

Abstract
BACKGROUND AND PURPOSE
Reduced cardiac contractility has been associated with disrupted myocardial Ca2+ signalling. The 1,4 benzothiazepine K201 (JTV-519) acts on several Ca2+ handling proteins and improves cardiac contractility in vivo in a variety of animal models of myocardial dysfunction. However, it is unclear whether this improvement depends on the systemic effects of K201 or if K201 reverses the effects of Ca2+ dysregulation, regardless of the cause.
EXPERIMENTAL APPROACH
The effect of K201 on cardiac mechanical function was assessed in isolated working hearts from adult rabbits, using a ventricular pressure-volume catheter. In separate experiments, the effect of K201 was investigated in hearts following pharmacologically induced Ca2+ overload using elevated extracellular [Ca2+] ([Ca2+]o) and β-adrenoceptor stimulation.
KEY RESULTS
K201 induced a concentration-dependent decline in systolic function (peak pressure, dP/dtmax and preload recruitable stroke work), lusitropy (reduced dP/dtmin and increased end diastolic pressure) and stroke volume, independent of decreased heart rate. In separate experiments, mechanical function in hearts exposed to 4.5 mmol·L−1[Ca2+]o and 150 nmol·L−1 isoprenaline declined until cessation of aortic flow (in 6 out of 11 hearts). However, all hearts perfused with the addition of 1 µmol·L−1 K201 maintained aortic flow and demonstrated significantly improved peak systolic pressures, dP/dtmax and dP/dtmin.
CONCLUSIONS AND IMPLICATIONS
K201 significantly improved mechanical function of the heart during Ca2+ overload. This suggests that K201 can limit the detrimental effects of elevated intracellular Ca2+ and exert beneficial effects on cardiac contractile function, independent of systemic effects previously observed in vivo.
Keywords: K201 (JTV-519), β-adrenoceptor stimulation, isolated working heart, pressure-volume catheter
Introduction
K201 (JTV-519) is a synthetic 1,4 benzothiazepine derivative originally described for its cardioprotective properties under conditions which promote cell death via Ca2+ overload (Kaneko, 1994). Subsequent molecular and cellular studies have highlighted the action of K201 on multiple targets involved in the process of excitation-contraction coupling in ventricular myocardium (Wehrens et al., 2005; Hunt et al., 2007; Loughrey et al., 2007). These include dose-dependent inhibition of (i) ryanodine receptor (RyR2) open probability (Oda et al., 2005; Wehrens et al., 2005); (ii) sarcoplasmic/endoplasmic reticulum Ca2+ ATPase (SERCA) pump activity (Loughrey et al., 2007); and (iii) the L-type Ca2+ channel current (ICa) (Inagaki et al., 2000) (receptor and channel nomenclature follows Alexander et al., 2011). Increased RyR2-mediated sarcoplasmic reticulum (SR) Ca2+ release during diastole is believed to play a role in cardiac dysfunction. Mutations in the RyR2 channels (Liu et al., 2008) as well as changes in channel function in various disease states such as congestive heart failure (Tateishi et al., 2009) result in an increased frequency of Ca2+ release during the diastolic period. Excessive spontaneous Ca2+ release during diastole has been identified as a potential mechanism in the genesis of triggered arrhythmias (Lederer and Tsien, 1976). Moreover, diastolic Ca2+ leak has also been proposed to lead to increased diastolic tone and incomplete relaxation, as well as potentially contributing to decreased SR content (Shannon et al., 2003). In the search for potential therapeutic strategies, attention has focused on compounds that can specifically target RyR2 and restore normal function.
The compound K201 has potential as both a cardioprotective and anti-arrhythmic agent (Inagaki et al., 2000; Hasumi et al., 2007). Most recently, in a mouse myocardial infarction model (Wehrens et al., 2005) and a canine model of pacing induced heart failure (Yano et al., 2003) K201 significantly attenuated development of cardiac dysfunction and ventricular remodelling. In these in vivo models, K201 was proposed to stabilize the RyR2 in the closed state, preventing SR Ca2+ leak and potentially arrhythmogenic events from occurring. However, several aspects of the effect of K201 on whole heart function remain unclear including the consequence of the inhibitory effect of K201 on multiple targets for non-diseased hearts and the degree to which K201 directly modifies heart function in animal models of cardiac disease, given the potential for the drug to indirectly improve heart function via beneficial effects on other tissues and organs, such as kidney or skeletal muscle (Wehrens et al., 2005; Lisy and Burnett, 2006).
To address these issues, the current study has simultaneously used a rabbit isolated working heart system and a pressure-volume (PV) catheter to characterize the direct effect of K201 on cardiac function. While such a combination of isolated working heart and micro-conductance catheter technology has been used in small rodents (Grieve et al., 2004), to the best of our knowledge this is the first time its use with rabbit hearts has been reported.
The isolated working heart preparation permits characterization of cardiac function without the compounding effects of (i) neurohumoral activity, (ii) anaesthesia, (iii) changes in loading parameters or (iv) influence of other organs, while the PV catheter provides detailed real-time characterization of cardiac function. These have been combined in the present study to assess the direct effect of K201 on the mechanical function of normal hearts and hearts exposed to an elevated extracellular Ca2+ concentration ([Ca2+]o) and β-adrenoceptor stimulation. Our data demonstrated that, under control conditions, 1 µmol·L−1 K201 decreased cardiac mechanical function in isolated working hearts to a small extent, but under conditions that induce Ca2+ overload and subsequent decrease in pump function, K201 substantially delayed the onset of detrimental effects.
Methods
Heart isolation
All animal care and experimental procedures conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication no. 85–23, revised 1996) and complied with the U K Animals (Scientific Procedures) Act 1986. A total of 41 male New Zealand White rabbits weighing 3.0–3.5 kg were used; they were injected with 0.5 mL heparin (5000 IU·mL−1; Leo Laboratories) and an overdose of sodium pentobarbitone (100.0 mg·kg−1; Euthatal, Merial). Hearts were excised via a thoracotomy and submerged in a modified Tyrode's solution (composition given below) at 4.0°C.
Working heart setup
The experimental setup used for the isolated working heart experiments was based on that described by Neely et al. (1967). A dual-headed peristaltic pump (Cole-Parmer Instruments; Vernon Hills, IL) controlled the rate of perfusion of modified Tyrode's solution (450 mL·min−1 with a 14.4 ± 0.5 s period delay between drug addition and perfusion of the heart). The system (a modified version from Radnoti, Monrovia, CA) was capable of perfusing hearts in Langendorff mode under constant pressure (55 mmHg) initially and then altered to establish non-recirculating and finally recirculating working heart mode with a 5 µm pore in-line filter (Millipore; Billerica, MA). The maximum recirculating volume of the system was 540 mL. Left atrial filling pressure was dictated by the height of a bubble trap (6 mmHg). A compliance chamber partially filled with 15 mL of air was located in the aortic outflow line, which provided an aortic Windkessel to compensate for the lack of elasticity of the outflow tubing. The afterload imposed on the heart was dictated by the height of the afterload chamber (55 mmHg).
Heart cannulation
In Langendorff mode the aorta was cannulated (tube diameter of 4.5 mm) as was the common pulmonary vein ostium. A pair of electrodes were positioned on the right atrium for pacing. These were connected to a constant voltage isolated stimulator and train/delay generator (Digitimer; Hertfordshire, UK). Where heart rate (HR) was required to be controlled over a wider range than permitted by the sinus rate, electrical pacing was applied following crushing of the sino-atrial node (to reduce the intrinsic HR). The system was then switched from Langendorff mode to working heart mode.
Pressure sensor calibration
A custom designed 3F PV catheter (Scisense, Canada) was equilibrated in saline (37.0°C) for 45 min prior to the experiment ensuring maximal stability of the pressure signal and prevention of drift over long periods (<1 mmHg·h−1). Signal calibration was based on a 2-point in-built linear calibration technique (0 mmHg: −2.86 V and 100.0 mmHg: −0.56 V), with subsequent balance adjustment to 0 mmHg with the sensor exposed to atmosphere. The voltage-pressure linear relationship for the Scisense pressure sensor was independently confirmed using a Deltacal (Utah Medical; Midvale, UT) pressure transducer tester.
Volume sensor calibration
The catheter was placed sequentially in two of a series of machined-drilled cuvette wells (radius of 4 mm and 7 mm respectively) filled with perfusate (37°C) to establish a linear relationship between the raw analogue signal and volume of well. The two cuvette wells were chosen to represent the highest and lowest volumes expected from the isolated working rabbit heart. A voltage swing of −2 V to +1 V was obtained by alteration of the volume signal gain and offset.
Calculation of volume
Using the two-point calibration described above and because solution resistivity remained constant in the working heart system and the catheter recording segment length was pre-determined, the following equation was used to calculate volume:
| 1 |
where VT is the true volume (mL), VC is the volume measured by the catheter (mL), Vp is the volume owed to structures other than the perfusate within the left ventricle (LV) (mL; parallel conductance) and α is a dimensionless constant used to correct the volume signal for non-uniformity of the electric field.
Calculation of alpha (α)
α is the ratio of the stroke volume (SV), measured by the catheter, to SV measured by another independent means. In the isolated working heart setup, α was ascertained by independent measurement of cardiac output (CO) defined as the sum of aortic flow (AoF) and coronary flow. Coronary flow was assessed by timed measurement of perfusate originating from the heart. AoF was measured continuously throughout the experiment using an in-line ultrasonic flowmeter (Malema; Florida, USA) located between the aortic cannula and the compliance chamber.
Calculation of parallel conductance (Vp)
A bolus of 20% saline solution was injected into the preload line. This resulted in a number of cardiac cycles (5–7 beats) where VC was changing without any alteration in LV pressure. Vp was estimated offline as previously published (Grieve et al., 2004).
Assessment of load-independent indices of cardiac function
Load-independent indices of cardiac function were assessed in the rabbit isolated working heart by performing transient occlusion of the left atrial inflow line to reduce preload. End diastolic pressure-volume relationships (EDPVRs) were assessed by fitting the appropriate curve to the end diastolic PV points of the resultant family of PV loops. The EDPVR was fitted to the following non-linear exponential equation:
| 2 |
where EDP and EDV are end diastolic pressure and volume, respectively, C is a curve-fitting constant and β is the diastolic stiffness constant. Preload occlusion also allowed for calculation of preload recruitable stroke work (PRSW), defined as the slope of the linear relationship between stroke work (SW) and end diastolic volume (Joho et al., 2007). PRSW was assessed by fitting a linear relationship through the individual SW, EDV points obtained during preload reduction and calculating the slope of the linear fit.
