII translocation and retention in spines. However, PSDs contain ∼20-fold more CaMKII holoenzymes than NMDAR subunits (20), suggesting that direct interactions with NMDARs account for only a fraction of spine-localized CaMKII holoenzymes (12). Thus, the overall dynamics of CaMKII targeting presumably reflect integration of NMDAR interactions with CaMKII binding to several other proteins (1). Indeed, CaMKII can simultaneously interact with α-actinin, GluN2B, and densin (21). Importantly, pharmacological disruption of F-actin interferes with synaptic clustering of CaMKIIα and actin-binding proteins such as α-actinin without changes in NMDAR localization (22, 23). Synaptic CaMKII targeting therefore depends on an intact F-actin cytoskeleton.
The four isoforms of α-actinin (1–4) link various signaling molecules to the cytoskeleton by virtue of conserved modular protein domains: an N-terminal F-actin-binding domain, four spectrin-like repeat domains (responsible for antiparallel dimerization), an EF hand domain (that binds divalent cations only in α-actinin-1 and α-actinin-4), and a C-terminal domain (CTD) (24, 25). For example, α-actinin links NMDAR GluN1 and GluN2B subunits to the actin cytoskeleton and modulates NMDAR activity by diverse mechanisms (26–30). CaMKII also associates with multiple α-actinin isoforms in brain (21, 31, 32). Our previous studies defined the CTD of α-actinin-2 (A2-CTD; residues 819–894) as the minimal CaMKII binding domain (21). In vitro, A2-CTD interaction with CaMKII is Ca2+-independent, competitive with Ca2+/CaM, and disrupted by Thr-305/6 autophosphorylation (18). However, the mechanism and functional consequences of CaMKII interaction with full-length α-actinin are poorly understood. Here, we show that α-actinin-2 mimics CaM in binding to the CaMKII regulatory domain, and targets CaMKII to the actin cytoskeleton. In vitro and intact cell studies show that α-actinin-2 is a Ca2+-independent, substrate-selective CaMKII activator, which also enhances CaMKII interaction with NMDARs containing GluN2B subunits.
EXPERIMENTAL PROCEDURES
Bacterial and Mammalian Expression Vectors
The cDNA encoding full-length (FL) α-actinin-2 (A2) (894 amino acids) in pCDNA3.1(+) was a gift from Dr. A. Beggs (Harvard). The GluA1 construct was provided by Dr. T. Soderling (Oregon Health Sciences University). Other mammalian expression vectors were previously described; full-length A2 with an N-terminal HA-tag and untagged CaMKIIα (33, 34); GluN1, GluN2A, and GluN2B (35). Expression of GST-A2-CTD, His6-A2-CTD, and His6-densin was described (21, 36). Site-directed mutagenesis was confirmed by sequencing. All constructs were purified using Maxi-Prep kits (Invitrogen, Carlsbad, CA).
Structural Alignment
The structure of CaM bound to CaMKII regulatory domain (PDB:1CM1) was aligned with that of α-actinin-2 bound to the titin z-repeat (1H8B) using the align command of PyMOL (DeLano Scientific LLC).
CaMKII Autophosphorylation
CaMKIIα (WT, T305A, or T306A) were purified from baculovirus-infected Sf9 cells and sequentially autophosphorylated essentially as described (37). Briefly, CaMKIIα (5 μm subunit) was incubated on ice for 90 s with 50 mm HEPES, pH 7.5, 10 mm magnesium acetate, 1.5 mm CaCl2, 10 μm CaM, 2 mm DTT, and 500 μm ATP. After 90 s, 4 mm EGTA was added and reaction continued for 2 min at 30 °C before termination using 25 mm EDTA. The efficacy of autophosphorylation of each protein was verified by assessing the impact on Ca2+/CaM binding (see “Results”).
GST Co-sedimentation Assays
Purified GST-fusion proteins or GST alone (≈250 nm full-length protein), CaMKII (WT or mutated) (≈250 nm subunit: sequentially autophosphorylated, or non-phosphorylated, as indicated) and glutathione-agarose (Sigma; 30 μl packed resin) were incubated at 4 °C for 2 h in 0.5 ml of PD buffer (50 mm Tris-HCl, pH 7.5, 200 mm NaCl, 0.5% (v/v) Triton X-100). After centrifugation, beads were washed with PD buffer and analyzed by immunoblotting. CaMKII bound to GST proteins was quantified using Image J from scans of Ponceau-S-stained membranes, immunoblotted membranes, or linearly exposed x-ray films, and normalized to the amount of recovered GST fusion protein (detected by Ponceau-S stain). Any background binding to GST alone was subtracted.
CaMKII Activation Assay
Purified CaMKIIα (10 nm) was incubated with indicated concentrations of CaM or His6-A2-CTD in presence of 50 mm HEPES pH 7.5, 1 mg/ml bovine serum albumin, 10 mm magnesium acetate, 0.5 mm CaCl2 or 1 mm EGTA, 1 mm DTT and a model peptide substrate (AC-2, syntide-2, or NR2B; 100 μm each). Reactions were started by adding [γ-32P]ATP (0.4 mm; 500–1000 cpm/pmol) and incubated at 30 °C for 10 min. Aliquots were spotted on phosphocellulose paper (Whatman P81), washed, and analyzed by liquid scintillation counting (38).
Cell Culture and Transfection
HEK293 cells maintained at 37 °C in 5% CO2 in modified Eagle's medium (MEM) containing 10% fetal bovine serum (Invitrogen, Carlsbad, CA), penicillin-streptomycin (Sigma) and 2 mm glutamate (Sigma) were transfected using Fugene (Roche, Indianapolis, IN) or PolyJet (SignaGen Laboratories, Gaithersburg, MD) (3 μl/μg DNA). Cells expressing NMDAR subunits were cultured with 1 mm (2R)-amino-5-phosphonovaleric acid; (2R)-amino-5-phosphonopentanoate (AP-5) (Ascent Scientific) or 500 μm AP-5 plus 5 mm MgCl2.
HEK293 Cell Lysis and Immunoprecipitation
Cell extracts were made 24–48 h after transfection (38). NaCl (150 mm final) was added to supernatants, and equal aliquots were incubated overnight at 4 °C with CaMKII IgG or normal IgG (2 μg each; goat). GammaBind Plus-Sepharose (Amersham Biosciences, Piscataway, NJ) (30 μl, 1:1 slurry) was added and incubations were continued for 2 h at 4 °C. Resin was washed ≥4 times with Buffer B (50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1% (v/v) Triton X-100) before analysis by immunoblotting.
Immunofluorescence Co-localization
HEK293 cells expressing TT305/6AA-CaMKIIα with or without WT or Y861R HA-α-actinin-2 were fixed 24 h after transfection, labeled with mouse CaMKIIα and goat HA antibody (Bethyl Labs). F-actin was detected using phalloidin conjugated to Alexafluor-647 (Invitrogen; 1:500, mixed with primary antibodies). Methods for cell imaging and intensity correlation analysis were described previously (33).
