

Abstract
BACKGROUND AND PURPOSE
The κ opioid receptor antagonists demonstrate potential for maintaining abstinence from psychostimulant abuse, but existing non-peptide κ-receptor selective antagonists show exceptionally long activity. We hypothesized that the L- and D-Trp isomers of CJ-15,208, a natural cyclic tetrapeptide reported to be a κ-receptor antagonist in vitro, would demonstrate short-acting, dose-dependent antagonism in vivo, preventing reinstatement of cocaine-seeking behaviour.
EXPERIMENTAL APPROACH
Affinity, selectivity and efficacy of the L-Trp and D-Trp isomers for opioid receptors were assessed in vitro in radioligand and GTPγS binding assays. Opioid receptor agonist and antagonist activities were characterized in vivo following i.c.v. administration with the 55°C warm water tail-withdrawal assay. The D-Trp isomer, which demonstrated primarily κ-receptor selective antagonist activity, was further evaluated for its prevention of stress- and drug-induced reinstatement of extinguished cocaine conditioned place preference (CPP).
KEY RESULTS
The two isomers showed similar affinity and selectivity for κ receptors (Ki 30–35 nM) as well as κ receptor antagonism in vitro. As expected, the D-Trp cyclic tetrapeptide exhibited minimal agonist activity and induced dose-dependent κ-receptor selective antagonism lasting less than 18 h in vivo. Pretreatment with this peptide prevented stress-, but not cocaine-induced, reinstatement of extinguished cocaine CPP. In contrast, the L-Trp cyclic tetrapeptide unexpectedly demonstrated mixed opioid agonist/antagonist activity.
CONCLUSIONS AND IMPLICATIONS
The L-Trp and the D-Trp isomers of CJ-15,208 demonstrate stereospecific opioid activity in vivo. The relatively brief κ opioid receptor antagonism, coupled with the prevention of stress-induced reinstatement of extinguished cocaine-seeking behaviour, suggests the D-Trp isomer could be used therapeutically to maintain abstinence from psychostimulant abuse.
Keywords: κ-opioid; agonist; antagonist; analgesic; antinociception; CJ-15,208; conditioned place preference; cocaine; cyclic tetrapeptide
Introduction
While κ opioid receptor-selective antagonists were initially developed as pharmacological tools, recent research suggests that these antagonists could offer therapeutic potential in the treatment of mood disorders and the relapse to cocaine abuse (Metcalf and Coop, 2005; Aldrich and McLaughlin, 2009). Several non-peptide ligands have demonstrated κ opioid receptor-selective antagonism in vivo, notably nor-binaltorphimine (nor-BNI; Portoghese et al., 1987) and the phenylpiperidine JDTic (Thomas et al., 2003). Pretreatment with these antagonists reduced immobility in the forced swimming assay similar to antidepressants (Mague et al., 2003; McLaughlin et al., 2003; Beardsley et al., 2005) and reduced behavioural measures of anxiety in rats (Knoll et al., 2007). Moreover, pretreatment with these antagonists prevented stress-induced reinstatement of extinguished cocaine-seeking behaviour (Beardsley et al., 2005; Redila and Chavkin, 2008), confirming that κ-receptor selective antagonists have broad therapeutic potential. However, these non-peptide antagonists demonstrate exceptionally long durations of action (Metcalf and Coop, 2005; Aldrich and McLaughlin, 2009), antagonizing κ opioid receptors for up to 28 days after a single dose (Horan et al., 1992; Carroll et al., 2005). This long duration of action of the non-peptide κ-receptor selective antagonists may have slowed their clinical development (F. Vocci and S. Wong; pers. comm.), spurring the search for shorter acting κ-receptor selective antagonists.
Peptides are another source of highly selective κ opioid receptor antagonists. A number of analogues of the endogenous dynorphin peptides have demonstrated κ opioid receptor antagonism (see, for example, Lu et al., 2001; Bennett et al., 2002; Patkar et al., 2005), and such selective peptide antagonists would be expected to exhibit a limited duration of activity in vivo due to metabolism by proteases. Modifications incorporated into these peptides (e.g. cyclization) can slow proteolytic cleavage, providing sufficient metabolic stability to facilitate CNS penetration and central activity. This was recently demonstrated for zyklophin, a dynorphin A analogue and κ-receptor selective antagonist (Patkar et al., 2005), which blocked κ opioid receptors for less than 12 h, and prevented stress-induced reinstatement of extinguished cocaine conditioned place preference (CPP) after systemic administration (Aldrich et al., 2009). These promising results suggest that peptide κ opioid receptor antagonists can provide a desirable finite duration of κ opioid receptor-selective antagonism for optimal therapeutic value.
The cyclic tetrapeptide CJ-15,208 was reported to preferentially bind to κ opioid receptors and antagonize the effects of a κ opioid receptor agonist in vitro (Saito et al., 2002), but the stereochemistry of the Trp residue in this natural product was not determined. Therefore, we recently synthesized both possible isomers of this novel κ-receptor ligand (Figure 1) (Kulkarni et al., 2009; Ross et al., 2010) and found that the optical rotation for the L-Trp isomer was consistent with that reported for the natural product. Unexpectedly, both isomers bound to κ receptors with similar affinity (Dolle et al., 2009; Kulkarni et al., 2009; Ross et al., 2010).
Figure 1.

The structures of the cyclic tetrapeptides containing L-Trp and D-Trp.
We hypothesized that both isomers of CJ-15,208 would demonstrate short-acting, dose-dependent κ-receptor selective antagonism in vivo, thereby preventing reinstatement of extinguished cocaine-CPP. Surprisingly, the isomers demonstrated stereospecific opioid activity in vivo, with the unnatural D-Trp peptide primarily exhibiting κ opioid receptor antagonism and preventing stress-induced reinstatement of eliminated cocaine-seeking behaviour, while the L-Trp isomer was a mixed agonist/antagonist in vivo in the antinociceptive assay.
Methods
Animals and chemicals
The L-Trp and D-Trp isomers of the cyclic tetrapeptide CJ-15,208 (Figure 1) were synthesized as previously described (Ross et al., 2010); zyklophin ([N-benzylTyr1,cyclo(D-Asp,5,Dap8)]dynorphin A-(1–11) amide, Dap = 2,3-diaminopropionic acid) was also synthesized as previously described (Patkar et al., 2005). The κ opioid receptor agonist (+/−)-trans-3,4-dichloro-N-methyl-N-[2-(1-pyrrolidinyl)cyclohexyl]benzeneacetamide (U50 488) was provided by the NIDA Drug Supply Program (Bethesda, MD, USA). All other compounds were obtained from Sigma (St Louis, MO, USA).
Three hundred and seventeen adult male C57Bl/6J mice weighing 20–25 g were obtained from Jackson Labs (Bar Harbor, ME, USA). All animal care and experimental procedures were in accordance with the 2002 National Institute of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care Committee. All mice were housed in groups, four to a cage, in self-standing plastic cages within the animal care facility. The colony room was illuminated on a 12-h light–dark cycle, with the lights on at 07 h 00 min. Food pellets and distilled water were available ad libitum. Note that C57Bl/6J mice were selected for this study because of their established responses to stress and cocaine place-conditioning (McLaughlin et al., 2003; Carey et al., 2007; Aldrich et al., 2009).
Radioligand opioid receptor binding to CHO membranes
Opioid binding studies were performed on membranes derived from CHO cells stably expressing cloned rat κ opioid receptors, rat µ opioid receptors or mouse δ opioid receptors as described previously (Arttamangkul et al., 1997). Incubations with isolated membrane protein were performed in triplicate with 12 different concentrations from 0.1 nM to 10 µM of the cyclic tetrapeptides for 90 min in 50 mM Tris, pH 7.4 at 22°C using [3H]-diprenorphine (Kd 0.45 nM), [3H]-DAMGO ([D-Ala2,NMePhe4,glyol]enkephalin, Kd 0.49 nM) and [3H]-DPDPE (cyclo[D-Pen2,D-Pen5]enkephalin, Kd 1.76 nM) as the radioligands for κ, µ and δ opioid receptors, respectively. Non-specific binding was determined in the presence of 10 µM unlabelled dynorphin A-(1–13) amide (Dyn A-(1–13) amide), DAMGO and DPDPE for κ, µ and δ opioid receptors, respectively. Reactions were terminated by rapid filtration over Whatman GF/B fibre filters using a Brandel M24-R cell harvester and the filters counted in 4 mL of Cytocint (ICN Radiochemicals, Irvine, CA, USA) using a Beckman LS6800 scintillation counter (Beckman Instruments, Fullerton, CA, USA).