Isolation of cardiomyocytes from adult rabbits
Adult male New Zealand White rabbits weighing 3.0–3.5 kg and maintained according to national guidelines were killed with an overdose of sodium pentobarbitone (100 mg·kg−1) containing 0.5 mL heparin (5000 IU·mL−1), administered via injection through the left marginal ear vein. Hearts were excised via a thoracotomy and submerged in a modified Krebs–Henseleit (KH) solution for isolation (for composition see below). Hearts were removed and perfused retrogradely at 25 mL·min−1 (37.0°C) with the same solution but with the addition of 0.75 mmol·L−1[Ca2+] for 5 min, followed by a nominally Ca2+-free KH solution with 0.1 mmol·L−1 EGTA for 5.0 min. Hearts were then perfused with modified KH solution containing 0.24 mmol·L−1[Ca2+], 1 mg·mL−1 collagenase [type I, Worthington Chemicals (New Jersey, USA)] and 0.06 mg·mL−1 protease [type XIV, Sigma Aldrich (Poole, UK)]. After ∼4–5 min, enzyme was removed and the LV free wall was then cut into strips in KH solution containing 1% BSA [Sigma Aldrich (Poole, UK)] before being mixed to yield a single cell suspension. The [Ca2+]o in the solution was raised to 1 mmol·L−1 via stepwise increments until use.
Simultaneous field stimulation of intact cardiomyocytes with whole cell epi-fluorescence and shortening measurements
Intact cardiomyocytes in a modified KH solution were loaded with a Ca2+ sensitive fluorophore [5 µmol·L−1 Fura-4F AM (Invitrogen, California, USA)] by incubation for ∼10 min. The incubation medium was removed and the cells resuspended in a modified KH solution (for composition see below) containing 1.8 mmol·L−1[Ca2+]. Cells were incubated for a further 30 min to ensure complete de-esterification. Cardiomyocytes were allowed to settle on a coverslip, placed on a bath (Cell Microcontrols, Virginia, USA) and superfused with the same modified KH solution at 37°C. Cells were field stimulated with 2 ms duration voltage pulses delivered through parallel platinum wires (stimulation voltage set to 1.5 times the threshold). The final concentration of K201 used during the protocol was 1 µmol·L−1. A parallel set of vehicle [dimethyl sulphoxide (DMSO)] time control experiments were performed. The Fura-4F fluorescence (340 and 380 nm excitation; R340/380 nm) was measured using a spinning wheel spectrophotometer (Cairn Research Ltd UK; sampling rate of 500 Hz) while cellular shortening was measured using a video edge detection system (IonOptix, Massachusetts, USA; sampling rate of 250 Hz). Data were analysed offline. Fura-4F fluorescence ratio was converted to intracellular [Ca2+] ([Ca2+]i) as previously described (Seidler et al., 2007). Diastolic cell length was measured during the protocol and data were expressed as the cell length (L)/quiescent cell length in 1.8 mmol·L−1[Ca2+]o (L0) prior to commencement of field stimulation. Mean data for [Ca2+]i and diastolic cell length were obtained by averaging 12 steady state transients using OriginPro v6.1 (OriginLab, Massachusetts, USA).
Data analysis and statistical procedures
Functional analysis of pressure and volume traces was performed using the BP module provided with LabChart v5.5.6 (AD Instruments; Colorado, USA) and Labscribe v2.0 (iWorx; New Hampshire, USA). Data are expressed as mean ± SEM. Data were analysed by unpaired or paired (where relevant) Student's t-test or repeated measures anova with Tukey's test for multiple comparisons performed as a post test. P < 0.05 was considered significant.
Materials
All chemicals were of analytical grade and were dissolved in double distilled water (Millipore). For isolated working heart experiments. the modified Tyrode's solution had the following composition (in mmol·L−1): NaCl (116), NaHCO3 (20), Na2HPO4 (0.4), MgSO4.H2O (1), KCl (4), CaCl2 (2.5) and glucose (11), gassed with a 95% O2–5% CO2 mixture to maintain pH at 7.4. For single myocyte isolation, the modified KH solution had the following composition (in mmol·L−1): NaCl (130), KCl (4.5), HEPES (5), NaH2PO4 (0.4), MgCl2 (3.5), CaCl2 (0.75) and glucose (10), pH 7.4 at 37°C with NaOH. For single myocyte perfusion experiments, the modified KH solution had the following composition (in mmol·L−1): NaCl (140), KCl (4), MgCl2 (1), HEPES (5), glucose (11.1), pH 7.4 at 37°C with NaOH.
Isoprenaline hydrochloride, supplied by Sigma Aldrich (Poole, UK), was dissolved and diluted to the appropriate concentration in distilled water. K201 (4-[3-(4-benzylpiperidin-1-yl) propionyl]-7-methoxy-2,3,4,5-tetrahydro-1, 4-benzothiazepine monohydrochloride), a gift from Aetas Pharma Ltd. (Japan), was dissolved in DMSO and diluted to a final stock concentration of 1 mmol·L−1 in double distilled water. K201 was used at final concentrations of 0.3–3 µmol·L−1 (0.003–0.03% DMSO) in working heart experiments.
Results
The effect of K201 on isolated working hearts
Working hearts from rabbits (n= 8) were perfused with a modified Tyrode's solution containing 0.3, 1 and 3 µmol·L−1 K201 for 4 min at each concentration. During perfusion with 3 µmol·L−1 K201, three of the eight hearts were not able to maintain a CO sufficient to sustain working heart function for 4 min. Mean data comparisons were therefore made after exposure to 3 µmol·L−1 K201 for 1.5 min, when all hearts produced a measurable AoF. For 0.3 and 1 µmol·L−1 K201, comparisons were made at 4 min exposure to K201. Representative LV pressure and volume recordings pre-K201 and at the time points stated above for each concentration of K201 are depicted in Figure 1A, with raw data values for each functional parameter displayed in Table 1. While 0.3 µmol·L−1 K201 (and all vehicle controls) did not significantly affect HR, 1 and 3 µmol·L−1 K201 decreased HR concentration-dependently (Figure 1B (i); P < 0.05). To enable assessment of the effect of K201 on cardiac functional parameters independent of HR, a subset of experiments (n= 4) were performed, which contained an equivalent concentration of vehicle and were paced at rates comparable to those perfused with K201 at each concentration (Figure 1B (i); vehicle control). Figure 1B (ii)–D show functional data from unpaced hearts perfused with K201 (K201 group; red circle) and paced control hearts perfused with vehicle (vehicle control; black square). Data were expressed as a % of steady state values prior to drug addition (baseline). Data from the K201 group were subsequently normalised to the functional parameters obtained from vehicle control at corresponding time points.
Figure 1.
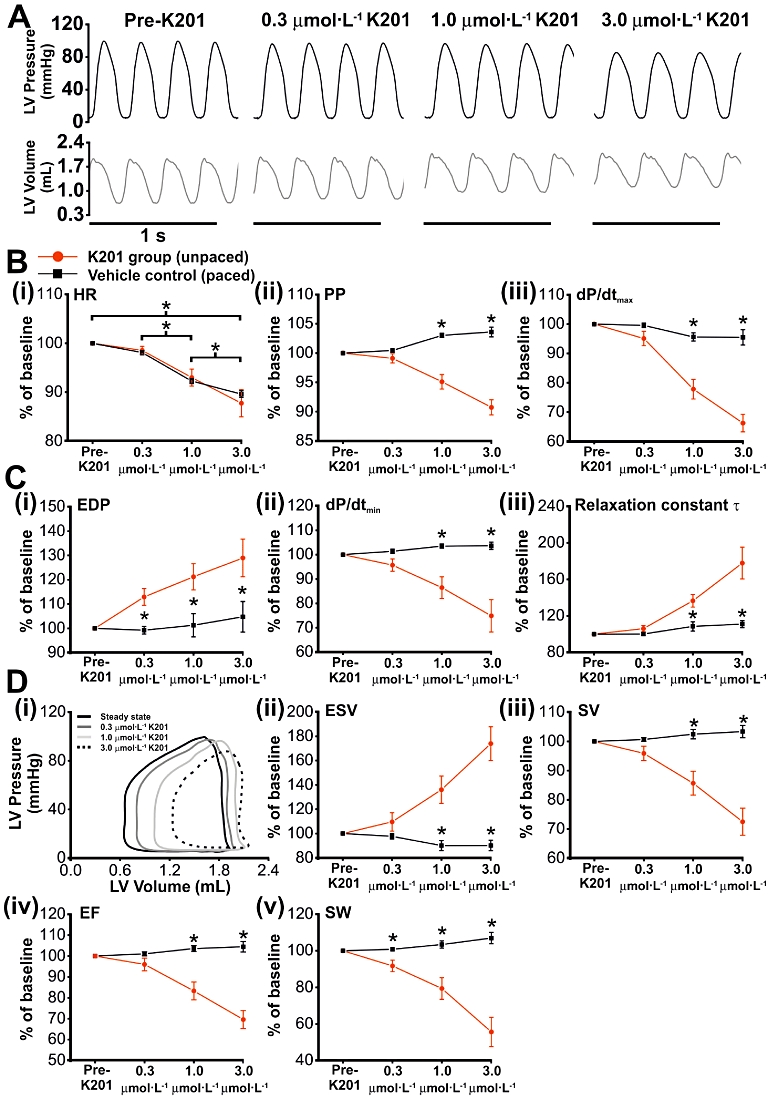
The effect of increasing concentrations of K201 on cardiac mechanical function. (A) Representative pressure and volume traces before and after perfusion with increasing concentrations (0.3, 1 and 3 µmol·L−1) of K201. (B) (i) HR in paced vehicle control (n= 4) and unpaced K201 (n= 8) hearts, expressed as a % of baseline values. Vehicle controls were paced to match HRs recorded at the end of the perfusion period for each concentration of K201. Indices of contractility [B (ii & iii)] and relaxation [C (iii)] are expressed as a % of baseline values. (D) (i) Representative PV loops from time points shown in (A), demonstrating concentration-dependent changes in ESV [D (ii)], SV [D (iii)], EF [D (iv)] and SW [D (v)]. *P < 0.05, significant differences between groups as shown.