Immunoprecipitation from Mouse Brain Extracts
Mouse forebrains were homogenized in low-ionic strength buffer (38), and supernatants were immunoprecipitated using goat CaMKIIα (2 μg) or rabbit α-actinin H-300 (Santa Cruz Biotechnology) (8 μg) antibodies, or an IgG control.
Immunoblotting
SDS-polyacrylamide gels were transferred to nylon-backed nitrocellulose membranes (38). Primary antibodies included: CaMKIIα (Affinity BioReagents, Golden, CO); HA (Vanderbilt Monoclonal antibody core, ICA5); GluN2B, GluN2A, and GluN1 (BD Biosciences); phospho-Ser1303 GluN2B (Millipore/Upstate 07–398); α-actinin (7A4, Abcam); total GluA1 (Abcam ab31232); phospho-Ser831 GluA1 (Phospho Solutions p1160–831). Secondary antibodies conjugated to alkaline phosphatase (Jackson ImmunoResearch, West Grove, PA), horseradish peroxidase (Promega or Santa Cruz), or infrared dyes (LiCor Biosciences, Lincoln, NE) were used for development with NBT/BCIP, enhanced chemiluminescence (Perkin-Elmer, Waltham, MA), or an Odyssey system (LiCor), respectively.
Statistics
Data were analyzed by one-way or two-way ANOVA with the specified post-hoc tests. CaMKII activation curves were fitted to one-site binding curves using GraphPad Prism.
RESULTS
Substrate-selective Activation of CaMKII by α-Actinin
The minimal CaMKII-binding A2-CTD (residues 819–894) (21) is highly conserved in all α-actinin isoforms and is similar to the C-terminal domain (lobe) of CaM (residues 73–149) (Fig. 1A and supplemental Fig. S1). Since the C-terminal lobe of CaM is sufficient to partially activate CaMKII in the presence of Ca2+ (39), we tested the effects of a His6-tagged A2-CTD on CaMKII activity. We detected a concentration-dependent activation of purified CaMKIIα using a model peptide substrate, autocamtide-2 (AC-2). This activation was Ca2+-independent (−Ca2+: Vmax 1.5 ± 0.2 μmol/min/mg, Ka 8.6 ± 1.6 μm; +Ca2+: Vmax 1.3 ± 0.2 μmol/min/mg, Ka 6.7 ± 1.7 μm), whereas CaM is a Ca2+-dependent activator (+Ca2+: Vmax 6.1 ± 0.1 μmol/min/mg, Ka 97 ± 17 nm) (Fig. 1B). Moreover, Ca2+/CaM stimulated the phosphorylation of all substrates tested, whereas CaMKII activation by the A2-CTD was detected with AC-2 and GluN2B-1290–1309, but not with syntide-2 (Fig. 1C). Thus, the A2-CTD is a Ca2+-independent and substrate-selective activator of CaMKII.
FIGURE 1.

Ca2+-independent activation of CaMKII by α-actinin-2. A, ClustalW2 alignment of the C-terminal domain (CTD) of human α-actinin-2 (A2; residues 819–894) with the CTDs of α-actinin-1, -3, and -4 (A1, A3, A4) and full-length human CaM (CM). Identical and similar residues are highlighted in black and gray, respectively. Asterisks indicate residues targeted for mutagenesis in α-actinin-2. B, concentration-dependent effects of CaM and His6-A2-CTD on CaMKII phosphorylation of autocamtide-2 in the presence of CaCl2 (+Ca; black/dashed lines) or EGTA (−Ca; gray/solid) (mean ± S.E., n = 3). C, Ca2+-independent activation of CaMKII by His6-A2-CTD (14 μm) is detected with autocamtide-2 (AC2) and GluN2B-(1290–1309), but not syntide-2. Data plotted as a percentage of maximum activation by Ca2+/CaM (mean ± S.E., n = 3).
Identification of CaMKII-binding Determinants in α-Actinin-2
We hypothesized that the different affinities and maximal activation by CaM and A2-CTD reflect distinct interactions with the α-helical CaMKII regulatory domain (residues 293–310). The N- and C-terminal lobes of full-length CaM interact with both sides of this helix (40) (supplemental Fig. S2A). Interestingly, the A2-CTD interacts with a single face of a regulatory domain helix in the giant muscle protein kinase, titin (41) (supplemental Fig. S2B). Given the amino acid sequence similarity between the A2-CTD and the C-lobe of CaM (Fig. 1A), we aligned these two structures (supplemental Fig. S2C). The alignment (RMS 2.97 Å) predicts a hydrophobic interface between one face of the CaMKII regulatory domain helix and a concave binding “pocket” in A2-CTD formed by Ser-834, Leu-854, Tyr-861, Leu-888, and other residues (Fig. 2A). To test this alignment as a model for CaMKII-binding to the A2-CTD, we introduced charge in the predicted hydrophobic binding pocket. S834R, L854R, and Y861R mutations completely abrogated CaMKII binding to GST-A2-CTD, and the L888R mutation reduced binding by ≈70% (Fig. 2B). However, none of these mutations affected binding of GST-A2-CTD to the densin PDZ domain in parallel experiments (supplemental Fig. S3A). Additional mutations reduced hydrophobicity without introducing charge; L854A and Y861A mutations completely abrogated CaMKII binding (like L854R and Y861R mutations), but S834A mutation had a minimal effect (supplemental Fig. S3B). In combination, these findings support our model for CaMKII interaction with the hydrophobic binding pocket in the CTD of α-actinin-2.
FIGURE 2.

α-Actinin mimics CaM in binding to the CaMKII regulatory domain. A and B, structural alignment of the A2-CTD (red) with the C-terminal lobe of CaM (blue) bound to CaMKII (from supplemental Fig. S2C). Residues in α-actinin-2 highlighted in green were mutated to arginine. Introduction of charge in the predicted hydrophobic binding pocket disrupted CaMKIIα binding, as seen by protein staining for GST proteins and immunoblotting (IB) for CaMKII. Binding is expressed as a percentage of binding to WT (mean ± S.E., n = 4. ***, p < 0.0001 versus WT; 1-way ANOVA, Bonferroni's post-test). C, close-up of CaMKII regulatory domain helix showing residues in CaMKII targeted for mutagenesis (cyan or magenta). D, effects of sequential autophosphorylation of WT, T305A-, or T306A-CaMKIIα (P) on binding to Ca2+/CaM or GST-A2-CTD. Levels of CaMKII in inputs were compared by immunoblot (top). Isolated complexes were analyzed by protein staining. Binding of autophosphorylated CaMKIIs was expressed as a percentage of binding of corresponding non-phosphorylated control CaMKII (mean ± S.E., n = 3. Two-way ANOVA, p < 0.0001. **, p < 0.001; ***, p < 0.0001 versus 100% by post-hoc 1-column t test).