[35S]-GTPγS binding to CHO membranes
The binding of the GTP analogue [35S]-GTPγS to membranes was assayed following the method described by Siebenallar and Murray (1999). Binding was determined in a volume of 500 µL. The assay mixture contained 50 mM HEPES, pH 7.4, 1 mM EDTA, 5 mM magnesium acetate, 1 µM GDP, 1 mM dithiothreitol, 100 mM NaCl, 1 mg BSA mL−1 and approximately 100 000 dpm [35S]-GTPγS (0.1 to 0.2 nM). Approximately 10 µg of κ or µ opioid receptor-expressing CHO cell membrane protein was used per tube. Following a 90 min incubation at 22°C, the assay was terminated by filtration under vacuum on a Brandel (Gaithersburg, MD, USA) model M-48R cell harvester using Schleicher and Schuell Inc. (Keene, NH, USA) number 32 glass fibre filters. The filters were rinsed with four 4 mL washes of ice-cold 50 mM Tris HCl pH 7.4 containing 5 mM MgCl at 5°C to remove unbound [35S]-GTPγS. Filter disks were then placed into counting vials to which 8 mL of Biocount scintillation fluid (Research Products International Corp., Mount Prospect, IL, USA) was added. Filter-bound radioactivity was determined by liquid scintillation spectrometry (Beckman Instruments) following overnight extraction at room temperature. Note that the amount of radioligand bound was less than 10% of the total added in all experiments. Specific binding was defined as total binding minus that occurring in the presence of 3 µM unlabelled GTPγS. Non-specific binding was approximately 1% of the total binding at 0.1 nM [35S]-GTPγS.
I.c.v. administration technique
The intracerebroventricular (i.c.v.) injections were made directly into the lateral ventricle according to the modified method of Haley and McCormick (1957). The volume of all i.c.v. injections was 5 µL. The mouse was lightly anaesthetized with isoflurane, an incision made in the scalp, and the injection made 2 mm lateral and 2 mm caudal to bregma at a depth of 3 mm using a 10 µL Hamilton syringe.
Antinociceptive testing
The 55°C warm-water tail-withdrawal assay was performed in C57Bl/6J mice as previously described (McLaughlin et al., 1999). Briefly, warm (55°C) water in a 2 L heated water bath was used as the thermal nociceptive stimulus, with the latency of the mouse to withdraw its tail from the water taken as the endpoint. After determination of baseline tail-withdrawal latencies (1.79 ± 0.03 s across all tested subjects), mice were administered a graded dose of the L-Trp or D-Trp isomer i.c.v.; the cyclic tetrapeptides were administered in 50% dimethyl sulphoxide (DMSO) in sterile saline (0.9%). To determine agonist activity, the tail-withdrawal latency was determined repeatedly every 10 min following administration of a cyclic tetrapeptide for 1 h or until latencies returned to baseline values. To determine antagonist activity, mice were pretreated with the L-Trp or D-Trp isomer 80 min before administration of the µ opioid receptor-preferring agonist morphine (10 mg·kg−1, i.p.), κ opioid receptor-selective agonist U50,488 (10 mg·kg−1, i.p.) or δ opioid receptor-selective agonist SNC-80 (100 nmol, i.c.v.). Antinociception produced by these established agonists was then measured 40 min after their administration. To determine the duration of κ opioid receptor antagonist activity, additional mice were pretreated for 7.3, 17.3, 23.3 or 47.3 h before the administration of U50,488 as described previously.
To determine the opioid receptor selectivity of the agonist activity of the L-Trp isomer, mice were pretreated with a single dose of β-FNA (5 mg·kg−1, s.c.) or nor-BNI (10 mg·kg−1, i.p.) 23.3 h in advance of administration of the L-Trp isomer (10 nmol, i.c.v). Additional mice were pretreated 30 min prior to the administration of the L-Trp isomer with the opioid-receptor non-selective antagonist naloxone (10 mg·kg−1, s.c.), μ opioid receptor-selective antagonist CTAP (1 nmol, i.c.v.), κ opioid receptor-selective antagonist zyklophin (3 mg·kg−1, s.c.) or δ opioid receptor-selective antagonist naltrindole (20 mg·kg−1, i.p.), with antinociceptive testing 40 min later. Reference agonists and antagonists were administered using sterile saline (0.9%) as the vehicle, except for SNC-80 which was dissolved in 35% DMSO in sterile saline (0.9%).
A cut-off time of 15 s was used in this study; if the mouse failed to display a tail-withdrawal response during that time, the tail was removed from the water and the animal was assigned a maximal antinociceptive score of 100%. At each time point, antinociception was calculated according to the following formula:
Cocaine CPP, extinction and reinstatement
Conditioned Place Preference (CPP)
Mice were conditioned based on a previously established biased cocaine CPP paradigm (Carey et al., 2007; Aldrich et al., 2009; see also Figure 7A). This has been demonstrated to be an effective protocol for the study of extinction and reinstatement (Szumlinski et al., 2002; Carey et al., 2007). Individual mice were allowed to run freely between the three linear compartments comprising the apparatus (Carey et al., 2007) for the 30 min testing period. Time spent in each compartment was measured, and the initial compartment preference of each mouse was determined. Notably, the mice initially demonstrated an equivalent amount of time spent on average in each of the two outer conditioning compartments (625 ± 16 and 586 ± 14 s in the left and right compartments, respectively, P= 0.07, n.s., Student's t-test). The mice were subsequently place-conditioned immediately following administration of cocaine (10 mg·kg−1, s.c.) and confined to the appropriate outer compartment starting on day 2. Place-conditioning in the opposite outer compartment initially preferred by the mouse was performed daily with vehicle (0.9% saline, 0.3 mL per 30 g body weight, s.c.) 4 h after the cocaine conditioning. This place-conditioning cycle was repeated once each day on days 3–5 (see Figure 7A), and on day 6 the animals were tested for a final place preference. Data are exhibited as the difference in time spent on cocaine- and vehicle-paired sides (Figure 7B). By convention, the initial bias generates a negative value, and a positive value represents a conditioned preference for the cocaine-paired side. Conditioned place aversion, where the animals avoid the drug-paired compartment and spend a significantly greater time in the saline-paired compartment than initially demonstrated, was not detected in this study under any conditions.
Figure 7.

Stress-induced reinstatement of cocaine-CPP prevented by pretreatment with the D-Trp cyclic tetrapeptide. (A) Schematic representation of reinstatement and testing protocol. Vehicle (0.9% saline) or the D-Trp cyclic tetrapeptide (3 nmol, i.c.v) were administered on days 28 and 29, 20 min before the initial exposure to forced swim stress; for cocaine place conditioning, the mice were also treated on days 28 and 29 with vehicle or peptide, followed by cocaine place conditioning 20 min after the injection on day 29. (B) Following 4 days of cocaine administration (10 mg·kg−1, s.c. daily), mice exhibited significant preference for the cocaine-paired environment, with extinction occurring 3 weeks later. Mice were then exposed to forced swim stress or another round of cocaine place conditioning, reinstating preference. Pretreatment with the D-Trp cyclic tetrapeptide (3 nmol, i.c.v.) prevented stress-induced reinstatement of place preference. However, pretreatment with the D-Trp peptide (3 nmol, i.c.v.) was ineffective at blocking cocaine-induced reinstatement. Columns represent means of n= 8–17 mice; cocaine place conditioning data in the left panel represents combined responses of all 81 mice tested. * Significantly different from preconditioning place preference response; † significantly different from post-conditioning place preference response; ‡ significantly different from stress-induced reinstatement of place preference response, Tukey's HSD post hoc test.
Extinction
Preference tests were completed twice weekly for 30 min until extinction was established (see Figure 7A). Extinction is defined as a statistically significant decrease in the time spent in the cocaine-paired compartment during the extinction trial as compared with the post-conditioning response after the initial 4 days of conditioning (Szumlinski et al., 2002; Brabant et al., 2005; Carey et al., 2007). As expected for the C57Bl/6J strain of mice, CPP responses showed extinction with biweekly testing over a 3 week period.