Table 1.
Left ventricular functional indices from unpaced isolated working rabbit hearts treated with K201
| Pre-K201 (n= 8) | 0.3 µmol·L−1K201 (n= 8) | 1 µmol·L−1K201 (n= 8) | 3 µmol·L−1K201 (n= 8) | |
|---|---|---|---|---|
| Parameter | ||||
| HR (beats·min−1) | 204 ± 3 | 201 ± 4 | 190 ± 5* | 177 ± 8** |
| Systolic | ||||
| PP (mmHg) | 98.1 ± 1.5 | 97.3 ± 2.1 | 93.4 ± 2.2* | 89.4 ± 1.9** |
| dP/dtmax (mmHg·s−1) | 1916 ± 73 | 1831 ± 110 | 1501 ± 115* | 1277 ± 93** |
| EF (%) | 66.3 ± 3.3 | 63.5 ± 3.5 | 56.3 ± 5.1* | 46.8 ± 4.9** |
| Diastolic | ||||
| EDP (mmHg) | 8.2 ± 1.4 | 9.1 ± 1.5 | 9.8 ± 1.6 | 10.2 ± 1.5 |
| dP/dtmin (mmHg·s−1) | −2077 ± 145 | −1987 ± 146 | −1787 ± 137 | −1523 ± 150 |
| τ (ms) | 27.4 ± 2.8 | 29.0 ± 3.1 | 37.7 ± 4.6* | 49.1 ± 7.1** |
| Volume | ||||
| AoF (mL·min−1) | 177 ± 14 | 166 ± 16 | 135 ± 21* | 100 ± 24** |
| CF (mL·min−1) | 67.5 ± 4.5 | 67.8 ± 4.6 | 67.9 ± 4.6 | 67.8 ± 4.7 |
| ESV (mL) | 0.63 ± 0.07 | 0.70 ± 0.10 | 0.87 ± 0.13 | 1.06 ± 0.14 |
| EDV (mL) | 1.88 ± 0.10 | 1.88 ± 0.11 | 1.94 ± 0.14 | 1.98 ± 0.16 |
| SV (mL) | 1.24 ± 0.09 | 1.19 ± 0.10 | 1.08 ± 0.12* | 0.90 ± 0.12** |
| SW (mmHg*mL) | 85.8 ± 7.1 | 79.8 ± 8.0 | 69.9 ± 10.1* | 50.8 ± 10.7** |
P < 0.05, 0.3 µmol·L−1 versus 1 µmol·L−1;
P < 0.05, 1 µmol·L−1 versus 3 µmol·L−1.
Effect of K201 on systolic functional parameters in normal hearts
Application of 0.3 µmol/L K201 did not alter any systolic functional parameter measured (Figure 1B(ii & iii)). However, 1.0 and 3.0 µmol/L K201 led to a significant concentration-dependent reduction in LV peak pressure (PP) which when normalised to vehicle control decreased to 92.2 ± 0.7 and 87.8 ± 1.0% (P < 0.05) of pre-K201 values, respectively (Figure 1B(ii)). Similarly, the maximum rate of rise of LV pressure (dP/dtmax) when normalised to vehicle control was significantly reduced to 80.9 ± 2.5 and 69.1 ± 2.7% (P < 0.05) of pre-K201 values respectively (Figure 1B(iii)).
Effect of K201 on diastolic functional parameters in normal hearts
The relaxation of the myocardium was impaired by 0.3, 1.0 and 3.0 µmol/L K201, as indicated by an increase in end diastolic pressure (EDP) to 113.9 ± 2.6, 119.7 ± 5.5 and 123.1 ± 7.4% of pre-K201 values, respectively (P < 0.05; Figure 1C(i)). Similarly, 1.0 and 3.0 µmol/L K201, when normalised to vehicle control decreased the maximum rate of LV pressure fall (dP/dtmin; Figure 1C(ii)) to 83.5 ± 2.5 and 71.7 ± 3.8% of the pre-K201 value respectively (P < 0.05) and increased the time for the relaxation constant τ to 125.2 ± 6.0 and 158.6 ± 10.5% of the pre-K201 value respectively (P < 0.05; Figure 1C(iii)).
Effect of K201 on volume parameters in normal hearts
As can be observed from the PV loops shown in Figure 1D(i) and mean data in Figure 1D(ii), 1.0 and 3.0 µmol/L K201 led to an increase in end-systolic volume (ESV) to 150.7 ± 9.8 and 185.6 ± 10.4% of pre-K201 values respectively when normalised to vehicle control (P < 0.05) without any significant alteration in end-diastolic volume (EDV; data not shown). The net effect was a decline in stroke volume (SV) by 1.0 and 3.0 µmol/L K201 to 83.6 ± 2.6 and 70.2 ± 3.0% of pre-K201 values respectively normalised to vehicle control (P < 0.05; Figure 1D(iii)). Ejection fraction (EF), normalised to vehicle control, was reduced by 1.0 and 3.0 µmol/L K201 to 80.4 ± 2.6 and 66.6 ± 2.9% of pre-K201 values respectively (P < 0.05; Figure 1D(iv)). Stroke work (SW) when normalised to vehicle control values, was significantly reduced by 0.3, 1.0 and 3.0 µmol/L K201 to 91.0 ± 1.9, 76.8 ± 3.6 and 52.0 ± 4.5% of pre-K201 values respectively (P < 0.05; Figure 1D(v)). K201 did not significantly alter coronary flow which at steady state was 67.5 ± 4.5 ml.min−1.
Load-independent indices of cardiac function
Assessment of EDPVR was performed in a subset of hearts to determine a load-independent index of diastolic cardiac function in paced hearts (200 beats·min−1) treated with 1 µmol·L−1 K201. Typical EDPVRs for working rabbit hearts before (steady state) and after treatment with K201 (1 µmol·L−1 K201) are shown in Figure 2A (i). Treatment with K201 did not significantly alter the diastolic stiffness constant, β (Figure 2A (ii); n= 5). Representative PRSW relationships from a single heart before and after treatment with K201 are depicted in Figure 2A (iii). While diastolic stiffness was shown to be unaltered, PRSW was reduced following treatment with 1 µmol·L−1 K201 (Figure 2A (iv); P < 0.05) indicating a decline in intrinsic systolic function at this concentration. The reversibility of the negative inotropic effects of K201 were assessed by a washout period of 30 min with normal Tyrode's solution. As shown from the experimental record in Figure 2B, contractility (dP/dtmax normalized to pre-K201 values) recovered towards pre-K201 values, following a washout period [mean recovery = 94 ± 2% of pre-K201 values (n= 3)].
Figure 2.
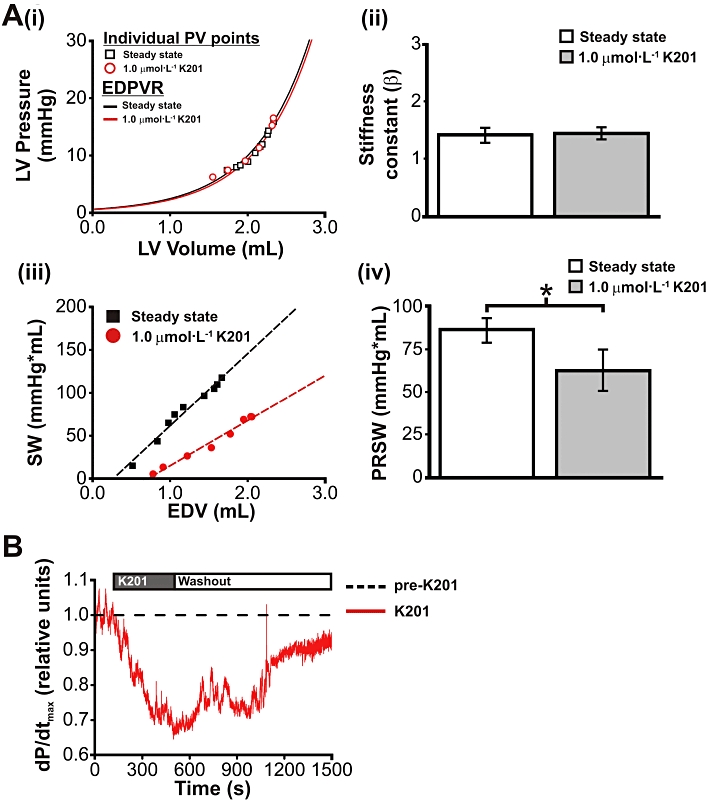
Pressure-volume relationships in the rabbit isolated working heart, following K201 treatment. (A) (i) EDPVRs before (Steady state) and after 1.0 µmol·L−1 K201. Individual end diastolic PV points for each group are indicated. (ii) Mean diastolic stiffness constant β was not significantly altered by K201 treatment. (iii) Representative PRSW relationships before (Steady state) and after 1.0 µmol·L−1 K201. (iv) PRSW was significantly reduced following K201 treatment. (B) The reversibility of K201's negative inotropic effects on the isolated working rabbit heart. Representative trace showing effects of K201 treatment and washout period on dP/dtmax, normalized to pre-K201 values. Dashed line indicates pre-K201 levels. Protocol bar indicates periods of K201 treatment and washout. *P < 0.05.
The effect of K201 on paced versus non-paced hearts
As increasing concentrations of K201 resulted in a negative chronotropic effect in unpaced rabbit working hearts, it was possible that this could be a significant factor in the negative inotropic effects of the drug. Furthermore, it was possible that the pacing in vehicle control hearts had altered the electrical activation pattern and therefore the subsequent mechanical function, making direct comparison more complex. To fully explore the nature of the interaction between the effects of K201 on HR and inotropy and, in addition, to ensure that the effects of K201 on systolic, diastolic and volume parameters were not significantly altered by pacing, a second subset of hearts were paced at a set rate (200 beats·min−1) and then perfused with 1 µmol·L−1 K201. Figure 3 shows the effects of exposure for 4 min to K201 in paced hearts (n= 4) and unpaced hearts (n= 8), as % of the corresponding pre-K201 values, for all pressure (Figure 3A) and volume derived parameters (Figure 3B). As can be seen from these data, pressure and volume parameters in the K201 paced group were altered significantly in the same direction and magnitude as the K201 unpaced group, demonstrating that the mechanical effects of K201 on the normal heart are due to a direct effect of K201 on the ventricular myocardium.