Determinants in CaMKII for Binding to α-Actinin
Ca2+-independent autophosphorylation blocks CaMKII binding to both GST-A2-CTD and Ca2+/CaM, and this loss of binding can be prevented by mutation of both Thr-305 and Thr-306 in CaMKIIα to Ala (21, 37). Our alignment model indicates that the Thr-306 side chain is buried in the A2-CTD interaction site, whereas the Thr-305 side chain is solvent exposed (Fig. 2C), suggesting that phosphorylation at Thr-306, but not Thr-305, should disrupt binding. Autophosphorylation of wild type (WT) CaMKIIα can occur at both Thr-305 and Thr-306, whereas T305A- and T306A-CaMKIIα mutants can be autophosphorylated at only one residue (Thr-306 and Thr-305, respectively) (37). Indeed, Ca2+-independent autophosphorylation reduced binding of WT, T305A-, and T306A-CaMKIIα to Ca2+/CaM-agarose by ≥80% (Fig. 2D), suggesting that the remaining native Thr residue in both mutated kinases was effectively autophosphorylated. In contrast, Ca2+-independent autophosphorylation had no effect on the binding of purified T306A-CaMKIIα to GST-A2-CTD, but reduced the binding of purified WT and T305A-CaMKIIα to GST-A2-CTD by ≥80% (Fig. 2D). Thus, CaMKIIα binding to α-actinin is sensitive to autophosphorylation at Thr-306, but not at Thr-305, consistent with our model for α-actinin binding to one face of the CaMKII regulatory domain helix.
To identify additional interaction determinants, we tested the binding of GST-A2-CTD to additional CaMKIIα mutants that had been expressed in HEK293 cells. Initial studies suggested that basal autophosphorylation of WT CaMKIIα at Thr-305/6 in HEK293 cells (supplemental Fig. S4A) suppressed interactions with both A2-CTD and Ca2+/CaM (supplemental Fig. S4B). Moreover, we found that levels of Thr-305/6 phosphorylation were sensitive to mutations within the CaMKII regulatory domain (data not shown). Thus, we analyzed the effects of regulatory domain mutations of CaMKII on binding to the A2-CTD on a background of a T305A/T306A double mutant kinase (TT305/6AA-CaMKIIα) to avoid possible indirect effects. A302R and I303R mutations had similar effects to reduce or block the binding of TT305/6AA-CaMKIIα to either A2-CTD or Ca2+/CaM (supplemental Fig. S4C), consistent with the hydrophobic interactions predicted by our alignment model. In contrast, Thr-310 in CaMKIIα contacts only the N-lobe of CaM (40), and a T310D mutation disrupted binding to Ca2+/CaM, with no apparent effect on binding to A2-CTD (supplemental Fig. S4B). In combination, these findings show that α-actinin interacts with a subset of residues in the CaMKIIα regulatory domain used for interaction with Ca2+/CaM.
CaMKII Association with the CTD in α-Actinin-2 Dimers
Our in vitro studies with the A2-CTD constructs suggest that α-actinin has the potential to profoundly affect CaMKII activity and/or localization in cells. However, CaMKII interactions with full-length, dimeric α-actinins have not been directly characterized. Therefore, we co-expressed full-length HA-tagged α-actinin-2 (HA-actinin-2) with CaMKIIα in HEK293 cells. HA-actinin-2 was readily detected in CaMKII immune complexes isolated from lysates of cells expressing WT or TT305/6AA-CaMKIIα, but mutation of Thr-305 and Thr-306 to Ala substantially enhanced the interaction (supplemental Fig. S4A). Subsequent studies used TT305/6AA-CaMKIIα to avoid potential confounding effects of variations in basal phosphorylation at Thr-305/6 (see above). Importantly, L854R or Y861R mutations in the CTD that prevent CaMKII binding in vitro (Fig. 2B) substantially reduced the amount of HA-actinin-2 that co-immunoprecipitated with TT305/6AA-CaMKIIα (Fig. 3A). Thus, the CTD of full-length α-actinin-2 appears to be critical for the association of CaMKII in intact cells.
FIGURE 3.

CaMKIIα is targeted to F-actin by the α-actinin-2 CTD in cells. A, CaMKII immune complexes isolated from extracts of HEK293 cells co-expressing TT305/6AA-CaMKIIα with full-length HA-actinin-2 (HA-A2; WT or mutated) were analyzed by protein stain for CaMKII and immunoblotting for HA. B, immunofluorescence microscopy of HEK293 cells expressing TT305/6AA-CaMKIIα (AA-CaMKII, red) alone (top) or with HA-actinin-2 (WT or Y861R; green). C, localization of AA-CaMKII and F-actin (phalloidin staining; blue) in the absence and presence of WT HA-actinin-2 (green). Arrows indicate colocalization of HA-A2 and AA-CaMKII alone (yellow) or HA-A2, AA-CaMKII and phalloidin (white). D, intensity correlation quotient (ICQ) analysis of colocalization between AA-CaMKII and WT or Y861R HA-actinin-2 (left) or AA-CaMKII and phalloidin in the presence and absence of WT HA-actinin-2 (right) (n = 8 cells; ***, p < 0.0001 versus WT by t test; **, p < 0.005 versus control by t test, n = 6 cells)
The cellular localization of WT/mutated HA-actinin-2 and TT305/6AA-CaMKIIα were also compared using immunofluorescence microscopy. In the absence of HA-actinin-2, TT305/6AA-CaMKIIα filled the cell cytoplasm relatively uniformly (Fig. 3B). Full length HA-actinin-2 was targeted to the submembrane cortical cytoskeleton (Fig. 3B), as shown previously (42). In the presence of co-expressed WT HA-actinin-2, the distribution of TT305/6AA-CaMKIIα became non-uniform. Moreover, there was a partial, but significant, co-localization of WT HA-actinin-2 with TT305/6AA-CaMKIIα, as revealed by yellow coloration in an overlay of images displayed in the red and green channels, respectively (Fig. 3B). Importantly, colocalization was disrupted by Y861R mutation of HA-actinin-2. Additional studies using phalloidin to label the F-actin cytoskeleton revealed little co-localization of TT305/6AA-CaMKIIα in the absence of HA-actinin-2. However, the co-expression of HA-actinin-2 significantly enhanced targeting of TT305/6AA-CaMKIIα to subcellular domains enriched in F-actin, primarily on the cell periphery (Fig. 3C). Analysis of images of multiple cells similar to those shown in Fig. 3, B and C by calculation of an intensity correlation quotient (ICQ) revealed significant colocalization of CaMKII with WT but not Y861R HA-actinin-2, and significant colocalization of CaMKII with F-actin in the presence but not the absence of WT HA-actinin-2 (Fig. 3D). Taken together, these data show that CaMKIIα is targeted to the F-actin cytoskeleton by binding to the CTD of full-length α-actinin-2 dimers in intact cells.