Reinstatement
Following the demonstration of extinction, reinstatement of cocaine CPP was examined after either exposure to forced swim stress (see below) or an additional cycle of cocaine place-conditioning (see Figure 7A) as described previously (Carey et al., 2007; Aldrich et al., 2009). Half the tested mice were pretreated with either vehicle (i.c.v.) or the D-Trp cyclic tetrapeptide (3 nmol, i.c.v.) daily 20 min prior to forced swimming (see below). Additional mice were also administered vehicle (i.c.v.) or the D-Trp cyclic tetrapeptide (3 nmol, i.c.v.) on days 28 and 29, and 20 min after the final administration of vehicle or the peptide on day 29 an additional session of cocaine place conditioning was performed. On the day following the completion of stress exposure or cocaine place-conditioning, mice were tested for place preference (see Figure 7A).
Forced swim stress
To produce stress-induced reinstatement of cocaine CPP, a 2 day forced swim stress protocol was used (McLaughlin et al., 2003; Carey et al., 2007; Aldrich et al., 2009). For each of the 2 days, mice were pretreated with either vehicle or the D-Trp cyclic tetrapeptide 20 min before exposure to forced swim stress (see Figure 7A). The day after the final exposure to swim stress, the place preference response of each mouse was tested to evaluate reinstatement of the extinguished cocaine CPP.
Statistical analysis
Radioligand equilibrium competition binding and agonist stimulated [35S]-GTPγS binding results represent the mean ± SEM obtained from three to five independent experiments each performed in triplicate. IC50 values were determined by non-linear regression analysis to fit a logistic equation to the competition data using GraphPad Prism software (GraphPad Software, La Jolla, CA, USA). The Ki values of unlabelled compounds were calculated from the Cheng and Prusoff equation Ki= IC50/(1 +S), where S= (concentration of radioligand)/(KD of radioligand) (Cheng and Prusoff, 1973), using Kd values of [3H]-diprenorphine, [3H]-DAMGO and [3H]-DPDPE as listed above. The KB values for the inhibition of opioid agonist-induced stimulation of [35S]-GTPγS binding by the cyclic tetrapeptides were calculated by Schild analysis using the equation: log (DR-l) = log [I]− log KB, where [I] is the concentration of peptide, KB is the equilibrium dissociation constant of the peptide and DR is the dose-ratio shift produced by the peptide (Arunlakshana and Schild, 1959).
Student's t-tests and anova (specifically using repeated measures anova for data in Figure 2) with Tukey's HSD post hoc tests were used to compare baseline and post-treatment tail-withdrawal latencies and to determine statistical significance for all tail-withdrawal data. Tail-withdrawal data points shown are the means of 7–12 mice, with SEM represented by error bars. Potency was quantitified by calculating ED50 values with Prism 5.0 software (GraphPad Software, La Jolla, CA, USA). Data for CPP experiments were analysed with anova using the SPSS 14.0 statistical package (Chicago, IL, USA). Analyses examined the main effect of CPP phase (e.g. post-conditioning, week of preference test, reinstatement) and the interaction of drug pretreatment (D-Trp cyclic tetrapeptide or vehicle) × reinstatement condition (stress or cocaine exposure). Significant effects were further analysed using Tukey's HSD post hoc test. All data are presented as mean ± SEM, with significance set at P < 0.05.
Figure 2.

The antinociceptive activity of the L-Trp cyclic tetrapeptide was assessed in vivo following i.c.v. (1–10 nmol) administration in the 55°C warm-water tail-withdrawal assay in C57Bl/6J mice with repeated measurement over time. The antinociceptive activity of the D-Trp isomer was also assessed in vivo following i.c.v. (3 and 10 nmol, circles) administration in the same assay. The i.c.v. administration of vehicle alone (50% DMSO in sterile saline) had no significant effect after an initial small response at 10 min post-injection. Antinociception was calculated at each time point according to the following formula: % antinociception = 100 × (test latency − control latency) / (15 − control latency). Mean % antinociception ± SEM from eight mice for each set is presented. *Significantly different from baseline response P < 0.05; repeated measures anova with Tukey's HSD post hoc test.
Results
Affinity and selectivity of L-Trp and D-Trp cyclic tetrapeptides for multiple opioid receptors in CHO membranes
The affinities of the L-Trp and D-Trp cyclic tetrapeptide isomers (Figure 1) for the multiple opioid receptors were measured in vitro by determining the Ki values for the inhibition of κ-, µ- and δ-opioid receptor binding in membranes from CHO cells stably expressing individual receptors (Table 1). Both isomers showed the highest (and equivalent) affinity for κ opioid receptors, with Ki values of 35.4 ± 3.6 and 30.6 ± 3.4 nM, respectively. The affinities for µ receptors were 17- and 8-fold lower than for κ receptors for the peptides containing L-Trp and D-Trp, respectively. Both peptides exhibited very low (micromolar) affinity for δ receptors (Table 1).
Table 1.
Opioid receptor binding affinities of the L-Trp and D-Trp cyclic tetrapeptide isomers for κ, µ and δ opioid receptors in CHO membranes
| Ki (nM ± SEM) | Ki ratio | |||
|---|---|---|---|---|
| Compound | κ | µ | δ | κ/µ/δ |
| cyclo[Trp-Phe-D-Pro-Phe] (L-Trp isomer) | 35.4 ± 3.6 | 619 ± 87 | 4150 ± 3020 | 1/17.5/117 |
| cyclo[D-Trp-Phe-D-Pro-Phe] (D-Trp isomer) | 30.6 ± 3.4 | 259 ± 29 | 2910 ± 1350 | 1/8.5/95 |
CHO membranes expressing cloned rat κ, rat µ or mouse δ opioid receptors were incubated for 90 min with 12 different concentrations of each cyclic tetrapeptide in the presence of [3H]-diprenorphine, [3H]-DAMGO or [3H]-DPDPE (for κ, µ and δ, respectively) in 50 mM Tris-HCl, pH 7.4, at 22°C as described in Methods. Data are listed as the mean Ki values ± SEM from three to five experiments, performed in triplicate.
Antagonist activity of L-Trp and D-Trp cyclic tetrapeptides at κ and µ receptors in CHO membranes in the [35S]-GTPγS binding assay
Neither cyclic tetrapeptide alone demonstrated appreciable agonist activity [<10% relative to the full agonists Dyn A(1–13) amide and DAMGO] at κ and µ receptors when tested at concentrations up to 1 µM in this assay (Table 2). In contrast, both the L-Trp and D-Trp isomers antagonized the agonist activity of Dyn A(1–13) amide, with KB values of 62.8 ± 0.8 and 22.8 ± 13.4 nM, respectively (Table 2). Interestingly, while the D-Trp isomer showed no antagonist activity at µ receptor, the L-Trp isomer demonstrated µ receptor antagonist activity, inhibiting DAMGO activity with a KB value of 10.3 ± 1.8 nM (Table 2). The relative potencies of the L-Trp isomer as an antagonist at κ and µ receptors, and the antagonist activity of the D-Trp isomer at κ receptors are in agreement with the results reported by Dolle et al. (2009) in this assay. These researchers, however, reported that the D-Trp isomer was also a potent antagonist at µ receptors. The reasons for this difference are unclear, but could be due to differences in the assay procedures, in particular species differences in the receptors and/or the agonist used to demonstrate µ receptor activity.
Table 2.
Opioid antagonist activity of the L-Trp and D-Trp cyclic tetrapeptide isomers at κ and µ opioid receptors in the [35S]-GTPγS assaya
| Compound | κKB (nM ± SEM) | µKB (nM ± SEM) |
|---|---|---|
| cyclo[Trp-Phe-D-Pro-Phe] (L-Trp isomer) | 62.8 ± 0.8 | 10.3 ± 1.8 |
| cyclo[D-Trp-Phe-D-Pro-Phe] (D-Trp isomer) | 22.8 ± 13.4 | b |
Neither the L-Trp nor the D-Trp isomer alone exhibited appreciable agonist activity (<10% efficacy relative to the full agonists dynorphin A-(1–13) amide and DAMGO, respectively) at κ or µ receptors. Note that evaluation of potential agonist activity of the cyclic tetrapeptides was performed in the absence of reference opioid agonists for κ and µ receptors.