Figure 3.
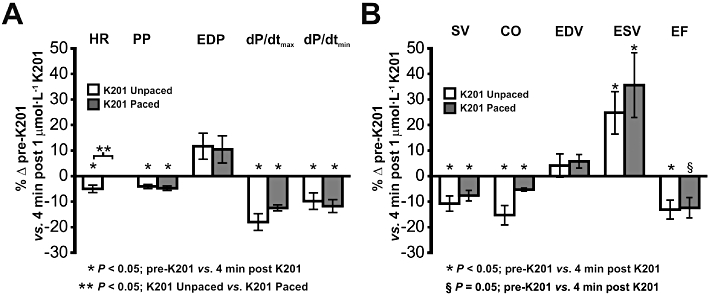
The influence of HR changes on negative inotropic effects of K201. The % change in contractility and relaxation indices (A) and volume indices (B) in paced and unpaced working hearts following 4 min of perfusion with 1.0 µmol·L−1 K201. *P < 0.05, significant effect of K201. §P= 0.05, significant effect of K201. **P < 0.05, significant effect of pacing.
2Effect of K201 during β-adrenoceptor stimulation and an elevated [Ca2+]o
In a separate set of experiments hearts were exposed to elevated [Ca2+]o and β-adrenoceptor stimulation, the combination of which in vivo leads to acute severe mechanical dysfunction (Kaneko et al., 2006b). While K201 in the in vivo model alleviated the dysfunction (Kaneko et al., 2006b), it was not known whether this was as a result of a direct effect on the heart; an indirect effect of other organs or an alteration of cardiac preload/afterload. To investigate this, all hearts were perfused with 4.5 mmol·L−1[Ca2+]o for 5 min. The effect of increasing [Ca2+]o on working rabbit heart function is summarized in Figure 4A–H. Switching from 2.5 mmol·L−1 to 4.5 mmol·L−1[Ca2+]o resulted in a sustained positive inotropic effect with significant increases in AoF, SV, PP and dP/dtmax (Figure 4A–D, respectively; n= 11), with no change in either HR or coronary flow (Figure 4E & F, respectively; n= 11). A significant decrease in ESV was also observed, with no change in EDV (Figure 4G & H respectively; n= 5). Hearts were then given one of two treatments; the first group were perfused with 150 nmol·L−1 isoprenaline while the second group were perfused with 1 µmol·L−1 K201, 30 s after addition of isoprenaline. The end of the experiment was reached when hearts were no longer able to produce an AoF (zero AoF), resulting in a decline in coronary perfusion pressure and consequently a precipitous decline in mechanical function. Figure 5A (i) shows typical PV loops in 4.5 mmol·L−1 Ca2+ and loops obtained 10 s and 50 s after perfusion with isoprenaline. The net effect of elevated [Ca2+]o and β-adrenoceptor stimulation was an impairment of cardiac function with only 5 out of 11 hearts still producing an AoF within the first 600 s (Figure 5A (ii)). In contrast, addition of 1 µmol·L−1 K201 enabled all hearts to maintain an AoF in the first 600 s and prolonged the time to zero AoF by 318% (Figure 5A (iii); P < 0.05).
Figure 4.
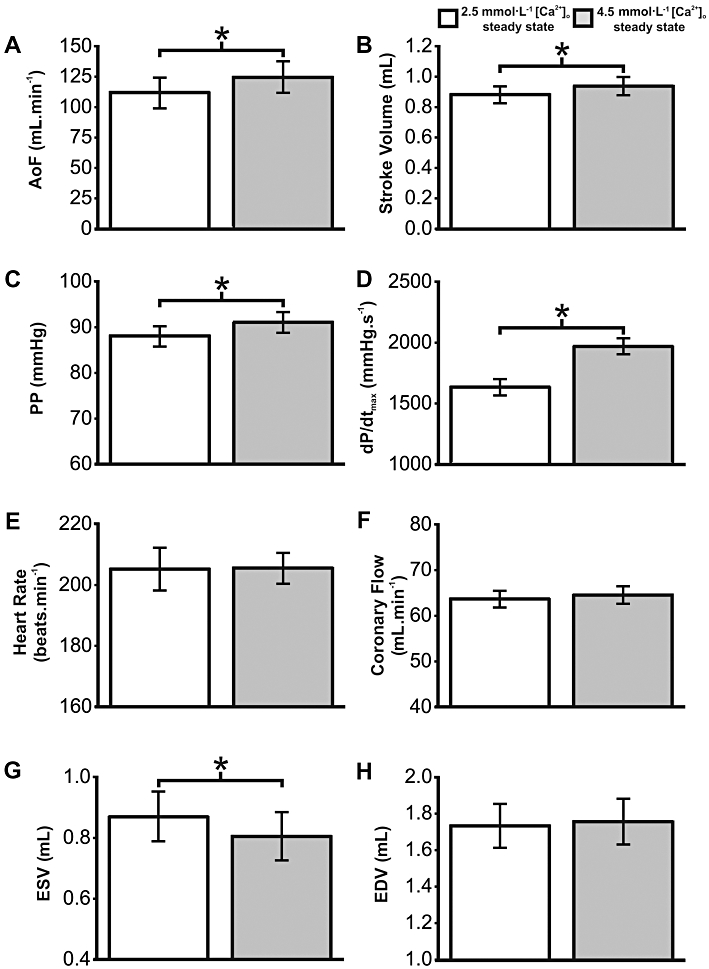
Influence of elevated [Ca2+]o on isolated working heart contractile function. Volume (A, B, G) and contractility indices (C, D) demonstrating sustained positive inotropic effect of increasing [Ca2+]o from 2.5 mmol·L−1 to 4.5 mmol·L−1. HR (E), coronary flow (F) and end diastolic volume (H) were not significantly altered. *P < 0.05, significant differences between groups as shown.
Figure 5.
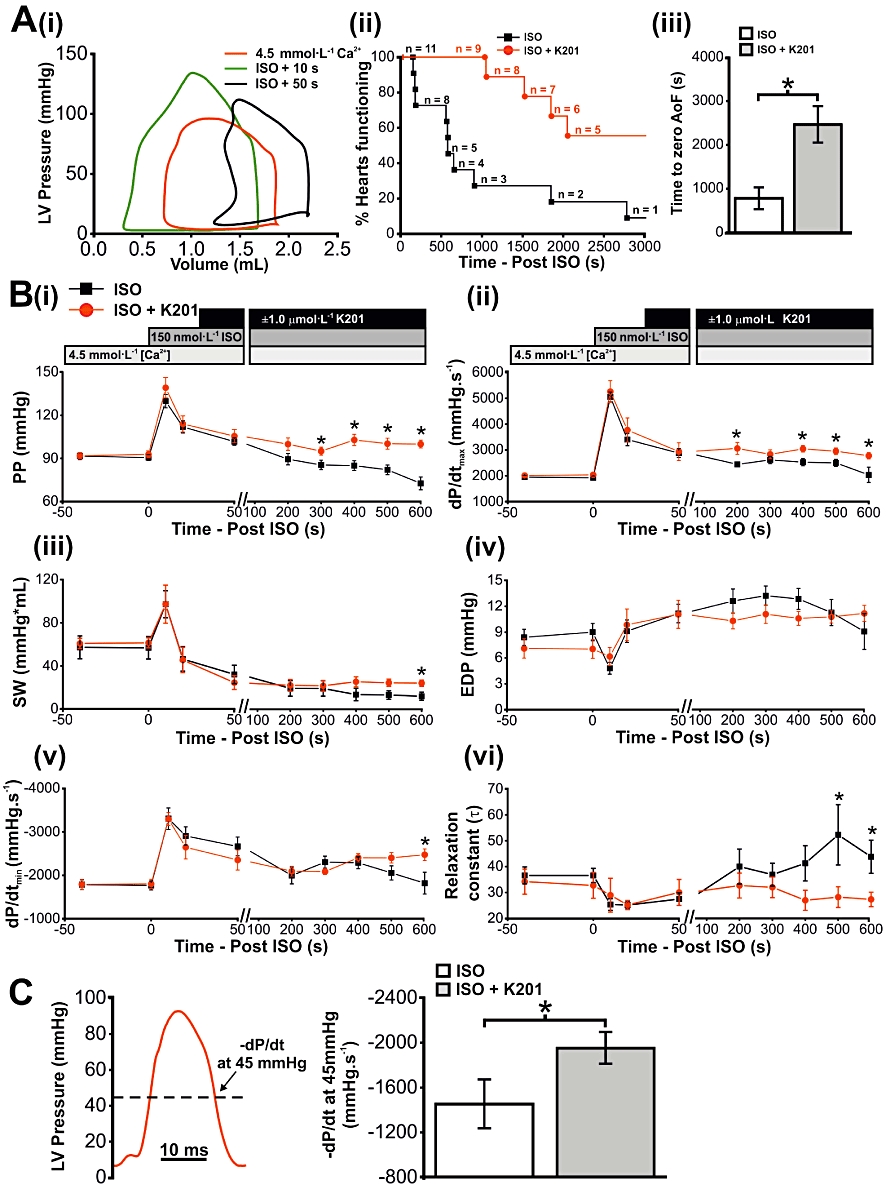
Elevated Ca2+ loading in the isolated working heart and the influence of K201. (A) (i) Representative PV loops in elevated [Ca2+]o alone, 10 s and 50 s after perfusion with isoprenaline (ISO). (ii) Line graph showing % of working hearts still producing an AoF up to 50 min after isoprenaline perfusion in untreated and K201-treated hearts, with mean time to zero AoF in both groups shown in histogram (iii). (B) Contractility (i–iii) and relaxation (iv–vi) indices up to 600 s after perfusion with isoprenaline in untreated (ISO) and K201-treated (ISO+K201) working hearts. (C) Measurement of –dP/dt at a fixed pressure of 45 mmHg. Left panel: representative LV pressure trace indicating where –dP/dt was measured. Right panel: mean data in untreated (ISO) and K201-treated (ISO+K201) hearts. *P < 0.05; isoprenaline versus isoprenaline + K201.