α-Actinin Modulation of CaMKII Interactions with NMDARs in Cells
Previous studies showed that CaMKII associates with multiple α-actinin isoforms in brain (21, 31, 32). We expanded these analyses to demonstrate that these complexes also contain NMDAR GluN1 and GluN2B subunits (Fig. 4). To determine the role of α-actinin-2 in formation of this complex, we co-expressed CaMKIIα phosphorylation site mutants with GluN1 and GluN2B in HEK293 cells, with or without HA-actinin-2. We again used TT305/6AA-CaMKIIα to avoid potential confounding effects of varying levels of Thr-305/6 phosphorylation (see above). Both NMDAR subunits associate with TT305/6AA-CaMKIIα in the absence of co-expressed HA-actinin-2 (Fig. 5A). This association was partially due to basal Thr-286 phosphorylation because additional mutation of Thr-286 to Ala (T286A/TT305/6AA-CaMKIIα) resulted in a significant ≈4-fold reduction in levels of co-precipitated NMDAR subunits (Fig. 5A). Co-expression of HA-actinin-2 significantly enhanced the associations of GluN1 and GluN2B with TT305/6AA-CaMKIIα by ≈1.8-fold, and with T286A/TT305/6AA-CaMKIIα by ≈4-fold, relative to corresponding levels in the absence of HA-actinin-2 (Fig. 5A). Indeed, co-expression of HA-actinin-2 largely rescued the deficit in association of GluN2B-NMDARs that was induced by T286A mutation. In contrast, there was a trend (p = 0.09) for HA-actinin-2 to decrease the association of GluN2A-NMDARs with TT305/6AA-CaMKIIα (Fig. 5B). Collectively, these data show that α-actinin-2 specifically enhances CaMKIIα targeting to GluN2B-NMDARs.
FIGURE 4.
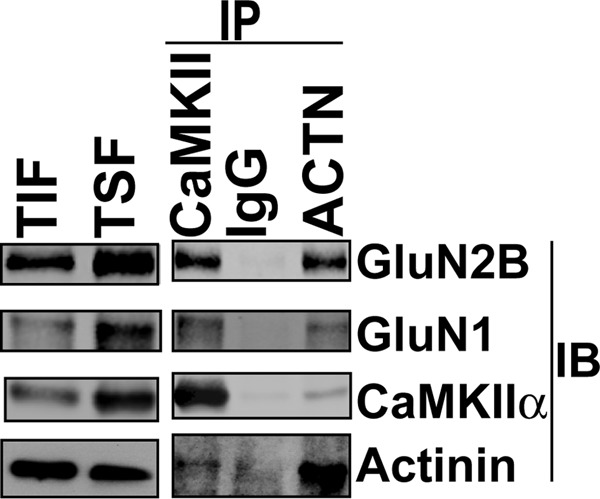
α-Actinin and CaMKII exist in complex with NMDARs in mouse brain. A mouse forebrain Triton-soluble fraction (TSF) was immunoprecipitated (IP) using CaMKII, α-actinin, or control IgG. The Triton-insoluble fraction (TIF), TSF and immune complexes were immunoblotted for GluN2B, GluN1, α-actinin, and CaMKII as indicated.
FIGURE 5.

α-Actinin-2 selectively enhances CaMKII association with GluN2B-NMDARs. A, CaMKII was immunoprecipitated from extracts of HEK293 cells (input) expressing GluN1, GluN2B, and CaMKIIα (TT305/6AA or T286A/TT305/6AA; AA-CaMKIIα or AAA-CaMKIIα, respectively) with or without HA-α-actinin-2. Input and CaMKII immune complexes were immunoblotted for GluN1, GluN2B, HA, CaMKII, and phospho-Thr286 (P-T286). Levels of co-immunoprecipitated GluN2B were normalized to input and plotted as mean ± S.E. (n = 5). *, p < 0.05 versus GluN1+GluN2B+AACK. ##, p < 0.01 versus GluN1+GluN2B+AACK+HA-A2. #, p < 0.05 versus GluN1+GluN2B+AAACK. B, similar to A, except GluN2B is replaced with GluN2A.
α-Actinin-2 Differentially Modulates CaMKII Phosphorylation of Glutamate Receptors in Cells
The CTD of α-actinin-2 activates CaMKII phosphorylation of AC2 or GluN2B-1290-1309 in the presence or absence of Ca2+ (Fig. 1, B and C), but competes with CaM to inhibit Ca2+-dependent phosphorylation of other substrates such as syntide-2 (18). These data lead us to hypothesize that α-actinin modulates the innate substrate-selectivity of CaMKII in cells. Therefore, we compared the effects of expressing HA-actinin-2 on CaMKII phosphorylation of two physiologically important substrates; Ser-831 in AMPA-type glutamate receptor GluA1 subunits and Ser-1303 in NMDAR GluN2B subunits (43–45). Phosphorylation of Ser-831 in GluA1 in HEK293 cells was enhanced ≈3-fold by co-expression of TT305/6AA-CaMKIIα, but additional expression of HA-actinin-2 blocked this increase (Fig. 6A). In contrast, phosphorylation of Ser-1303 in GluN2B by TT305/6AA-CaMKII was significantly enhanced by co-expression of HA-actinin-2 (Fig. 6B). Thus, α-actinin-2 modifies the innate substrate selectivity of CaMKII in intact cells, favoring phosphorylation of GluN2B over GluA1.
FIGURE 6.