No antagonist activity observed at concentrations up to 300 nM.
CHO membranes expressing cloned rat κ or µ receptors were incubated for 90 min with a range of dynorphin A-(1–13) amide or DAMGO concentrations, respectively, at 22°C either alone or in the presence of one of four different concentrations of each cyclic tetrapeptide, as described in Methods. These data represent the mean KB values ± SEM from two experiments, each performed in triplicate, which were derived from a Schild regression.
Agonist activity of L-Trp and D-Trp cyclic tetrapeptides in the 55°C warm-water tail-withdrawal assay
The in vivo effects of the L-Trp and D-Trp isomers of CJ-15,208 were examined in C57Bl/6J mice using the 55°C warm-water tail-withdrawal assay. Intracerebroventricular administration of the D-Trp isomer (3 nmol) did not significantly change the tail-withdrawal latency from baseline [F(4,28)= 0.17, P= 0.95; repeated measures one-way anova with Tukey's HSD post hoc test], although a higher dose (10 nmol) slightly (≤10%) but significantly increased the tail-withdrawal latency above baseline for up to 50 min (Figure 2, F(5,35)= 14.7, P < 0.0001; repeated measures one-way anova with Tukey's HSD post hoc test). Notably, i.c.v. administration of vehicle alone produced a slight antinociceptive effect (3.9%) that was significant (F(6,42)= 3.71, P= 0.005; repeated measures one-way anova with Tukey's HSD post hoc test) only in the first 10 min following administration (Figure 2, grey diamonds). In contrast, administration of the L-Trp isomer (1, 3 or 10 nmol, i.c.v.) produced statistically significant (F(7,49)= 5.39 and 8.35, P < 0.0001 for 1 and 3 nmol doses, respectively, and F(11,77)= 155.2, P < 0.0001 for the 10 nmol dose; all repeated measures one-way anova with Tukey's HSD post hoc test), dose-dependent antinociception (Figure 2). The L-Trp isomer produced antinociception with an ED50 value (and 95% CI) of 3.71 (0.90–6.54) nmol that lasted for up to 100 min. The surprising antinociceptive effects of the L-Trp isomer appeared to be mediated by opioid receptors, as a 20 min pretreatment with the non-selective opioid receptor antagonist naloxone (10 mg·kg−1, s.c.) significantly reduced the tail-withdrawal latency of mice treated 20 min before testing with a 10 nmol dose of the L-Trp cyclic tetrapeptide (from 14.1 ± 0.9 to 2.14 ± 0.22 s; P= 0.002).
As the L-Trp isomer produced a robust antinociceptive effect, the opioid receptor mediation and selectivity of the L-Trp isomer-induced antinociception was determined following pretreatment with opioid receptor-selective antagonists (Figure 3). Pretreatment with the κ-receptor selective antagonists nor-BNI or zyklophin (Patkar et al., 2005; Aldrich et al., 2009) each significantly antagonized the L-Trp isomer-mediated antinociception (Figure 3). In addition, pretreatment with the µ-receptor selective antagonists β-FNA or CTAP both partially reduced the antinociception induced by the L-Trp cyclic tetrapeptide. Together, these results suggest that the L-Trp isomer acts as a combined κ- and µ-receptor agonist. Notably, pretreatment of the mice with the δ-receptor selective antagonist naltrindole did not significantly change the antinociceptive response of the L-Trp isomer (Figure 3).
Figure 3.

κ- and µ-receptor selective agonism by the L-Trp cyclic tetrapeptide. Antinociceptive activity of the L-Trp isomer (10 nmol, i.c.v.) was assessed 24 h after pretreatment of the mice with β-FNA (5 mg·kg−1, s.c.) or nor-BNI (10 mg·kg−1, i.p.). Additional mice were pretreated with CTAP (1 nmol, i.c.v., −25 min), zyklophin (3 mg·kg−1, s.c., −75 min) or naltrindole (20 mg·kg−1, i.p., −15 min) before administration of the L-Trp cyclic tetrapeptide. Tail-withdrawal latencies were measured in the mouse 55°C warm-water tail-withdrawal test 40 min after injection of the L-Trp peptide (10 nmol, i.c.v.). Mean % antinociception ± SEM from 8–12 mice is presented. * Significantly different from baseline response P < 0.05; † significant difference from the response of the L-Trp isomer alone (P < 0.05); Student's t-test.
Antagonist activity of the L-Trp and D-Trp cyclic tetrapeptides against the antinociceptive activity of opioid-receptor selective agonists
The in vivo effects of the L-Trp and D-Trp isomers on antinociception mediated by opioid-receptor selective agonists were next evaluated at time points after the dissipation of agonist activity. Intracerebroventricular pretreatment with either cyclic tetrapeptide (3 nmol) 80 min before agonist administration significantly antagonized the antinociceptive effects of the κ-receptor selective agonist U50,488, but not of the µ-receptor-preferring agonist morphine or the δ-receptor- selective agonist SNC-80 (Figure 4A,B). Both cyclic tetrapeptides antagonized κ opioid receptors in a dose-dependent manner (Figure 5) that was statistically significant (F(4,39)= 27.7, P < 0.0001 and F(3,32)= 19.7, P < 0.0001 for the L-Trp and D-Trp isomers, respectively; both one-way anova with Tukey's HSD post hoc test), although the D-Trp isomer (Figure 5B) demonstrated greater potency as an antagonist than the L-Trp peptide (Figure 5A).
Figure 4.

κ opioid receptor-selective antagonism by the L-Trp cyclic tetrapeptide (A) or D-Trp isomer (B). Antinociceptive activity of morphine (10 mg·kg−1, i.p.) and SNC-80 (100 nmol, i.c.v.) was not reduced by pretreatment of the mice for 80 min with the L-Trp cyclic tetrapeptide (3 nmol, i.c.v., A) or the D-Trp isomer (3 nmol, i.c.v., B), whereas the antinociceptive effect of U50,488 (10 mg·kg−1, i.p.) was significantly antagonized by pretreatment of the mice for 80 min with both the L-Trp peptide (3 nmol, i.c.v., A) and the D-Trp isomer (3 nmol, i.c.v., B). Tail-withdrawal latencies were measured in the mouse 55°C warm-water tail-withdrawal test 40 min after injection of the known selective agonists. Whether the listed agent was administered or not is indicated by plus and minus signs under the columns. Mean % antinociception ± SEM from 8–12 mice is presented. * Significantly different from response of agonist alone, P < 0.05; Student's t-test.
Figure 5.
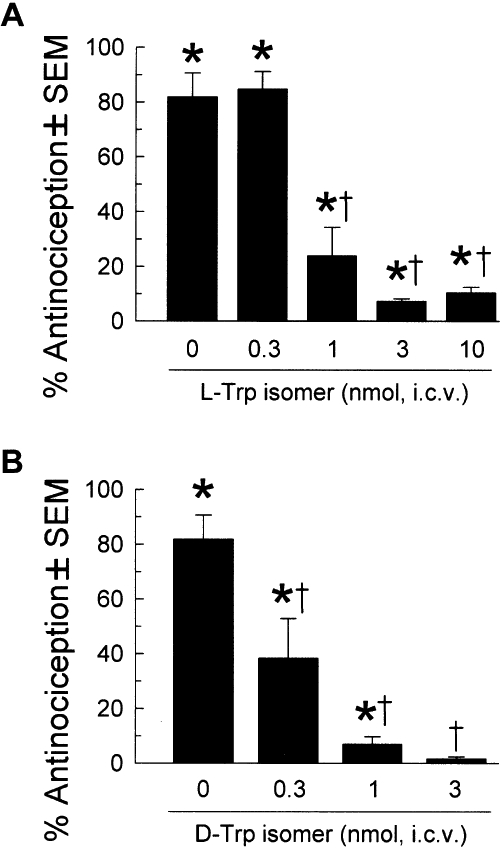
Dose-dependent antagonism of U50,488-induced antinociception by (A) the L-Trp cyclic tetrapeptide or (B) the D-Trp isomer. The antinociceptive effects of U50,488 (10 mg·kg−1, i.p.) were determined 40 min after administration in mice pretreated for 2 h with vehicle, L-Trp cyclic tetrapeptide (0.3–10 nmol, i.c.v., A) or the D-Trp isomer (0.3–10 nmol, i.c.v., B). Mean % antinociception ± SEM from 7–12 mice is presented. * Significantly different from baseline response (P < 0.05; Student's t-test); † significant difference from the response of U50,488 alone (P < 0.05; one-way anova with Tukey's HSD post hoc test).