Systolic parameters during β-adrenoceptor stimulation and elevated [Ca2+]o
K201-treated hearts demonstrated significantly greater systolic function 600 s after the addition of isoprenaline to elevated [Ca2+]o, compared with hearts perfused with elevated [Ca2+]o and isoprenaline only. PP and dP/dtmax (Figure 5B (i & ii) respectively) were elevated in the K201-treated group (n= 9), compared with with the hearts not treated with K201 (n= 5), following 600 s of perfusion with isoprenaline (P < 0.05, for both). At the same time, SW was significantly higher in K201-treated hearts (n= 4), compared with the isoprenaline only hearts (n= 4; Figure 5B (iii); P < 0.05).
Diastolic parameters during β-adrenoceptor stimulation and elevated [Ca2+]o
End diastolic pressure was not significantly altered in either group [Figure 5B (iv)], but K201-treated hearts (n= 9) demonstrated improved relaxation at 600 s after the addition of isoprenaline, compared with untreated hearts (n= 5), as measured by dP/dtmin (Figure 5B (v); P < 0.05). At the same time, the relaxation constant τ (Figure 5B (vi); P < 0.05) was shorter in the K201-treated hearts, compared with the isoprenaline only hearts (n= 5). dP/dtmin can be altered independently of PP due to changes in relaxation time course. However, the rate of fall in pressure (−dP/dt) was also measured at a fixed pressure (depicted in the left-hand panel of Figure 5C with mean values for −dP/dt at 45 mmHg displayed in the right-hand panel). 45 mmHg was chosen as it represented a pressure value lower than the afterload of 55 mmHg, and therefore a point after which the aortic valves had closed. At 600 s after the addition of isoprenaline, −dP/dt at 45 mmHg was significantly greater in the K201-treated hearts, compared with the isoprenaline only group (P < 0.05). As −dP/dt at 45 mmHg was altered in proportion to PP, it appeared that the two events were related.
Heart rate was not significantly different between groups at any stage of the protocol (at 600 s after the addition of isoprenaline, 218 ± 31 beats·min−1 in the isoprenaline only group (n= 5) and 244 ± 12 beats·min−1 in the isoprenaline + K201 group (n= 9; P > 0.05).
Volume parameters during β-adrenoceptor stimulation and elevated [Ca2+]o
In K201-treated hearts (n= 9), AoF (Figure 6A) was higher in comparison with untreated hearts (n= 5) at 600 s after the addition of isoprenaline (P < 0.05). A similar difference was seen with SV (Figure 6B), ESV (Figure 6C) and EDV (Figure 6D; P < 0.05 for all).
Figure 6.
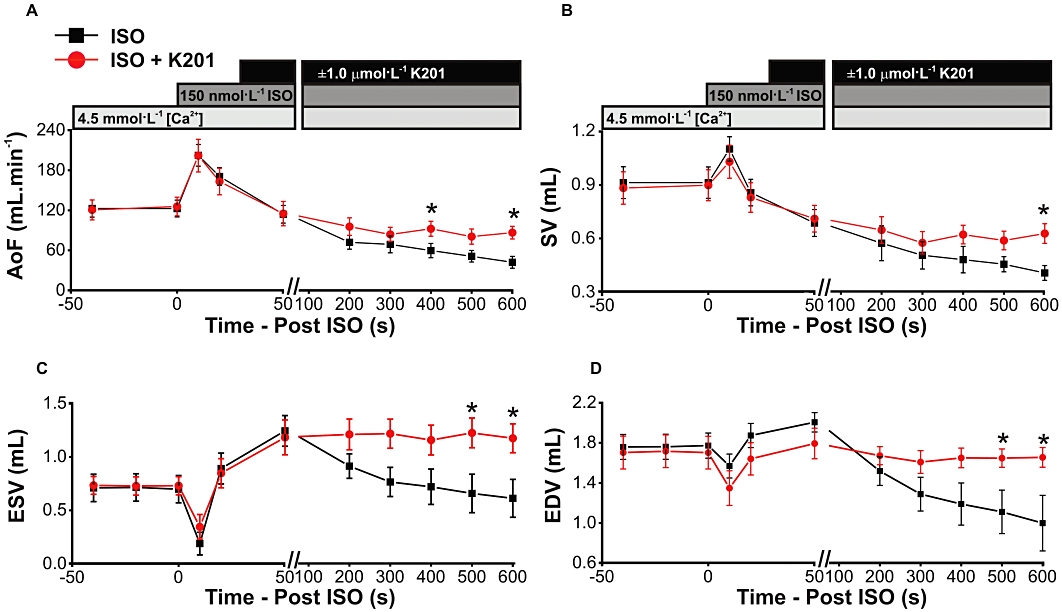
(A–D) Volume indices up to 600 s after perfusion with isoprenaline in untreated (ISO) and K201-treated (ISO+K201) working hearts. *P < 0.05, significant effect of K201.
Functional effects of K201 when added later in the protocol
To further investigate the nature of the contractile dysfunction induced by elevated [Ca2+]o and β-adrenoceptor stimulation, a further set of experiments was performed with addition of K201 at 300 s after isoprenaline, instead of 30 s. It was hypothesized that if the elevated [Ca2+]o and isoprenaline protocol was causing Ca2+-induced cellular dysfunction, rather than irreversible cellular damage, then addition of K201 at a later time point within the protocol would still result in a significant rescue of contractile function. A schematic representation of the protocol used in this subset of experiments is illustrated in Figure 7A, while Figure 7B shows a number of functional indices, measured 600 s after the addition of isoprenaline, in three separate groups; hearts receiving no K201 treatment (isoprenaline only, n= 5), hearts perfused with 1 µmol·L−1 K201, 30 s after isoprenaline (Group 1, n= 9) and hearts perfused with 1 µmol·L−1 K201, 300 s after isoprenaline (Group 2, n= 3). All data in Figure 7B are expressed as % of baseline function which, as indicated on the protocol timeline in Figure 7A, was measured 20 s after the addition of isoprenaline. Compared with the isoprenaline only group, PP was higher in both Group 1 and Group 2 (Figure 7B (i); P < 0.05, for both). The same was true for developed pressure, AoF and SV (Figures 7B, (ii), (iii) and (iv) respectively; P < 0.05 throughout). These data demonstrate that when K201 is added at a later time in the Ca2+ overload protocol, a significant rescue of function can still be achieved.
Figure 7.
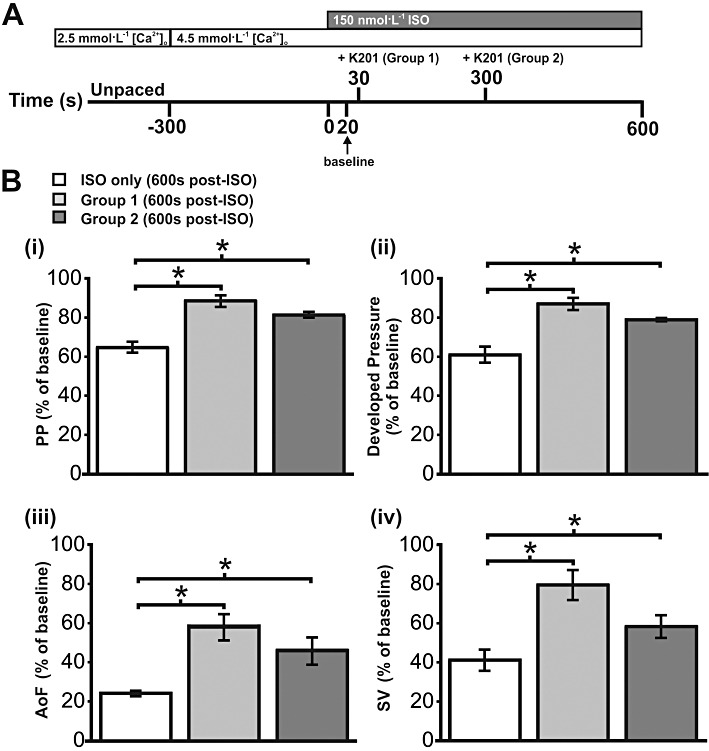
Effect of K201 following extended period of elevated Ca2+ loading. (A) Schematic representation of Ca2+ loading protocol to induce contractile dysfunction. Timeline indicates addition of K201 in Group 1 (30 s after isoprenaline (ISO) addition) and Group 2 (300 s after isoprenaline addition). (B) (i–iv) Functional indices at 600 s after isoprenaline addition expressed relative to baseline in three groups; hearts receiving no K201 treatment (ISO only), hearts treated with K201 30 s after isoprenaline addition (Group 1) and hearts treated with K201 300 s after isoprenaline addition (Group 2). *P < 0.05, significant differences between groups as shown.