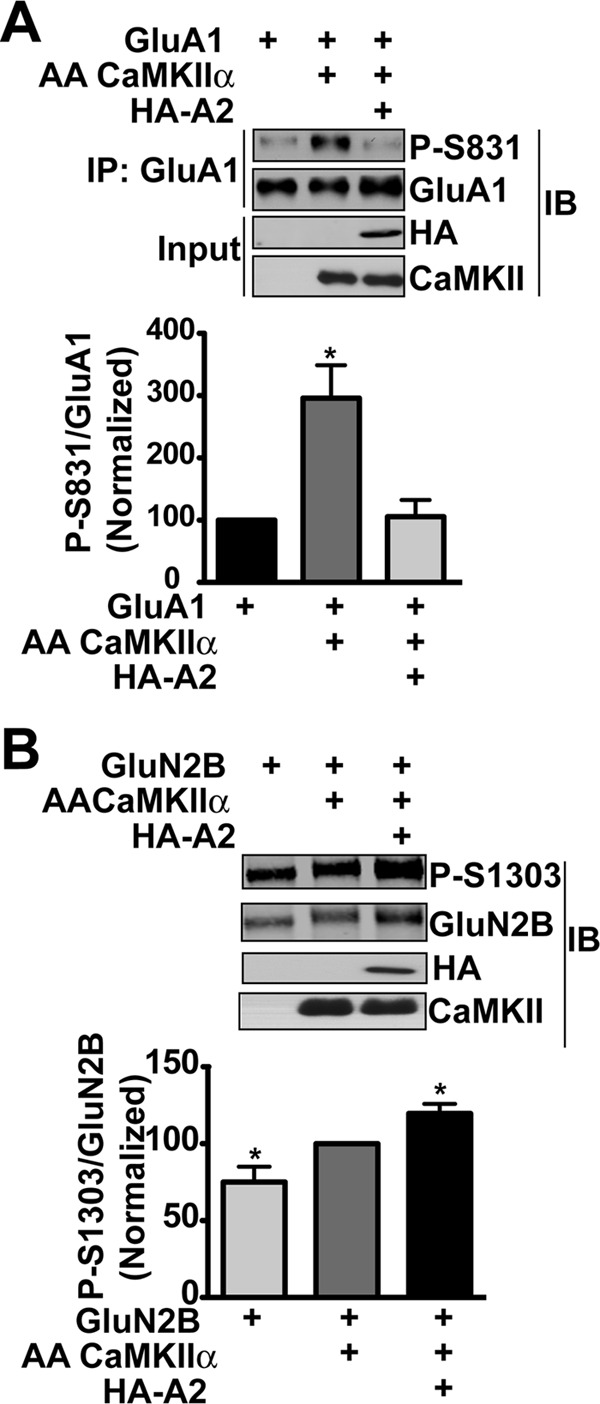
α-Actinin modulates phosphorylation of GluA1 and GluN2B by CaMKII. A, GluA1 was immunoprecipitated from lysates of HEK293 cells expressing GluA1 with or without TT305/6AA-CaMKIIα and HA-actinin-2 (WT) and immunoblotted for total GluA1 and phospho-Ser831 (P-S831). B, lysates of HEK293 cells expressing GluN2B with or without TT305/6AA-CaMKIIα and HA-actinin-2 were directly immunoblotted for total GluN2B, phospho-Ser1303 (P-S1303), CaMKII and HA as indicated. Graphs plot phospho-protein levels as a ratio to corresponding total protein after normalization to total GluA1 alone (A) or total GluN2B+AACK (B) (mean ± S.E., n = 4. *, p < 0.01 by 1-way ANOVA with Bonferoni post-test).
DISCUSSION
CaMKII interactions with several synaptic proteins are dynamically regulated by CaMKII activation and autophosphorylation, but their specific contributions to overall targeting and modulation of kinase activity are poorly understood (see Introduction). The present findings show that α-actinin can function as a novel modulator of CaMKII targeting and kinase activity by mimicking CaM.
α-Actinin as a Surrogate for CaM
In the presence of Ca2+, the two lobes of CaM wrap around the α-helical regulatory domain to activate CaMKII (40). However, each lobe of CaM can independently activate CaMKII, albeit with weaker affinity and to reduced levels (39, 46–48). The current data show that Ca2+-independent interaction of A2-CTD with one side of the regulatory domain helix can activate CaMKII (Fig. 1B). The weaker apparent affinity and partial activation by A2-CTD compared with full-length Ca2+/CaM (see “Results”) presumably results from occupation of a reduced surface area on the regulatory domain by the A2-CTD causing only partial displacement of the regulatory domain from the catalytic site. This may also account for the fact that activation by A2-CTD is selective for substrates with primary sequences resembling that surrounding the Thr-286 autophosphorylation site, such as GluN2B Ser-1303 and the AC-2 model peptide (Fig. 1C). These peptide substrates also mimic the regulatory domain in binding to a modulatory “T-site” on the catalytic domain (49), which can competitively displace the regulatory domain to help “open” the catalytic site for substrate and ATP binding (50, 51). Thus, we posit that binding of α-actinin allows for peptide binding at the T-site, thereby selectively enhancing access of these substrates to the catalytic site. However, the regulatory domain is not displaced to the same extent as Ca2+/CaM, accounting for the reduced Vmax. This cooperation may relate to a previous report that GluN2B can “trap” CaMKII in an autonomously active conformation in the absence of Thr-286 autophosphorylation (16). This mechanism may also explain why we have been unable to detect A2-CTD-dependent autophosphorylation at Thr-286 (data not shown), although we cannot exclude the possibility that the low binding affinity limits simultaneous activation of adjacent CaMKII subunits that is required for Thr-286 autophosphorylation.
Role of Thr-305/Thr-306 Autophosphorylation
Most prior studies have treated Thr-305 and Thr-306 as functionally redundant because autophosphorylation at either Thr-305 or Thr-306 can block Ca2+/CaM binding (37). Thr-305 and Thr306 are both strictly conserved among all CaMKII isoforms from Hydra to Homo sapiens (Fig. S5), perhaps suggesting that these sites have unique functional roles or that dual phosphorylation of both sites is functionally important. The current data favor the first possibility; unlike Thr-306 autophosphorylation, which prevents binding of both Ca2+/CaM and α-actinin, Thr-305 phosphorylation prevents binding of Ca2+/CaM, but not α-actinin (Fig. 2D). Unfortunately, the specific role of each site in controlling interactions with α-actinin is not recapitulated by T305D and T306D mutations, which individually disrupt CaMKII binding to both Ca2+/CaM and A2-CTD.4 Consequently disruption of CaMKII targeting to dendritic spines by a Thr-305 to Asp mutation (7, 13), may be due to loss of interaction with α-actinin, especially because Thr-305/6 phosphorylation appears to have relatively modest effects on direct interactions with GluN2B or other binding partners (21, 52). Moreover, the effects of mutating Thr-305 and Thr-306 to Ala (or Val) to remove both autophosphorylation sites and stabilize synaptic targeting, which appears to dictate the role of CaMKII in regulating synaptic strength and plasticity (5–7, 13), may result from stabilized interactions with Ca2+/CaM and/or α-actinin. Our data suggest that Thr-306 phosphorylation makes a specific contribution to modulating CaMKII that is mediated by disruption of α-actinin interactions and that is distinct from the effects of phosphorylation at Thr-305. Thus, it will be important to assess the specific phosphorylation at both Thr-305 and Thr-306 and the interactions with α-actinin to understand CaMKII regulation in cells.
CaMKII Targeting to Actin Cytoskeleton and NMDA-type Glutamate Receptors
Unlike certain CaMKIIβ splice variants (53–56), CaMKIIα homomers are not directly targeted to F-actin (54). Nevertheless, postsynaptic localization of CaMKIIα, the major isoform present in many forebrain neurons (57), is dependent on an intact F-actin cytoskeleton (23). This is consistent with the α-actinin-dependent targeting of CaMKIIα to F-actin (Fig. 3) and the fact that CaMKIIα associates with multiple α-actinin isoforms in brain (21, 31, 32) (Fig. 4). Thus, α-actinin plays an important role in targeting homomeric CaMKIIα holoenzymes that is independent of direct interactions between CaMKII and F-actin. However, a recent study from our laboratory indicates that at least one more F-actin-binding protein (spinophilin) is involved in CaMKII targeting to F-actin (58).