The duration of κ opioid receptor antagonism produced by a single dose of either isomer (3 nmol, i.c.v.) was next determined (Figure 6). Mice were pretreated with one of the two isomers for 1.3 to 47.3 h before administration of U50,488 (10 mg·kg−1, i.p.), and antinociception was measured in the mouse 55°C warm-water tail withdrawal assay. Pretreatment with the L-Trp isomer significantly (F(5,52)= 24.7, P < 0.0001; one-way anova with Tukey's HSD post hoc test) antagonized U50,488-induced antinociception for at least 24 h, but for less than 48 h (Figure 6A). The D-Trp isomer exhibited a shorter duration of activity, with significant (F(4,38)= 11.7, P < 0.0001; one-way anova with Tukey's HSD post hoc test) antagonism of U50,488-induced antinociception for at least 8 h, but for less than 18 h (Figure 6B). These data demonstrate a reversible, short duration of κ opioid receptor antagonism mediated by the cyclic tetrapeptides. These results are similar to those reported with the dynorphin A- based antagonist zyklophin (Aldrich et al., 2009), but contrast with the exceptionally long activity observed from a single dose of the established κ-receptor selective non-peptide antagonists (Metcalf and Coop, 2005).
Figure 6.

Time course of (A) the L-Trp cyclic tetrapeptide or (B) the D-Trp isomer-mediated antagonism of U50,488-induced antinociception in the mouse 55°C warm-water tail-withdrawal test. Antinociception of U50,488 (10 mg·kg−1, i.p.) in mice pretreated for 1–48 h with the L-Trp cyclic tetrapeptide (3 nmol, i.c.v., A) or the D-Trp isomer (3 nmol, i.c.v., B). Tail-withdrawal latencies were determined 40 min after agonist administration. Mean % antinociception ± SEM from 7–12 mice is presented. * Significantly different from the baseline tail-withdrawal latency (P < 0.05; Student's t-test); † significantly different from U50,488 alone (P < 0.05; one-way anova with Tukey's HSD post hoc test).
Activity of the D-Trp cyclic tetrapeptide in a CPP model of stress-induced reinstatement of cocaine-seeking behaviour
Previously, we demonstrated that the selective peptide κ-receptor antagonists arodyn (Ac[Phe1,2,3,Arg4,D-Ala8]dynorphin A-(1–11) amide; Carey et al., 2007) and zyklophin (Aldrich et al., 2009) can suppress stress-induced (but not drug primed) reinstatement of cocaine-seeking behaviour previously extinguished in mice. Therefore, we expected that the selective κ opioid receptor antagonism with minimal agonist activity produced by pretreatment with the D-Trp isomer would likewise block stress-induced reinstatement. To test this hypothesis, C57Bl/6J mice were first place conditioned over 4 days with cocaine (see Figure 7A). Mice demonstrated a cocaine CPP that was significantly greater than that of the initial preference (Figure 7B, lefthand columns; F(4,290)= 20.73, P < 0.0001; one-way anova with Tukey's HSD post hoc test). This place preference lasted more than 2 weeks (Figure 7B, dark grey column, left), demonstrating extinction after 3 weeks with a place preference response similar to the initial preference response, but significantly less than the preference immediately after place conditioning (Figure 7B, light grey bar, P < 0.01).
Following the extinction of cocaine CPP, mice were administered vehicle or the D-Trp cyclic tetrapeptide (3 nmol, i.c.v.) daily for 2 days (see Figure 7A) and exposed 20 min later to repeated forced swim stress. The day following the completion of exposure to stress (i.e. day 30, see Figure 7A), mice were tested for place preference to examine possible reinstatement of drug-seeking behaviour. The vehicle-pretreated mice exposed to swim stress subsequently demonstrated the expected reinstatement of CPP (Figure 7B, striped purple column, centre, F(4,202)= 20.5, P < 0.0001; one-way anova with Tukey's HSD post hoc test). In contrast, pretreatment with the D-Trp isomer prevented stress-induced reinstatement. Mice pretreated daily with the D-Trp isomer prior to exposure to forced swimming demonstrated place preference responses that were not significantly different from preconditioning or extinguished responses (Figure 7B, orange column, centre). Additional mice demonstrating extinction of CPP were exposed to a single cycle of cocaine conditioning before the place preference testing (see Figure 7A). Cocaine-exposed mice pretreated with vehicle exhibited reinstatement of place preference (Figure 7B, thatched right column, F(4,196)= 28.88, P < 0.0001; one-way anova with Tukey's HSD post hoc test). Mice treated daily for 2 days with the D-Trp isomer before exposure to the additional cycle of cocaine place-conditioning still displayed a significantly greater preference for the cocaine-paired compartment as compared with pre-conditioning and extinguished preferences (Figure 7B, rightmost column, P < 0.01). Moreover, the reinstated preference of the mice pretreated with the D-Trp isomer was not significantly different from the response of vehicle pretreated mice (Figure 7B, rightmost columns).
Discussion and conclusions
The cyclic tetrapeptide CJ-15,208 was of interest due to its reported κ opioid receptor antagonist activity in vitro (Saito et al., 2002). This peptide was found to preferentially bind to κ receptors and antagonize the κ-receptor agonist asimadoline in vitro in the electrically stimulated rabbit vas deferens, but the stereochemistry of the Trp residue in this natural product was not determined. While there was a brief reference to the cyclic tetrapeptide containing L-Trp in a conference proceedings (Seale et al., 2000), no evidence was provided to establish whether this isomer was the natural product. In order to determine conclusively the structure of the natural product, we synthesized both possible isomers varying the stereochemistry of the Trp residue (Kulkarni et al., 2009; Ross et al., 2010) and found that the optical rotation for the L-Trp isomer was consistent with that reported for the natural product (Ross et al., 2010). Therefore, we expected the L-Trp isomer to exhibit κ opioid receptor affinity and antagonist activity, while the unnatural D-Trp isomer was expected to display much lower or negligible activity. However, both isomers bound to κ receptors with similar affinities (Dolle et al., 2009; Kulkarni et al., 2009; Ross et al., 2010). This was not due to epimerization of the Trp residue during synthesis; the L- and D-Trp cyclic tetrapeptides were easily separated by HPLC, and the epimer was not detected following cyclization when Trp was the C-terminal residue (Ross et al., 2010). Interestingly, these two peptides were recently reported to be antagonists of both κ and µ opioid receptors in the GTPγS assay (Dolle et al., 2009), with substantially lower IC50 values for antagonism of µ than κ receptors in spite of their higher affinity for κ receptors in binding assays.
Not only did both peptides exhibit opioid activity in vitro, but here we report that they also unexpectedly exhibited stereospecific differences in vivo. In contrast to the reported activity in vitro (Table 2, and also Saito et al., 2002 and Dolle et al., 2009), the L-Trp isomer demonstrated mixed agonist activity in vivo mediated by both κ and µ receptors in addition to κ-receptor selective antagonism. Interestingly, the unnatural D-Trp isomer produced the expected selective κ-receptor antagonist activity with minimal agonist activity in vivo. While changes in stereochemistry have been shown in a number of cases to affect the potency of opioid ligands [e.g. in the κ-receptor agonist salvinorin A (Beguin et al., 2006)], changes in the type of activity observed (agonism vs. antagonism) are rare (Schiller et al., 1992). Thus, the differences in the pharmacological profiles of the two peptides with different stereochemistries at the Trp residue, with only one isomer exhibiting appreciable agonist activity in the antinociceptive assay, are unusual among opioid ligands.