The effect of K201 on SR-mediated Ca2+ release in rabbit ventricular cardiomyocytes
In order to assess the effect of K201 on the stimulated Ca2+ transient and spontaneous SR Ca2+ release events during β-adrenoceptor stimulation with elevated [Ca2+]o, a similar protocol to that used in the working rabbit heart was utilized in isolated rabbit cardiomyocytes. Cardiomyocytes (n= 8) were perfused with a modified KH solution containing 4.75 mmol·L−1[Ca2+]o and 150 nmol·L−1 isoprenaline. Cells were field stimulated at 2.5 Hz in order to obtain a sufficient length of time between stimulations in which to characterize diastolic Ca2+ events under these conditions. When cells were perfused with 4.75 mmol·L−1[Ca2+]o and 150 nmol·L−1 isoprenaline, stimulated Ca2+ transients were accompanied by diastolic Ca2+ release events [Figure 8A (i)]. Addition of DMSO (as a vehicle control) did not significantly alter any of the stimulated Ca2+ transient parameters including: the Ca2+ transient peak [Figure 8A (ii) & C (i)], the Ca2+ transient amplitude [Figure 8A (ii) & C (iii)] or other Ca2+ transient parameters (Tau, dCa/dtmax, dCa/dtmin– data not shown). In a separate subset of cells (n= 6) the same protocol was applied [Figure 8B (i)] but with the addition of 1 µmol·L−1 K201 instead of DMSO [Figure 8B (ii)]. Again no significant alteration to the stimulated Ca2+ transient parameters were detected post-K201 with the exception of a significant reduction in the diastolic [Ca2+]i to 59 ± 4% of pre-K201 levels (Figure 8C (ii); P < 0.05); where diastolic [Ca2+]i is defined as the minimum [Ca2+]i in the inter-stimulus interval. DMSO had no significant effect on spontaneous diastolic Ca2+ event amplitude or frequency [Figure 8A (i & ii) & D (i & ii)] but K201 significantly decreased spontaneous diastolic Ca2+ event amplitude by 100% [Figure 8B (i & ii) & D (i & ii)]. Cell shortening was also assessed during this protocol. While all cells visibly contracted when paced under β-adrenoceptor stimulation and an elevated [Ca2+]o, the edge detection system failed to routinely track the large and fast contractions in the presence of diastolic Ca2+ events and therefore provide a reliable systolic measurement. However, diastolic cell length was accurately measured. The diastolic cell length data demonstrated that cells perfused with DMSO contracted over time to 93 ± 2% of pre-DMSO levels, (Figure 8E; P < 0.05). This was in contrast to K201, which enabled the cell to maintain its diastolic cell length (99 ± 1% of pre-K201 values; Figure 8E).
Figure 8.
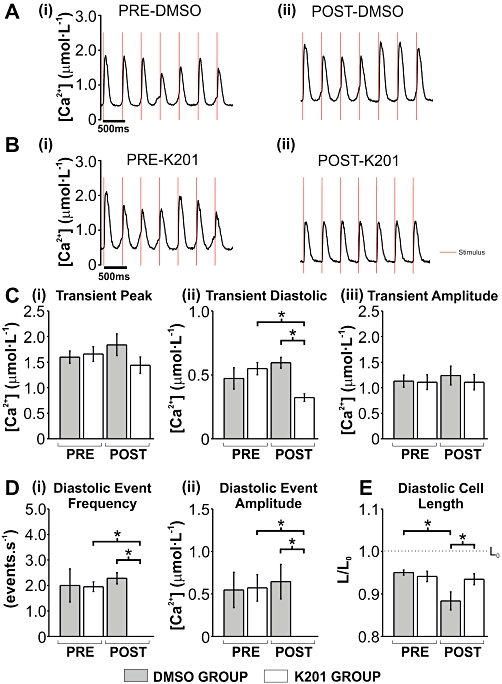
The effect of 1 µmol·L−1 K201 on [Ca2+]i and diastolic cell length in isolated rabbit cardiomyocytes under conditions of elevated Ca2+ loading. (A, B) Typical traces of [Ca2+]i under conditions indicated above each (red line denotes stimulus mark). (C) Mean ± SEM values for stimulated Ca2+ transient (i) peak, (ii) diastolic and (iii) amplitude. (D) Mean ± SEM values for diastolic event (i) frequency and (ii) amplitude. (E) Mean ± SEM values for diastolic cell length (L/L0). DMSO group (n= 8) and K201 group (n= 6); *P < 0.05, significant differences between groups as shown.
Discussion
The in vivo use of PV catheters in small rodents is considered a comprehensive technique for assessing various indices of cardiac mechanical function, particularly in genetically altered mice (Pacher et al., 2008). While in vivo measurements provide valuable, integrated biological data, there are situations where it is necessary to assess cardiac function directly without the influence of anaesthesia, neurohumoral activity or variable cardiac-related loading conditions. This study is the first to combine PV catheters and a rabbit isolated working heart preparation. This approach permits characterization of the direct effect of K201 on working hearts under constant loading conditions independent of the action of the drug on other organs in a species with excitation-contraction coupling similar to human myocardium. This study highlights for the first time the extent to which the negative inotropic effects of K201 depressed the mechanical function of an intact, ejecting whole heart. Moreover, the results demonstrated a clear protective influence of K201 on working heart function under conditions of combined elevated [Ca2+]o loading and β-adrenoceptor stimulation.
K201 has a negative inotropic effect on the intact isolated working heart
The cardioprotective effects of K201 have been demonstrated in a number of studies utilizing different stress-inducing protocols (Kaneko, 1994; Inagaki et al., 2000; Ito et al., 2000); however, little is known about the effects of K201 on contractile function in the normal functioning heart. The results from this study demonstrated that 1 µmol·L−1 K201 depresses, to a small yet significant degree, a number of contractile indices, including dP/dtmax, LV developed pressure, SV and EF. This negative effect was more pronounced with 3 µmol·L−1 K201, such that three of the eight hearts used were not able to sustain AoF less than 4 min after perfusion with K201.
The influence of HR on the negative inotropic effect of K201
K201 dose-dependently decreased HR which in itself may have altered cardiac function in the rabbit heart. However, using a subset of experiments where HR was fixed (paced) throughout the protocol, the direction of the change in the functional parameters was the same as in hearts not paced. Furthermore, there were no significant differences in the magnitude of change in any parameter between these two groups. This suggests that the decline in HR caused by K201 does not explain the negative inotropic action at 1 µmol·L−1.
What are the possible factors contributing to the negative inotropic effect of K201?
Previous studies investigating the mechanism of action of K201 in normally functioning heart preparations are limited. K201 inhibited the Ca2+-dependent binding of Annexin-V to F-actin, a protein that comprises the microfilaments of the cytoskeleton. This effect, however, requires concentrations of 10 µmol·L−1 or higher (Kaneko et al., 1994; 1997). K201 is also known to inhibit sodium (INa) (Kimura et al., 1999) and potassium currents (IK1) (Kimura et al., 1999; Kiriyama et al., 2000) in guinea pig ventricular cardiomyocytes and leads to a shortening of action potential duration. The effect of K201 in rabbit cardiomyocytes on these currents is unknown and warrants further investigation. The authors have extensively investigated the effect of K201 on Ca2+ handling proteins involved in the process of excitation-contraction coupling in rabbit ventricular cardiomyocytes in a previous study (Loughrey et al., 2007). Two of the relevant key findings of this study were that:
1 µmol·L−1 K201 had no effect on ICa,L while 3 µmol·L−1 K201 reduced ICa,L by ∼33% (Loughrey et al., 2007). This is in contrast to the finding in rat and guinea pig ventricular cardiomyocytes where K201 inhibited ICa,L by ∼20% and ∼50% at 1 and 3 µmol·L−1 respectively (Kimura et al., 1999; Inagaki et al., 2000). It is conceivable therefore that reductions in ICa,L in rabbit hearts may contribute to the negative inotropic response observed with 3 µmol·L−1 K201 but not with 1 µmol·L−1 K201.
1.0 µmol·L−1 K201 decreased the Ca2+ transient amplitude in normal rabbit ventricular cardiomyocytes by ∼17% and that this may have been the result of a dose-dependent inhibition of SR Ca2+ uptake via the sarcoplasmic/endoplasmic Ca2+ ATPase pump, SERCA (Loughrey et al., 2007). It is possible that the negative inotropic action of K201 in the whole heart may relate to an action on intracellular Ca2+ handling within ventricular cardiomyocytes. The observation that load-independent contractility, as assessed by PRSW, was reduced by 1 µmol·L−1 K201 could be explained by an inhibitory effect of K201 on SERCA resulting in a reduction of Ca2+ availability to the myofilaments under any given set of loading conditions and, in turn, a decline in intrinsic contractility. Inhibition of SR Ca2+ uptake via SERCA may be expected to contribute to delayed relaxation and an increase in diastolic stiffness (Kass et al., 2004). However, the results of Loughrey et al. (2007) demonstrated that at 1 µmol·L−1 K201 (in the absence of Ca2+ overload), the mean diastolic Ca2+ levels were not significantly altered and this may explain the lack of effect on load-independent diastolic stiffness found in the current study. These observations are further supported by additional data, which include: (i) the dP/dtmax–EDV relationship, an index of intrinsic contractility (Little, 1985; Joho et al., 2007), which was (like PRSW) significantly reduced in the presence of 1 µmol·L−1 K201; (ii) measuring EDPVR using a previously published alternative elastic model (Kawaguchi et al., 2003), which was not significantly altered; and (iii) dP/dtmin–EDV, a comparable diastolic functional index (Wallace et al., 1995), which was also not significantly altered (data not shown). While dP/dtmin was significantly decreased and the relaxation constant τ significantly elevated following application of 1 µmol·L−1 K201, these parameters are not load-insensitive, do not reflect the intrinsic diastolic function of the ventricle and are most likely to be reduced because of the decrease in PP by K201.
It is interesting to note that higher doses of K201 (∼13 µmol·L−1, although the exact systemic concentrations are unclear given the uncertainties of the degree of renal clearance or degradation of K201) have been infused intra-renally in dogs. While the infusion had a beneficial effect on glomerular filtration rate, the net effect included a decrease of CO, mean arterial BP and HR the mechanism of which was attributed to a possible sympatho-inhibitory action of K201. Given the results of the current study, an alternative explanation is that the observed decrease in CO and HR demonstrated during intra-renal infusion was a direct effect of K201 on the heart.
The effect of K201 during β-adrenoceptor stimulation and an elevated [Ca2+]o
K201 was initially associated with an ability to suppress cardiomyocyte death via Ca2+ overload induced by combined application of adrenaline and Ca2+ loading (Kaneko, 1994). More recently, an in vivo study demonstrated that a combination of elevated [Ca2+]o and β-adrenoceptor stimulation resulted in diastolic dysfunction in rats (Kaneko et al., 2006b). These effects were not apparent when Ca2+ loading or adrenaline was applied individually. These effects can be limited by K201 (Kaneko et al., 2006a); however, the possibility that this improvement of cardiac function is primarily due to a systemic effect of K201 remains unknown. Using the isolated working heart model, perfusion with isoprenaline following 5 min of an increased [Ca2+]o ultimately resulted in a decline in function. Perfusion with K201 30 s after isoprenaline attenuated this decline allowing more hearts to maintain AoF for a significantly longer period of time.