Activated CaMKII translocates to dendritic spines and binds NMDAR GluN2B subunits (16, 49, 59). CaMKII interactions with GluN2B are critical for synaptic plasticity, learning, and memory (14, 15, 17, 19). NMDAR GluN1 and GluN2B subunits, but not GluN2A, also interact with α-actinin (27), tethering NMDARs to the actin cytoskeleton to modulate channel activity (29, 30). Pharmacological destabilization of F-actin disrupts the colocalization of both α-actinin and CaMKII with synaptic NMDARs (22, 23). We found that α-actinin selectively stabilizes CaMKII targeting to GluN2B-NMDARs (Fig. 5A), apparently by the combined effects of activating CaMKII to interact directly with GluN2B and of bridging indirect interactions with GluN2B and/or GluN1. The trend for α-actinin to reduce CaMKII targeting to GluN2A-NMDARs may be explained by the lack of α-actinin interaction with GluN2A, the low affinity of direct CaMKII-GluN2A interactions, and/or competition between CaMKII and α-actinin for binding GluN1 (27, 59, 60). Thus, α-actinin may selectively target CaMKII to specific subpopulations of neuronal NMDARs.
Differential Modulation of Glutamate Receptor Phosphorylation
In addition to targeting CaMKII, the CTD of α-actinin has complex effects on CaMKII activity, not only in vitro (Fig. 1) but also toward physiologically relevant substrates in intact cells (Fig. 6). Phosphorylation of Ser-831 in GluA1 subunits increases the conductance of synaptic AMPARs during LTP (61, 62), and we found that α-actinin suppressed CaMKII phosphorylation of Ser-831 (Fig. 6A). GluA1 is considered to be a “pure” S-site substrate and α-actinin failed to directly activate S-site substrate phosphorylation (Fig. 1C). Therefore, we posit that α-actinin suppresses GluA1 phosphorylation by competing for Ca2+/CaM binding to the regulatory domain, in agreement with prior findings (18). In contrast, CaMKII is a negative-feedback modulator of Ca2+ influx via GluN2B-NMDARs (35) and α-actinin enhanced CaMKII phosphorylation of Ser-1303 in GluN2B NMDAR subunits (Fig. 6B). Interestingly, α-actinin can form a ternary complex with CaMKII and densin (21, 32), and densin contains a novel CaMKII inhibitory domain that effectively blocks phosphorylation of GluA1 but not GluN2B (38). Thus, densin and α-actinin may collaborate to target neuronal CaMKII, favoring phosphorylation of the associated GluN2B-NMDARs at the expense of GluA1-AMPARs. In this way, α-actinin may act as a PSD “phosphostat”, maintaining basal phosphorylation of a subset of CaMKII targets to control synaptic activity. It will be important to investigate the impact of α-actinin isoforms on the phosphorylation of CaMKII substrates at synapses and in other subcellular compartments.
Supplementary Material
Acknowledgments
We thank Dr. Y. Jiao for help with α-actinin plasmids and Martha Bass for expression and purification of CaMKII. We are grateful to Drs. D. Webb, A.J. Robison, B. Wadzinski, and Y. Jiao for critical evaluations of drafts of this manuscript.
This work was supported, in whole or in part by National Institutes of Health Grants R01-MH063232 (to R. J. C.) and T32-DK07563 (to R. K. B.), and AHA Grants 0815090E (to N. J.-S.) and 0625289B (to R. K. B.). Fluorescence microscopy was performed in the VUMC Cell Imaging Shared Resource, supported by NIH grants CA68485, DK20593, DK58404, HD15052, DK59637, and EY08126.

This article contains supplemental Figs. S1–S5.
N. Jalan-Sakrikar, unpublished observations.
- CaM
- calmodulin.
- CaMKII
- Ca2+/calmodulin-dependent protein kinase II.
- NMDAR
- N-methyl d-Aspartate receptor.
- PSD
- postsynaptic density
- AMPAR
- α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionic acid receptor.
- CaMKAP
- CaMKII-anchoring protein.
- CTD
- C-terminal domain.
REFERENCES
- 1. Colbran R. J. (2004) Targeting of calcium/calmodulin-dependent protein kinase II. Biochem. J. 378, 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hudmon A., Schulman H. (2002) Neuronal Ca2+/calmodulin-dependent protein kinase II: the role of structure and autoregulation in cellular function. Annu. Rev. Biochem. 71, 473–510 [DOI] [PubMed] [Google Scholar]
- 3. Swulius M. T., Waxham M. N. (2008) Ca(2+)/calmodulin-dependent protein kinases. Cell Mol. Life Sci. 65, 2637–2657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Soderling T. R., Stull J. T. (2001) Structure and regulation of calcium/calmodulin-dependent protein kinases. Chem. Rev. 101, 2341–2352 [DOI] [PubMed] [Google Scholar]
- 5. Pi H. J., Otmakhov N., Lemelin D., De Koninck P., Lisman J. (2010b) Autonomous CaMKII can promote either long-term potentiation or long-term depression, depending on the state of T305/T306 phosphorylation. J. Neurosci. 30, 8704–8709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pi H. J., Otmakhov N., El Gaamouch F., Lemelin D., De Koninck P., Lisman J. (2010a) CaMKII control of spine size and synaptic strength: role of phosphorylation states and nonenzymatic action. Proc. Natl. Acad. Sci. U.S.A. 107, 14437–14442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Elgersma Y., Fedorov N. B., Ikonen S., Choi E. S., Elgersma M., Carvalho O. M., Giese K. P., Silva A. J. (2002) Inhibitory autophosphorylation of CaMKII controls PSD association, plasticity, and learning. Neuron 36, 493–505 [DOI] [PubMed] [Google Scholar]
- 8. Giese K. P., Fedorov N. B., Filipkowski R. K., Silva A. J. (1998) Autophosphorylation at Thr-286 of the α calcium-calmodulin kinase II in LTP and learning. Science 279, 870–873 [DOI] [PubMed] [Google Scholar]
- 9. Griffith L. C., Lu C. S., Sun X. X. (2003) CaMKII, an enzyme on the move: regulation of temporospatial localization. Mol. Interv 3, 386–403 [DOI] [PubMed] [Google Scholar]
- 10. Bayer K. U., Schulman H. (2001) Regulation of signal transduction by protein targeting: the case for CaMKII. Biochem. Biophys. Res. Commun. 289, 917–923 [DOI] [PubMed] [Google Scholar]
- 11. Yuste R., Majewska A., Holthoff K. (2000) From form to function: calcium compartmentalization in dendritic spines. Nat. Neurosci. 3, 653–659 [DOI] [PubMed] [Google Scholar]
- 12. Feng B., Raghavachari S., Lisman J. (2011) Quantitative estimates of the cytoplasmic, PSD, and NMDAR-bound pools of CaMKII in dendritic spines. Brain Res. 1419, 46–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shen K., Meyer T. (1999) Dynamic control of CaMKII translocation and localization in hippocampal neurons by NMDA receptor stimulation. Science 284, 162–166 [DOI] [PubMed] [Google Scholar]