The development of peptide ligands selective for κ-receptors potentially offers several advantages, including high activity and specificity, low toxicity, and the minimization of drug–drug interactions (Marx, 2005). A number of peptides with favourable pharmacokinetic properties have been developed successfully as therapeutic agents (Vlieghe et al., 2010), and alternative delivery methods [such as inhalation, which has been utilized to administer dynorphin A-(1–13) derivatives to rats (Brugos et al., 2004)] could enhance the clinical acceptance of peptides. While endogenous peptides such as dynorphin A undergo rapid metabolism (Reed et al., 2003; Brugos and Hochhaus, 2004; Klintenberg and Andren, 2005) that typically preclude their therapeutic use, structural modifications (e.g. D- and unnatural amino acids) can result in peptides with sufficient metabolic stability to allow penetration of the CNS and production of pharmacological effects. This has been demonstrated with E-2078, a metabolically stable analogue of dynorphin A-(1–8) (Terasaki et al., 1991; Yu et al., 1997) which exhibits analgesic activity in humans (Fujimoto and Momose, 1995). Recently, we have demonstrated that the dynorphin A-(1–11) analogue zyklophin (Patkar et al., 2005) is a systemically active κ-receptor selective antagonist that appears to penetrate the CNS (Aldrich et al., 2009).
While peptide antagonists that are derivatives of the endogenous opioid peptide dynorphin A such as arodyn (Bennett et al., 2002) and zyklophin (Patkar et al., 2005) are stabilized to metabolism compared with the parent peptide (Aldrich et al., 2007), they remain subject to eventual proteolytic degradation. Such degradation is expected to generate peptides with a much shorter κ-receptor antagonism compared with the exceptionally long duration of activity seen with the established κ-receptor selective non-peptide antagonists (Metcalf and Coop, 2005). A single dose of the non-peptide κ-receptor selective antagonist nor-BNI (1 nmol, i.c.v.) blocked the antinociceptive effects of κ-receptor agonists U69,593 and bremazocine for up to 28 days (Horan et al., 1992). In contrast, arodyn produced potent κ-receptor selective antagonism lasting for up to 3 days (Carey et al., 2007), and zyklophin (Aldrich et al., 2009) produced κ-receptor selective antagonism that lasted for up to only 12 h in the mouse 55°C warm-water withdrawal assay.
Head-to-tail cyclic peptides such as CJ-15,208 are expected to exhibit significantly enhanced pharmacokinetic properties compared with linear peptides, especially resistance to proteolytic degradation (Delaforge et al., 1997). A key question for this study was whether the cyclic tetrapeptide κ-receptor antagonists would behave more like the derivatives of the linear peptide dynorphin or the non-peptide antagonists. The data demonstrate that both the L-Trp and D-Trp cyclic tetrapeptides produced relatively short durations of κ-receptor antagonist activity (Figure 4), with the κ-receptor selective antagonism mediated by the D-Trp isomer lasting approximately 8 h. Thus, the time course of the κ-receptor antagonist activity of these peptides is comparable with that of the other selective peptide κ-receptor antagonists, making these compounds potentially favourable for clinical development as therapeutic agents. The present data justify further examination of the distribution and pharmacokinetics of these cyclic tetrapeptides to determine other parameters affecting their activity.
The ability of the D-Trp isomer to block stress-induced, but not drug-primed, reinstatement of extinguished cocaine CPP in mice (Figure 7B) is also consistent with the results with other κ-receptor selective antagonists, including arodyn (Carey et al., 2007) and zyklophin (Aldrich et al., 2009). Increasing evidence suggests that the endogenous activation of κ receptors modulates an organism's response to stress (Takahashi et al., 1990; McLaughlin et al., 2003; Valdez et al., 2007), probably due to the stress-induced release of dynorphin peptides (Nabeshima et al., 1992; Shirayama et al., 2004). Stress is known to potentiate the rewarding properties of drugs of abuse (Piazza et al., 1990; Covington and Miczek, 2001) and promotes the reinstatement of drug-seeking behaviour in abstinent subjects (McMahon, 2001). Prevention of κ opioid receptor signalling upon exposure to stress, either by κ-receptor selective antagonist pretreatment or disruption of the κ opioid receptor gene reduced immobility in a mouse forced swim stress test (Mague et al., 2003; McLaughlin et al., 2006) and blocked both stress-induced potentiation of cocaine CPP (McLaughlin et al., 2003; 2006) and stress-induced reinstatement of extinguished cocaine CPP (Beardsley et al., 2005; Carey et al., 2007; Redila and Chavkin, 2008; Aldrich et al., 2009). In our reinstatement model, this antagonism of κ receptors results in the blockade of stress-induced, but not drug-primed, reinstatement of CPP previously extinguished in mice. These promising results add to existing data to suggest that peptide κ-receptor selective antagonists may serve as clinically relevant therapeutics for the maintenance of abstinence from cocaine abuse.
Results from the mouse 55°C warm-water tail-withdrawal assay indicated that the L-Trp isomer initially displayed opioid agonist activity. The antinociceptive activity, which was both time- and dose-dependent, was mediated by both κ and, apparently more modestly, by µ opioid receptors. Subsequently, the L-Trp isomer selectively reversed the antinociceptive activity of U50,488 for up to 48 h. This profile of activity is of considerable interest. Ligands with mixed opioid agonist/antagonist activity have been reported to produce antinociception with lower abuse liability. Nalbuphine, a mixed-action κ-receptor agonist/µ-receptor antagonist analgesic which is approximately equipotent to morphine for pain control (Miller, 1980), has shown lower abuse liability when tested in human populations (Schmidt et al., 1985). It was also recently reported that the mixed κ/µ receptor agonist nalbuphine may decrease cocaine intake in animal models of self-administration (Mello et al., 2005). Thus, compounds such as the L-Trp cyclic tetrapeptide with a mixed opioid activity profile could have clinically relevant application as low abuse liability analgesics, although more study is required. Further ongoing studies of this novel cyclic tetrapeptide analgesic are expected to provide insight into the mechanisms governing its activity.
In conclusion, these studies demonstrate that the Trp isomers of CJ-15,208 have applications as both pharmacological tools and as lead compounds for potential therapeutic development. The D-Trp isomer represents a significant advance in the development of κ-receptor selective antagonists. The κ receptor selectivity and relatively short duration of antagonist activity produced by this cyclic tetrapeptide make it a promising lead compound for maintaining abstinence from substance abuse. Given the lack of Food and Drug Administration-approved medications for the treatment of cocaine abuse or to prevent relapse to cocaine use in abstinent former users, the findings here add to a growing body of evidence (Beardsley et al., 2005; Carey et al., 2007; Aldrich et al., 2009), suggesting the value offered by κ-receptor selective antagonists to maintain abstinence from cocaine abuse. The L-Trp isomer represents a novel lead compound for the potential development of low abuse liability analgesics. Additional studies of both of these cyclic tetrapeptides are ongoing in our laboratories.
Acknowledgments
We gratefully acknowledge the synthesis of the L-Trp and D-Trp isomers by Santosh S. Kulkarni and Sanjeewa N. Senadheera. This work was supported by NIDA grants R01 DA018832 and DA023924 (to JVA) and the State of Florida, Executive Office of the Governor's Office of Tourism, Trade, and Economic Development (to JPM).
Glossary
- Arodyn
Ac[Phe1,2,3,Arg4,D-Ala8]dynorphin A-(1-11) amide CHO cells
- β-FNA
β-funaltrexamine
- CPP
conditioned place preference
- CTAP
H-D-Phe-cyclo[Cys-Tyr-D-Trp-Arg-Thr-Pen]-Thr-NH2
- DAMGO
[D-Ala2,NMePhe4,glyol]enkephalin
- DMSO
dimethyl sulphoxide
- DPDPE
cyclo[D-Pen2,D-Pen5]enkephalin (Pen = penicillamine)
- JDTic
(3R)-7-hydroxy-N-((1S)-1-[[(3R,4R)-4-(3-hydroxyphenyl)-3,4-dimethyl-1-piperidinyl] methyl]-2-methylpropyl)-1,2,3,4-tetrahydro-3-isoquinoline-carboxamide
- nor-BNI
nor-binaltorphimine
- SNC-80
(+)-4-[(αR)-α-(2S,5R)-4-allyl-2,5-dimethyl-1-piperazinyl)-3-methoxybenzyl]-N,N-diethylbenzamide
- Trp
tryptophan
- U50,488
trans-(±)-3,4-dichloro-N-methyl-N-[2-(1-pyrrolidinyl)cyclohexyl]-benzeneacetamide
- zyklophin
[N-benzylTyr1,cyclo(D-Asp,5,Dap8)]dynorphin A-(1-11) amide (Dap = 2,3-diaminopropionic acid). Note that amino acids are the L-isomer unless otherwise specified; abbreviations for amino acids follow IUPAC-IUB Joint Commission of Biochemical Nomenclature [Eur J Biochem (1984) 138: 9–37].