Possible effects of K201 during β-adrenoceptor stimulation and elevated [Ca2+]o
There are a number of possible mechanisms by which K201 may have elicited its cardioprotective effect in this setting. One possibility is that K201 limited the formation of myocardial lesions and subsequent contracture induced by isoprenaline and elevated [Ca2+]o (Kaneko, 1994). The decrease in EDV (a change that is prevented by addition of K201) may support this theory. However, K201 resulted in a significant ‘rescue’ of mechanical functional parameters including PP and SV when applied at a later stage of the protocol [300 s vs. 30 s post-ISO; Figure 7B (i–iv)]. This suggests that (at least up to 5 min after isoprenaline) irreversible cellular damage, for example cellular apoptosis and necrosis, may not be the primary mechanism for LV dysfunction. Another explanation for the protective effect of K201 during β -adrenoceptor stimulation is the possibility that K201 interacts directly with the β-adrenoceptor receptor to inhibit the action of isoprenaline. It is unlikely that this mechanism plays a major role as 1 µmol·L−1 K201 prevented cell apoptosis and necrosis in adult rat ventricular cardiomyocytes exposed to 10 µmol·L−1 isoprenaline without significantly altering the isoprenaline-induced activation of protein kinase A (Ellison et al., 2007).
The effect of K201 to reduce isoprenaline-induced diastolic Ca2+ events
Another key finding of a previous publication by the authors is that K201 limits spontaneous SR-mediated diastolic Ca2+ events (Ca2+ waves) in isolated rabbit ventricular cardiomyocytes (Loughrey et al., 2007); a finding that has been confirmed in other species (Hunt et al., 2007; Sedej et al., 2010). Ca2+ waves in ventricular cardiomyocytes can be produced via increases in [Ca2+]i (Ca2+ overload) enabling the SR Ca2+ content to reach threshold for spontaneous diastolic Ca2+ release. In the study by Loughrey et al. (2007), diastolic Ca2+ events in rabbit cardiomyocytes were induced by Ca2+ overload achieved via a raised [Na+]i and [Ca2+]o (Loughrey et al., 2007). The mechanism by which K201 limits diastolic Ca2+ events in rabbit cardiomyocytes is attributed to a co-inhibition of RyR2 and SERCA (Loughrey et al., 2007). Although it is known that during the conditions used in the current study (4.5 mmol·L−1[Ca2+]o and β-adrenoceptor stimulation K201 limits the production of diastolic Ca2+ events in rat cardiomyocytes (Elliott et al., 2008) the effect on rabbit cardiomyocytes under these same conditions is unknown. Figure 8 demonstrates that perfusion with 4.75 mmol·L−1[Ca2+]o and β-adrenoceptor stimulation induced spontaneous diastolic Ca2+ events, which were prevented by K201. The SR Ca2+ content is an important determinant of the propensity for diastolic Ca2+ events to occur and the amplitude of the Ca2+ transient. One mechanism by which K201 may reduce diastolic Ca2+ release events is by a reduction in the SR Ca2+ content therefore preventing the SR Ca2+ content from reaching threshold for spontaneous SR-mediated Ca2+ release (Kashimura et al., 2010). Alternatively, it is also possible that the compound acts solely to inhibit RyR2, which would limit diastolic Ca2+ events but increase SR Ca2+ content (Venetucci et al., 2006). Similar to the previous study that used a raised [Na+]i to induce Ca2+ overload (Loughrey et al., 2007), K201 markedly reduced the diastolic events induced by β-adrenoceptor stimulation with little effect on the stimulated Ca2+ transient amplitude. Therefore, as K201 did not affect Ca2+ efflux from the cell via the sodium-calcium exchanger (Loughrey et al., 2007) the dual inhibition of RyR2 and SERCA is again the most plausible explanation for the action of K201 on diastolic Ca2+ events during Ca2+ overload.
Loughrey et al. (2007) also demonstrated in permeabilized isolated rabbit cardiomyocytes that K201 limited the localized spontaneous SR-mediated diastolic Ca2+ events (Ca2+ sparks). While the conditions used within this study prohibit accurate measurement of Ca2+ sparks while ensuring a constant SR Ca2+ load, it is likely that the known effect of K201 to reduce Ca2+ spark amplitude and frequency (Loughrey et al., 2007) limits diastolic cellular events observed in this study. There are at least two main reasons why it is difficult to measure Ca2+ sparks in this study. First, the conditions used in the rabbit isolated cardiomyocytes are known to result in one or more Ca2+ waves appearing post-stimulation, which will act to empty the SR Ca2+ content to below that present during pacing. Second, the high pacing rates required would limit the diastolic time in which to measure a sufficient number of Ca2+ sparks.
The effect of K201 on diastolic cell length and [Ca2+]i
In isolated whole heart models of ischaemia-reperfusion injury, the beneficial effect of K201 has been attributed to its ability to attenuate a rise of [Ca2+]i associated with reperfusion (Hachida et al., 1999; Inagaki et al., 2000). The current study is the first to measure the effect of K201 on diastolic [Ca2+]i and diastolic cell length in rabbit cardiomyocytes during β-adrenoceptor stimulation. K201 was able to reduce diastolic [Ca2+]i and prevent the diastolic cell shortening, which occurred over the time the cell was exposed to an elevated [Ca2+]o and β-adrenoceptor stimulation. Given the potent effect of K201 in experimental models associated with elevation of [Ca2+]i it is likely that some of its cardioprotective effects in the current study were attributable to a reduction in cellular Ca2+ overload and subsequent maintenance of EDV in working hearts and diastolic cell length in cardiomyocytes. Indeed, under pathological conditions where Ca2+ overload (ischaemia-reperfusion injury) or high circulating catecholamine levels (‘stress’ cardiomyopathy) are thought to be responsible for poor contractile performance, K201 may prove to be an effective treatment.
Effect of K201 on intracellular energy stores
Mitochondria are susceptible to Ca2+ overload, resulting in a reduction in high energy phosphate production, intracellular ATP levels and exhaustion of intracellular energy stores (Fleckenstein et al., 1983; Miyata et al., 1992). In the current study, there were no detectable differences in lactate concentrations between groups during the protocol (results not shown). K201 (1 µmol·L−1) preserves ATP levels during ischaemia and reperfusion, as well as limiting rising Pi during ischaemia (Kawabata et al., 2000). This cardioprotective effect may relate to activation of PKC and mitochondrial KATP channels (Ito et al., 2000). It is possible that lactate release was too indirect a measure of the respiratory state of the muscle and that alternative markers of energy metabolism may have revealed a difference between the groups and this warrants further study.
In conclusion, this study is the first to provide a detailed characterization of the acute effects of K201 on isolated working hearts. It is clear that K201 preserved function during conditions of pharmacologically induced Ca2+ overload by a direct action on the heart. Ca2+ overload was induced in normal hearts without any specific initial pathology, for example myocardial hypertrophy or infarction. This suggests that K201 limits the detrimental effects of Ca2+ overload, regardless of the cause, and therefore use of this drug to reverse Ca2+ dysfunction may be applicable to a range of human myocardial diseases.
Acknowledgments
This work was supported by Medical Research Scotland (grant number 95 FRG to CML & GLS), The Royal Society (grant number 2005/R1 to CML), Tenovus Scotland (grant number SO5/20 to CML) and Heart Research UK (grant number RG2545 to CML & GLS). Aetas Pharma Ltd. for the kind gift of K201.