- 14. Zhou Y., Takahashi E., Li W., Halt A., Wiltgen B., Ehninger D., Li G. D., Hell J. W., Kennedy M. B., Silva A. J. (2007) Interactions between the NR2B receptor and CaMKII modulate synaptic plasticity and spatial learning. J Neurosci. 27, 13843–13853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Barria A., Malinow R. (2005) NMDA receptor subunit composition controls synaptic plasticity by regulating binding to CaMKII. Neuron 48, 289–301 [DOI] [PubMed] [Google Scholar]
- 16. Bayer K. U., De Koninck P., Leonard A. S., Hell J. W., Schulman H. (2001) Interaction with the NMDA receptor locks CaMKII in an active conformation. Nature 411, 801–805 [DOI] [PubMed] [Google Scholar]
- 17. Sanhueza M., Fernandez-Villalobos G., Stein I. S., Kasumova G., Zhang P., Bayer K. U., Otmakhov N., Hell J. W., Lisman J. (2011) Role of the CaMKII/NMDA receptor complex in the maintenance of synaptic strength. J. Neurosci. 31, 9170–9178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Robison A. J., Bartlett R. K., Bass M. A., Colbran R. J. (2005) Differential modulation of Ca2+/calmodulin-dependent protein kinase II activity by regulated interactions with N-methyl-d-aspartate receptor NR2B subunits and α-actinin. J. Biol. Chem. 280, 39316–39323 [DOI] [PubMed] [Google Scholar]
- 19. Halt A. R., Dallapiazza R. F., Zhou Y., Stein I. S., Qian H., Juntti S., Wojcik S., Brose N., Silva A. J., Hell J. W. (2012) CaMKII binding to GluN2B is critical during memory consolidation. EMBO J. 31, 1203–1216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cheng D., Hoogenraad C. C., Rush J., Ramm E., Schlager M. A., Duong D. M., Xu P., Wijayawardana S. R., Hanfelt J., Nakagawa T., Sheng M., Peng J. (2006) Relative and absolute quantification of postsynaptic density proteome isolated from rat forebrain and cerebellum. Mol. Cell Proteomics 5, 1158–1170 [DOI] [PubMed] [Google Scholar]
- 21. Robison A. J., Bass M. A., Jiao Y., MacMillan L. B., Carmody L. C., Bartlett R. K., Colbran R. J. (2005) Multivalent interactions of calcium/calmodulin-dependent protein kinase II with the postsynaptic density proteins NR2B, densin-180, and α-actinin-2. J. Biol. Chem. 280, 35329–35336 [DOI] [PubMed] [Google Scholar]
- 22. Allison D. W., Gelfand V. I., Spector I., Craig A. M. (1998) Role of actin in anchoring postsynaptic receptors in cultured hippocampal neurons: differential attachment of NMDA versus AMPA receptors. J. Neurosci. 18, 2423–2436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Allison D. W., Chervin A. S., Gelfand V. I., Craig A. M. (2000) Postsynaptic scaffolds of excitatory and inhibitory synapses in hippocampal neurons: maintenance of core components independent of actin filaments and microtubules. J. Neurosci. 20, 4545–4554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Blanchard A., Ohanian V., Critchley D. (1989) The structure and function of α-actinin. J. Muscle Res. Cell Motil. 10, 280–289 [DOI] [PubMed] [Google Scholar]
- 25. Sjöblom B., Salmazo A., Djinovi-Carugo K. (2008) α-Actinin structure and regulation. Cell Mol. Life Sci. 65, 2688–2701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wyszynski M., Kharazia V., Shanghvi R., Rao A., Beggs A. H., Craig A. M., Weinberg R., Sheng M. (1998) Differential regional expression and ultrastructural localization of α-actinin-2, a putative NMDA receptor-anchoring protein, in rat brain. J. Neurosci. 18, 1383–1392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wyszynski M., Lin J., Rao A., Nigh E., Beggs A. H., Craig A. M., Sheng M. (1997) Competitive binding of α-actinin and calmodulin to the NMDA receptor. Nature 385, 439–442 [DOI] [PubMed] [Google Scholar]
- 28. Krupp J. J., Vissel B., Thomas C. G., Heinemann S. F., Westbrook G. L. (1999) Interactions of calmodulin and α-actinin with the NR1 subunit modulate Ca2+-dependent inactivation of NMDA receptors. J. Neurosci. 19, 1165–1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang S., Ehlers M. D., Bernhardt J. P., Su C. T., Huganir R. L. (1998) Calmodulin mediates calcium-dependent inactivation of N-methyl-d-aspartate receptors. Neuron 21, 443–453 [DOI] [PubMed] [Google Scholar]
- 30. Michailidis I. E., Helton T. D., Petrou V. I., Mirshahi T., Ehlers M. D., Logothetis D. E. (2007) Phosphatidylinositol-4,5-bisphosphate regulates NMDA receptor activity through α-actinin. J. Neurosci. 27, 5523–5532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dhavan R., Greer P. L., Morabito M. A., Orlando L. R., Tsai L. H. (2002) The cyclin-dependent kinase 5 activators p35 and p39 interact with the α-subunit of Ca2+/calmodulin-dependent protein kinase II and α-actinin-1 in a calcium-dependent manner. J. Neurosci. 22, 7879–7891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Walikonis R. S., Oguni A., Khorosheva E. M., Jeng C. J., Asuncion F. J., Kennedy M. B. (2001) Densin-180 forms a ternary complex with the (α)-subunit of Ca2+/calmodulin-dependent protein kinase II and (α)-actinin. J. Neurosci. 21, 423–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Baucum A. J., 2nd, Jalan-Sakrikar N., Jiao Y., Gustin R. M., Carmody L. C., Tabb D. L., Ham A. J., Colbran R. J. (2010) Identification and validation of novel spinophilin-associated proteins in rodent striatum using an enhanced ex vivo shotgun proteomics approach. Mol. Cell Proteomics 9, 1243–1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jiao Y., Robison A. J., Bass M. A., Colbran R. J. (2008) Developmentally regulated alternative splicing of densin modulates protein-protein interaction and subcellular localization. J. Neurochem. 105, 1746–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sessoms-Sikes S., Honse Y., Lovinger D. M., Colbran R. J. (2005) CaMKIIα enhances the desensitization of NR2B-containing NMDA receptors by an autophosphorylation-dependent mechanism. Mol. Cell Neurosci. 29, 139–147 [DOI] [PubMed] [Google Scholar]
- 36. Strack S., Robison A. J., Bass M. A., Colbran R. J. (2000) Association of calcium/calmodulin-dependent kinase II with developmentally regulated splice variants of the postsynaptic density protein densin-180. J. Biol. Chem. 275, 25061–25064 [DOI] [PubMed] [Google Scholar]
- 37. Colbran R. J. (1993) Inactivation of Ca2+/calmodulin-dependent protein kinase II by basal autophosphorylation. J. Biol. Chem. 268, 7163–7170 [PubMed] [Google Scholar]