Conflicts of interest
The authors state no conflict of interest.
References
- Aldrich JV, McLaughlin JP. Peptide kappa opioid receptor ligands: potential for drug development. AAPS J. 2009;11:312–322. doi: 10.1208/s12248-009-9105-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldrich JV, Patkar KA, Chappa AK, Fang W, Audus KL, Lunte SM, et al. Development of centrally acting peptide analogs: structural transport studies and pharmacological evaluation of analogs of the opioid peptide dynorphin A. In: Wilce J, editor. Proceedings of the 4th International Peptide Symposium. 2007. http://www.peptideoz.org/docs/M_64_Jane_Aldrich.pdf. [Google Scholar]
- Aldrich JV, Patkar KA, McLaughlin JP. Zyklophin, a systemically active selective kappa opioid receptor peptide antagonist with short duration of action. Proc Natl Acad Sci USA. 2009;106:18396–18401. doi: 10.1073/pnas.0910180106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arttamangkul S, Ishmael JE, Murray TF, Grandy DK, DeLander GE, Kieffer BL, et al. Synthesis and opioid activity of conformationally constrained dynorphin A analogues. 2. Conformational constraint in the ‘address’ sequence. J Med Chem. 1997;40:1211–1218. doi: 10.1021/jm960753p. [DOI] [PubMed] [Google Scholar]
- Arunlakshana O, Schild HO. Some quantitative uses of drug antagonists. Br J Pharmacol Chemother. 1959;14:48–58. doi: 10.1111/j.1476-5381.1959.tb00928.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beardsley PM, Howard JL, Shelton KL, Carroll FI. Differential effects of the novel kappa opioid receptor antagonist, JDTic, on reinstatement of cocaine-seeking induced by footshock stressors vs cocaine primes and its antidepressant-like effects in rats. Psychopharmacology (Berl) 2005;183:118–126. doi: 10.1007/s00213-005-0167-4. [DOI] [PubMed] [Google Scholar]
- Beguin C, Richards MR, Li JG, Wang Y, Xu W, Liu-Chen LY, et al. Synthesis and in vitro evaluation of salvinorin A analogues: effect of configuration at C(2) and substitution at C(18) Bioorg Med Chem Lett. 2006;16:4679–4685. doi: 10.1016/j.bmcl.2006.05.093. [DOI] [PubMed] [Google Scholar]
- Bennett MA, Murray TF, Aldrich JV. Identification of arodyn, a novel acetylated dynorphin A-(1-11) analogue, as a kappa opioid receptor antagonist. J Med Chem. 2002;45:5617–5619. doi: 10.1021/jm025575g. [DOI] [PubMed] [Google Scholar]
- Brabant C, Quertemont E, Tirelli E. Influence of the dose and the number of drug-context pairings on the magnitude and the long-lasting retention of cocaine-induced conditioned place preference in C57BL/6J mice. Psychopharmacology (Berl) 2005;180:33–40. doi: 10.1007/s00213-004-2138-6. [DOI] [PubMed] [Google Scholar]
- Brugos B, Hochhaus G. Metabolism of dynorphin A(1-13) Pharmazie. 2004;59:339–343. [PubMed] [Google Scholar]
- Brugos B, Arya V, Hochhaus G. Stabilized dynorphin derivatives for modulating antinociceptive activity in morphine tolerant rats: effect of different routes of administration. AAPS J. 2004;6:e36. doi: 10.1208/aapsj060436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey AN, Borozny K, Aldrich JV, McLaughlin JP. Reinstatement of cocaine place-conditioning prevented by the peptide kappa-opioid receptor antagonist arodyn. Eur J Pharmacol. 2007;569:84–89. doi: 10.1016/j.ejphar.2007.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll FI, Harris LS, Aceto MD. Effects of JDTic, a selective kappa-opioid receptor antagonist, on the development and expression of physical dependence on morphine using a rat continuous-infusion model. Eur J Pharmacol. 2005;524:89–94. doi: 10.1016/j.ejphar.2005.09.013. [DOI] [PubMed] [Google Scholar]
- Cheng YC, Prusoff WH. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Covington HE, Miczek KA. Repeated social-defeat stress, cocaine or morphine. Effects on behavioral sensitization and intravenous cocaine self-administration ‘binges’. Psychopharmacology (Berl) 2001;158:388–398. doi: 10.1007/s002130100858. [DOI] [PubMed] [Google Scholar]
- Delaforge M, Andre F, Jaouen M, Dolgos H, Benech H, Gomis J-M, et al. Metabolism of tentoxin by hepatic cytochrome P-450 3A isozymes. Eur J Biochem. 1997;250:150–157. doi: 10.1111/j.1432-1033.1997.00150.x. [DOI] [PubMed] [Google Scholar]
- Dolle RE, Michaut M, Martinez-Teipel B, Seida PR, Ajello CW, Muller AL, et al. Nascent structure-activity relationship study of a diastereomeric series of kappa opioid receptor antagonists derived from CJ-15,208. Bioorg Med Chem Lett. 2009;19:3647–3650. doi: 10.1016/j.bmcl.2009.04.105. [DOI] [PubMed] [Google Scholar]
- Fujimoto K, Momose T. Analgesic efficacy of E-2078 (dynorphin analog) in patients following abdominal surgery. Jpn J Anesth. 1995;44:1233–1237. [PubMed] [Google Scholar]
- Haley TJ, McCormick WG. Pharmacological effects produced by intracerebral injections of drugs in the conscious mouse. Br J Pharmacol Chemother. 1957;12:12–15. doi: 10.1111/j.1476-5381.1957.tb01354.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horan P, Taylor J, Yamamura HI, Porreca F. Extremely long-lasting antagonistic actions of nor-binaltorphimine (nor-BNI) in the mouse tail-flick test. J Pharmacol Exp Ther. 1992;260:1237–1243. [PubMed] [Google Scholar]
- Klintenberg R, Andren PE. Altered extracellular striatal in vivo biotransformation of the opioid neuropeptide dynorphin A(1-17) in the unilateral 6-OHDA rat model of Parkinson's disease. J Mass Spectrom. 2005;40:261–270. doi: 10.1002/jms.754. [DOI] [PubMed] [Google Scholar]
- Knoll AT, Meloni EG, Thomas JB, Carroll FI, Carlezon WA., Jr Anxiolytic-like effects of kappa-opioid receptor antagonists in models of unlearned and learned fear in rats. J Pharmacol Exp Ther. 2007;323:838–845. doi: 10.1124/jpet.107.127415. [DOI] [PubMed] [Google Scholar]
- Kulkarni SS, Ross NC, McLaughlin JP, Aldrich JV. Synthesis of cyclic tetrapeptide CJ 15,208: a novel kappa opioid receptor antagonist. Adv Exp Med Biol. 2009;611:269–270. doi: 10.1007/978-0-387-73657-0_121. [DOI] [PubMed] [Google Scholar]
- Lu Y, Nguyen TM-D, Weltrowska G, Berezowska I, Lemieux C, Chung NN, et al. [2′,6′-Dimethyltyrosine]dynorphin A(1-11)NH2 analogues lacking an N-terminal amino group: potent and selective κ opioid antagonists. J Med Chem. 2001;44:3048–3053. doi: 10.1021/jm0101186. [DOI] [PubMed] [Google Scholar]
- McLaughlin JP, Hill KP, Jiang Q, Sebastian A, Archer S, Bidlack JM. Nitrocinnamoyl and chlorocinnamoyl derivatives of dihydrocodeinone: in vivo and in vitro characterization of mu-selective agonist and antagonist activity. J Pharmacol Exp Ther. 1999;289:304–311. [PubMed] [Google Scholar]