Glossary
- AoF
aortic flow
- [Ca2+]i
intracellular calcium concentration
- [Ca2+]o
extracellular calcium concentration
- CO
cardiac output
- dP/dtmax
maximum rate of rise in pressure
- dP/dtmin
maximum rate of fall in pressure
- −dP/dt
rate of fall in pressure
- EDP
end diastolic pressure
- EDPVR
end diastolic pressure-volume relationship
- ICa
L-type Ca2+ channel current
- INa
Inward Na+ channel current
- IK1
inward K+ channel current
- LV
left ventricle
- PP
peak pressure
- PRSW
preload recruitable stroke work
- PV
pressure-volume
- RyR2
ryanodine receptor
- SERCA
sarcoplasmic/endoplasmic Ca2+ ATPase pump
- SR
sarcoplasmic reticulum
- SV
stroke volume
- SW
stroke work
Conflicts of interest
NK is employed in the position of Chairman by Aetas Pharma Co. Ltd. Aetas Pharma Co. Ltd. agree with this submission for the British Journal of Pharmacology and believe no conflict of interest arises from it.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th edn. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott E, Matsuda T, Kaneko N, Loughrey C, Smith G. K201 inhibits diastolic Ca2+ release and contractions in isolated rat cardiomyocytes without increase in subsequent transient amplitude. J Biophys. 2008;487:12 B331. [Google Scholar]
- Ellison GM, Torella D, Karakikes I, Purushothaman S, Curcio A, Gasparri C, et al. Acute beta-adrenergic overload produces myocyte damage through calcium leakage from the ryanodine receptor 2 but spares cardiac stem cells. J Biol Chem. 2007;282:11397–11409. doi: 10.1074/jbc.M607391200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleckenstein A, Frey M, Fleckenstein-Grun G. Consequences of uncontrolled calcium entry and its prevention with calcium antagonists. Eur Heart J. 1983;4(Suppl. H):43–50. doi: 10.1093/eurheartj/4.suppl_h.43. [DOI] [PubMed] [Google Scholar]
- Grieve DJ, Cave AC, Byrne JA, Layland J, Shah AM. Analysis of ex vivo left ventricular pressure-volume relations in the isolated murine ejecting heart. Exp Physiol. 2004;89:573–582. doi: 10.1113/expphysiol.2004.027573. [DOI] [PubMed] [Google Scholar]
- Hachida M, Lu H, Kaneko N, Nonoyama M, Koyanagi H. Protective effect of JTV519 on prolonged myocardial preservation. Transplant Proc. 1999;31:1094. doi: 10.1016/s0041-1345(98)02104-6. [DOI] [PubMed] [Google Scholar]
- Hasumi H, Matsuda R, Shimamoto K, Hata Y, Kaneko N. K201, a multi-channel blocker, inhibits clofilium-induced torsades de pointes and attenuates an increase in repolarization. Eur J Pharmacol. 2007;555:54–60. doi: 10.1016/j.ejphar.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Hunt DJ, Jones PP, Wang R, Chen W, Bolstad J, Chen K, et al. K201 (JTV519) suppresses spontaneous Ca2+ release and [3H]ryanodine binding to RyR2 irrespective of FKBP12.6 association. Biochem J. 2007;404:431–438. doi: 10.1042/BJ20070135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki K, Kihara Y, Izumi T, Sasayama S. The cardioprotective effects of a new 1,4-benzothiazepine derivative, JTV519, on ischemia/reperfusion-induced Ca2+ overload in isolated rat hearts. Cardiovasc Drugs Ther. 2000;14:489–495. doi: 10.1023/a:1007884905461. [DOI] [PubMed] [Google Scholar]
- Ito KI, Shigematsu S, Sato T, Abe T, Li Y, Arita M. JTV-519, a novel cardioprotective agent, improves the contractile recovery after ischaemia-reperfusion in coronary perfused guinea-pig ventricular muscles. Br J Pharmacol. 2000;130:767–776. doi: 10.1038/sj.bjp.0703373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joho S, Ishizaka S, Sievers R, Foster E, Simpson PC, Grossman W. Left ventricular pressure-volume relationship in conscious mice. Am J Physiol Heart Circ Physiol. 2007;292:369–397. doi: 10.1152/ajpheart.00704.2006. [DOI] [PubMed] [Google Scholar]
- Kaneko N. New 1,4-benzothiazepine derivative, K201, demonstrates cardio-protective effects against sudden cardiac cell death and intracellular calcium blocking action. Drug Dev Res. 1994;33:429–438. [Google Scholar]
- Kaneko N, Matsuda R, Chiwaki F, Hosoda S. Purification of cardiac annexin V from the beagle dog heart and changes in its localization in the ischemic rat heart. Heart Vessels. 1994;9:148–154. doi: 10.1007/BF01745240. [DOI] [PubMed] [Google Scholar]
- Kaneko N, Matsuda R, Toda M, Shimamoto K. Inhibition of annexin V-dependent Ca2+ movement in large unilamellar vesicles by K201, a new 1,4-benzothiazepine derivative. Biochim Biophys Acta. 1997;1330:1–7. doi: 10.1016/s0005-2736(97)00132-6. [DOI] [PubMed] [Google Scholar]
- Kaneko N, Matsuda R, Nakajima T, Shinozaki T, Ohtani N, Oda K, et al. Norepinephrine-induced diastolic dysfunction with aortic valve opening under calcium-loading in rats. Drug Dev Res. 2006a;67:511–518. [Google Scholar]
- Kaneko N, Matsuda R, Ohtani N, Nakajima T, Arikawa T, Suzuki H, et al. K201 improves norepinephrine-induced diastolic dysfunction with preserved ejection fraction. Drug Dev Res. 2006b;67:852–861. [Google Scholar]
- Kashimura T, Briston SJ, Trafford AW, Napolitano C, Priori SG, Eisner DA, et al. In the RyR2R4496C mouse model of CPVT, β-adrenergic stimulation induces Ca waves by increasing SR Ca content and not by decreasing the threshold for Ca waves. Circ Res. 2010;107:1483–1489. doi: 10.1161/CIRCRESAHA.110.227744. [DOI] [PubMed] [Google Scholar]
- Kass DA, Bronzwaer JG, Paulus WJ. What mechanisms underlie diastolic dysfunction in heart failure. Circ Res. 2004;94:1533–1542. doi: 10.1161/01.RES.0000129254.25507.d6. [DOI] [PubMed] [Google Scholar]
- Kawabata H, Ryomoto T, Ishikawa K. Effect of a novel cardioprotective agent, JTV-519, on metabolism, contraction and relaxation in the ischemia-reperfused rabbit heart. Jpn Circ J. 2000;64:772–776. doi: 10.1253/jcj.64.772. [DOI] [PubMed] [Google Scholar]
- Kawaguchi M, Hay I, Fetics B, Kass D. Combined ventricular systolic and arterial stiffening in patients with heart failure and preserved ejection fraction. Circulation. 2003;107:714–720. doi: 10.1161/01.cir.0000048123.22359.a0. [DOI] [PubMed] [Google Scholar]
- Kimura J, Kawahara M, Sakai E, Yatabe J, Nakanishi H. Effects of a novel cardioprotective drug, JTV-519, on membrane currents of guinea pig ventricular myocytes. Jpn J Pharmacol. 1999;79:275–281. doi: 10.1254/jjp.79.275. [DOI] [PubMed] [Google Scholar]
- Kiriyama K, Kiyosue T, Wang JC, Dohi K, Arita M. Effects of JTV-519, a novel anti-ischaemic drug, on the delayed rectifier K+ current in guinea-pig ventricular myocytes. Naunyn Schmiedebergs Arch Pharmacol. 2000;361:646–653. doi: 10.1007/s002100000230. [DOI] [PubMed] [Google Scholar]
- Lederer WJ, Tsien RW. Transient inward current underlying arrhythmogenic effects of cardiotonic steroids in Purkinje fibres. J Physiol. 1976;263:73–100. doi: 10.1113/jphysiol.1976.sp011622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisy O, Burnett JC., Jr New cardioprotective agent K201 is natriuretic and glomerular filtration rate enhancing. Circulation. 2006;113:246–251. doi: 10.1161/CIRCULATIONAHA.105.558213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little WC. The left ventricular dP/dtmax– end-diastolic volume relation in closed chest dogs. Circ Res. 1985;56:808–815. doi: 10.1161/01.res.56.6.808. [DOI] [PubMed] [Google Scholar]
- Liu N, Ruan Y, Priori SG. Catecholaminergic polymorphic ventricular tachycardia. Prog Cardiovasc Dis. 2008;51:23–30. doi: 10.1016/j.pcad.2007.10.005. [DOI] [PubMed] [Google Scholar]
- Loughrey CM, Otani N, Seidler T, Craig MA, Matsuda R, Kaneko N, et al. K201 modulates excitation-contraction coupling and spontaneous Ca2+ release in normal adult rabbit ventricular cardiomyocytes. Cardiovasc Res. 2007;76:236–246. doi: 10.1016/j.cardiores.2007.06.014. [DOI] [PubMed] [Google Scholar]
- Miyata H, Lakatta EG, Stern MD, Silverman HS. Relation of mitochondrial and cytosolic free calcium to cardiac myocyte recovery after exposure to anoxia. Circ Res. 1992;71:605–613. doi: 10.1161/01.res.71.3.605. [DOI] [PubMed] [Google Scholar]
- Neely JR, Liebermeister H, Battersby EJ, Morgan HE. Effect of pressure development on oxygen consumption by isolated rat heart. Am J Physiol. 1967;212:804–814. doi: 10.1152/ajplegacy.1967.212.4.804. [DOI] [PubMed] [Google Scholar]
- Oda T, Yano M, Yamamoto T, Tokuhisa T, Okuda S, Doi M, et al. Defective regulation of interdomain interactions within the ryanodine receptor plays a key role in the pathogenesis of heart failure. Circulation. 2005;111:3400–3410. doi: 10.1161/CIRCULATIONAHA.104.507921. [DOI] [PubMed] [Google Scholar]
- Pacher P, Nagayama T, Mukhopadhyay P, Batkai S, Kass DA. Measurement of cardiac function using pressure-volume conductance catheter technique in mice and rats. Nat Protoc. 2008;3:1422–1434. doi: 10.1038/nprot.2008.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedej S, Heinzel FR, Walther S, Dybkova N, Wakula P, Groborz J, et al. Na+-dependent SR Ca2+ overload induces arrhythmogenic events in mouse cardiomyocytes with a human CPVT mutation. Cardiovasc Res. 2010;87:50–59. doi: 10.1093/cvr/cvq007. [DOI] [PubMed] [Google Scholar]
- Seidler T, Loughrey CM, Zibrova D, Kettlewell S, Teucher N, Kogler H, et al. Overexpression of FK-506 binding protein 12.0 modulates excitation contraction coupling in adult rabbit ventricular cardiomyocytes. Circ Res. 2007;101:1020–1029. doi: 10.1161/CIRCRESAHA.107.154609. [DOI] [PubMed] [Google Scholar]
- Shannon TR, Pogwizd SM, Bers DM. Elevated sarcoplasmic reticulum Ca2+ leak in intact ventricular myocytes from rabbits in heart failure. Circ Res. 2003;93:592–594. doi: 10.1161/01.RES.0000093399.11734.B3. [DOI] [PubMed] [Google Scholar]
- Tateishi H, Yano M, Mochizuki M, Suetomi T, Ono M, Xu X, et al. Defective domain-domain interactions within the ryanodine receptor as a critical cause of diastolic Ca2+ leak in failing hearts. Cardiovasc Res. 2009;81:536–545. doi: 10.1093/cvr/cvn303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venetucci LA, Trafford AW, Diaz ME, O'Neill SC, Eisner DA. Reducing ryanodine receptor open probability as a means to abolish spontaneous Ca2+ release and increase Ca2+ transient amplitude in adult ventricular myocytes. Circ Res. 2006;98:1299–1305. doi: 10.1161/01.RES.0000222000.35500.65. [DOI] [PubMed] [Google Scholar]
- Wallace A, Lam H-W, Mangano DT. Linearity, load dependence, hysteresis and clinical associations of systolic and diastolic indices of left ventricular function in man. J Card Surg. 1995;10:460–467. doi: 10.1111/j.1540-8191.1995.tb00678.x. [DOI] [PubMed] [Google Scholar]
- Wehrens XHT, Lehnart SE, Reiken S, van der Nagel R, Morales R, Sun J, et al. Enhancing calstabin binding to ryanodine receptors improves cardiac and skeletal muscle function in heart failure. Proc Natl Acad Sci USA. 2005;102:9607–9612. doi: 10.1073/pnas.0500353102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano M, Kobayashi S, Kohno M, Doi M, Tokuhisa T, Okuda S, et al. FKBP12.6-mediated stabilization of calcium-release channel (ryanodine receptor) as a novel therapeutic strategy against heart failure. Circulation. 2003;107:477–484. doi: 10.1161/01.cir.0000044917.74408.be. [DOI] [PubMed] [Google Scholar]