- 38. Jiao Y., Jalan-Sakrikar N., Robison A. J., Baucum A. J., 2nd, Bass M. A., Colbran R. J. (2011) Characterization of a central Ca2+/calmodulin-dependent protein kinase IIα/β binding domain in densin that selectively modulates glutamate receptor subunit phosphorylation. J. Biol. Chem. 286, 24806–24818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shifman J. M., Choi M. H., Mihalas S., Mayo S. L., Kennedy M. B. (2006) Ca2+/calmodulin-dependent protein kinase II (CaMKII) is activated by calmodulin with two bound calciums. Proc. Natl. Acad. Sci. U.S.A. 103, 13968–13973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Meador W. E., Means A. R., Quiocho F. A. (1993) Modulation of calmodulin plasticity in molecular recognition on the basis of x-ray structures. Science 262, 1718–1721 [DOI] [PubMed] [Google Scholar]
- 41. Atkinson R. A., Joseph C., Kelly G., Muskett F. W., Frenkiel T. A., Nietlispach D., Pastore A. (2001) Ca2+-independent binding of an EF-hand domain to a novel motif in the α-actinin-titin complex. Nat. Struct. Biol. 8, 853–857 [DOI] [PubMed] [Google Scholar]
- 42. Khoory W., Wu E., Svoboda K. K. (1993) Intracellular relationship between actin and α-actinin in a whole corneal epithelial tissue. J. Cell Sci. 106, 703–717 [DOI] [PubMed] [Google Scholar]
- 43. Barria A., Derkach V., Soderling T. (1997) Identification of the Ca2+/calmodulin-dependent protein kinase II regulatory phosphorylation site in the α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate-type glutamate receptor. J. Biol. Chem. 272, 32727–32730 [DOI] [PubMed] [Google Scholar]
- 44. Strack S., McNeill R. B., Colbran R. J. (2000) Mechanism and regulation of calcium/calmodulin-dependent protein kinase II targeting to the NR2B subunit of the N-methyl-d-aspartate receptor. J. Biol. Chem. 275, 23798–23806 [DOI] [PubMed] [Google Scholar]
- 45. O'Leary H., Liu W. H., Rorabaugh J. M., Coultrap S. J., Bayer K. U. (2011) Nucleotides and phosphorylation bi-directionally modulate Ca2+/calmodulin-dependent protein kinase II (CaMKII) binding to the N-methyl-d-aspartate (NMDA) receptor subunit GluN2B. J. Biol. Chem. 286, 31272–31281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Forest A., Swulius M. T., Tse J. K., Bradshaw J. M., Gaertner T., Waxham M. N. (2008) Role of the N- and C-lobes of calmodulin in the activation of Ca(2+)/calmodulin-dependent protein kinase II. Biochemistry 47, 10587–10599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jama A. M., Gabriel J., Al-Nagar A. J., Martin S., Baig S. Z., Soleymani H., Chowdhury Z., Beesley P., Török K. (2011) Lobe-specific functions of Ca2+·calmodulin in αCa2+·calmodulin-dependent protein kinase II activation. J. Biol. Chem. 286, 12308–12316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Evans T. I., Shea M. A. (2009) Energetics of calmodulin domain interactions with the calmodulin binding domain of CaMKII. Proteins 76, 47–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bayer K. U., LeBel E., McDonald G. L., O'Leary H., Schulman H., De Koninck P. (2006) Transition from reversible to persistent binding of CaMKII to postsynaptic sites and NR2B. J. Neurosci. 26, 1164–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hoffman L., Stein R. A., Colbran R. J., McHaourab H. S. (2011) Conformational changes underlying calcium/calmodulin-dependent protein kinase II activation. EMBO J. 30, 1251–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pradeep K. K., Cheriyan J., Suma Priya S. D., Rajeevkumar R., Mayadevi M., Praseeda M., Omkumar R. V. (2009) Regulation of Ca2+/calmodulin-dependent protein kinase II catalysis by N-methyl-d-aspartate receptor subunit 2B. Biochem. J. 419, 123–132 [DOI] [PubMed] [Google Scholar]
- 52. Leonard A. S., Bayer K. U., Merrill M. A., Lim I. A., Shea M. A., Schulman H., Hell J. W. (2002) Regulation of calcium/calmodulin-dependent protein kinase II docking to N-methyl-d-aspartate receptors by calcium/calmodulin and α-actinin. J. Biol. Chem. 277, 48441–48448 [DOI] [PubMed] [Google Scholar]
- 53. Shen K., Teruel M. N., Subramanian K., Meyer T. (1998) CaMKIIβ functions as an F-actin targeting module that localizes CaMKIIα/β heterooligomers to dendritic spines. Neuron 21, 593–606 [DOI] [PubMed] [Google Scholar]
- 54. Fink C. C., Bayer K. U., Myers J. W., Ferrell J. E., Jr., Schulman H., Meyer T. (2003) Selective regulation of neurite extension and synapse formation by the β but not the α isoform of CaMKII. Neuron 39, 283–297 [DOI] [PubMed] [Google Scholar]
- 55. Okamoto K., Narayanan R., Lee S. H., Murata K., Hayashi Y. (2007) The role of CaMKII as an F-actin-bundling protein crucial for maintenance of dendritic spine structure. Proc. Natl. Acad. Sci. U.S.A. 104, 6418–6423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lin Y. C., Redmond L. (2008) CaMKIIβ binding to stable F-actin in vivo regulates F-actin filament stability. Proc. Natl. Acad. Sci. U.S.A. 105, 15791–15796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Erondu N. E., Kennedy M. B. (1985) Regional distribution of type II Ca2+/calmodulin-dependent protein kinase in rat brain. J. Neurosci. 5, 3270–3277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Baucum A. J., 2nd, Strack S., Colbran R. J. (2012) Age-dependent targeting of protein phosphatase 1 to Ca2+/calmodulin-dependent protein kinase II by spinophilin in mouse striatum. PLoS One 7, e31554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Strack S., Colbran R. J. (1998) Autophosphorylation-dependent targeting of calcium/ calmodulin-dependent protein kinase II by the NR2B subunit of the N-methyl- d-aspartate receptor. J. Biol. Chem. 273, 20689–20692 [DOI] [PubMed] [Google Scholar]
- 60. Merrill M. A., Malik Z., Akyol Z., Bartos J. A., Leonard A. S., Hudmon A., Shea M. A., Hell J. W. (2007) Displacement of α-actinin from the NMDA receptor NR1 C0 domain By Ca2+/calmodulin promotes CaMKII binding. Biochemistry 46, 8485–8497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Barria A., Muller D., Derkach V., Griffith L. C., Soderling T. R. (1997) Regulatory phosphorylation of AMPA-type glutamate receptors by CaM-KII during long-term potentiation. Science 276, 2042–2045 [DOI] [PubMed] [Google Scholar]
- 62. Kristensen A. S., Jenkins M. A., Banke T. G., Schousboe A., Makino Y., Johnson R. C., Huganir R., Traynelis S. F. (2011) Mechanism of Ca2+/calmodulin-dependent kinase II regulation of AMPA receptor gating. Nat. Neurosci. 14, 727–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.