- McLaughlin JP, Marton-Popovici M, Chavkin C. Kappa opioid receptor antagonism and prodynorphin gene disruption block stress-induced behavioral responses. J Neurosci. 2003;23:5674–5683. doi: 10.1523/JNEUROSCI.23-13-05674.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin JP, Land BB, Li S, Pintar JE, Chavkin C. Prior activation of kappa opioid receptors by U50,488 mimics repeated forced swim stress to potentiate cocaine place preference conditioning. Neuropsychopharmacology. 2006;31:787–794. doi: 10.1038/sj.npp.1300860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon RC. Personality, stress, and social support in cocaine relapse prediction. J Subst Abuse Treat. 2001;21:77–87. doi: 10.1016/s0740-5472(01)00187-8. [DOI] [PubMed] [Google Scholar]
- Mague SD, Pliakas AM, Todtenkopf MS, Tomasiewicz HC, Zhang Y, Stevens WC, Jr, et al. Antidepressant-like effects of kappa-opioid receptor antagonists in the forced swim test in rats. J Pharmacol Exp Ther. 2003;305:323–330. doi: 10.1124/jpet.102.046433. [DOI] [PubMed] [Google Scholar]
- Marx M. Watching peptide drugs grow up. Chem Eng News. 2005;83:17–24. [Google Scholar]
- Mello NK, Mendelson JH, Sholar MB, Jaszyna-Gasior M, Goletiani N, Siegel AJ. Effects of the mixed mu/kappa opioid nalbuphine on cocaine-induced changes in subjective and cardiovascular responses in men. Neuropsychopharmacology. 2005;30:618–632. doi: 10.1038/sj.npp.1300631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metcalf MD, Coop A. Kappa opioid antagonists: past successes and future prospects. AAPS J. 2005;7:E704–E722. doi: 10.1208/aapsj070371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RR. Evaluation of nalbuphine hydrochloride. Am J Hosp Pharm. 1980;37:942–949. [PubMed] [Google Scholar]
- Nabeshima T, Katoh A, Wada M, Kameyama T. Stress-induced changes in brain Met-enkephalin, Leu-enkephalin and dynorphin concentrations. Life Sci. 1992;51:211–217. doi: 10.1016/0024-3205(92)90077-3. [DOI] [PubMed] [Google Scholar]
- Patkar KA, Yan X, Murray TF, Aldrich JV. [Nα-benzylTyr1cyclo(D-Asp5,Dap8)]- dynorphin A-(1-11)NH2 cyclized in the ‘address’ domain is a novel kappa-opioid receptor antagonist. J Med Chem. 2005;48:4500–4503. doi: 10.1021/jm050105i. [DOI] [PubMed] [Google Scholar]
- Piazza PV, Deminiere JM, LeMoal M, Simon H. Stress- and pharmacologically-induced behavioral sensitization increases vulnerability to acquisition of amphetamine self-administration. Brain Res. 1990;514:22–26. doi: 10.1016/0006-8993(90)90431-a. [DOI] [PubMed] [Google Scholar]
- Portoghese PS, Lipkowshi AW, Takemori AE. Bimorphinans as highly selective, potent κ opioid receptor antagonists. J Med Chem. 1987;30:238–239. doi: 10.1021/jm00385a002. [DOI] [PubMed] [Google Scholar]
- Redila VA, Chavkin C. Stress-induced reinstatement of cocaine seeking is mediated by the kappa opioid system. Psychopharmacology (Berl) 2008;200:59–70. doi: 10.1007/s00213-008-1122-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed B, Zhang Y, Chait BT, Kreek MJ. Dynorphin A(1-17) biotransformation in striatum of freely moving rats using microdialysis and matrix-assisted laser desorption/ionization mass spectrometry. J Neurochem. 2003;86:815–823. doi: 10.1046/j.1471-4159.2003.01859.x. [DOI] [PubMed] [Google Scholar]
- Ross NC, Kulkarni SS, McLaughlin JP, Aldrich JV. Synthesis of CJ-15,208, a potent κ-opioid receptor antagonist. Tetrahedron Lett. 2010;51:5020–5023. doi: 10.1016/j.tetlet.2010.07.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T, Hirai H, Kim YJ, Kojima Y, Matsunaga Y, Nishida H, et al. CJ-15,208, a novel kappa opioid receptor antagonist from a fungus, Ctenomyces serratus ATCC15502. J Antibiot (Tokyo) 2002;55:847–854. doi: 10.7164/antibiotics.55.847. [DOI] [PubMed] [Google Scholar]
- Schiller PW, Nguyen TM-D, Weltrowska G, Wilkes BC, Marsden BJ, Lemieux C, et al. Differential stereochemical requirements of µ vs δ opioid receptors for ligand binding and signal transduction: development of a class of potent and highly δ-selective peptide antagonists. Proc Natl Acad Sci USA. 1992;89:11871–11875. doi: 10.1073/pnas.89.24.11871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt WK, Tam SW, Shotzberger GS, Smith DH, Jr, Clark R, Vernier VG. Nalbuphine. Drug Alcohol Depend. 1985;14:339–362. doi: 10.1016/0376-8716(85)90066-3. [DOI] [PubMed] [Google Scholar]
- Seale PW, Stead P, Jaxa-Chamiec A. Cyclic tetrapepides – Novel scaffolds for pharmacophore design. In: Martinez J, Fehrentz J-A, editors. Peptides 2000. Paris, France: EDK; 2000. pp. 271–272. [Google Scholar]
- Shirayama Y, Ishida H, Iwata M, Hazama GI, Kawahara R, Duman RS. Stress increases dynorphin immunoreactivity in limbic brain regions and dynorphin antagonism produces antidepressant-like effects. J Neurochem. 2004;90:1258–1268. doi: 10.1111/j.1471-4159.2004.02589.x. [DOI] [PubMed] [Google Scholar]
- Siebenallar JF, Murray TF. Hydrostatic pressure alters the time course of GTP [S] binding to G proteins in brain membranes from two congeneric marine fishes. Biol Bull. 1999;197:388–394. doi: 10.2307/1542793. [DOI] [PubMed] [Google Scholar]
- Szumlinski KK, Price KL, Frys KA, Middaugh LD. Unconditioned and conditioned factors contribute to the ‘reinstatement’ of cocaine place conditioning following extinction in C57BL/6 mice. Behav Brain Res. 2002;136:151–160. doi: 10.1016/s0166-4328(02)00102-x. [DOI] [PubMed] [Google Scholar]
- Takahashi M, Senda T, Tokuyama S, Kaneto H. Further evidence for the implication of a kappa-opioid receptor mechanism in the production of psychological stress-induced analgesia. Jpn J Pharmacol. 1990;53:487–494. doi: 10.1254/jjp.53.487. [DOI] [PubMed] [Google Scholar]
- Terasaki T, Deguchi Y, Sato H, Hirai K, Tsuji A. In vivo transport of a dynorphin-like analgesic peptide, E-2078, through the blood-brain barrier: an application of brain microdialysis. Pharm Res. 1991;8:815–820. doi: 10.1023/a:1015882924470. [DOI] [PubMed] [Google Scholar]
- Thomas JB, Atkinson RN, Vinson NA, Catanzaro JL, Perretta CL, Fix SE, et al. Identification of (3R)-7-hydroxy-N-((1S)-1-[[(3R,4R)-4-(3-hydroxyphenyl)- 3,4-dimethyl-1-piperidinyl]methyl]-2-methylpropyl)-1,2,3,4-tetrahydro-3-isoquinolinecarboxamide as a novel potent and selective opioid kappa receptor antagonist. J Med Chem. 2003;46:3127–3137. doi: 10.1021/jm030094y. [DOI] [PubMed] [Google Scholar]
- Valdez GR, Platt DM, Rowlett JK, Ruedi-Bettschen D, Spealman RD. Kappa agonist-induced reinstatement of cocaine seeking in squirrel monkeys: a role for opioid and stress-related mechanisms. J Pharmacol Exp Ther. 2007;323:525–533. doi: 10.1124/jpet.107.125484. [DOI] [PubMed] [Google Scholar]
- Vlieghe P, Lisowski V, Martinez J, Khrestchatisky M. Synthetic therapeutic peptides: science and market. Drug Disc Today. 2010;15:40–56. doi: 10.1016/j.drudis.2009.10.009. [DOI] [PubMed] [Google Scholar]
- Yu J, Butelman ER, Woods JH, Chait BT, Kreek MJ. Dynorphin A (1-8) analog, E-2078, is stable in human and rhesus monkey blood. J Pharmacol Exp Ther. 1997;280:1147–1151. [PubMed] [Google Scholar]