

Abstract
BACKGROUND AND PURPOSE
Oxidative stress plays a critical role in liver fibrogenesis. Reactive oxygen species (ROS) stimulate hepatic stellate cells (HSCs), and ROS-mediated increases in calcium influx further increase ROS production. Azelnidipine is a calcium blocker that has been shown to have antioxidant effects in endothelial cells and cardiomyocytes. Therefore, we evaluated the anti-fibrotic and antioxidative effects of azelnidipine on liver fibrosis.
EXPERIMENTAL APPROACH
We used TGF-β1-activated LX-2 cells (a human HSC line) and mouse models of fibrosis induced by treatment with either carbon tetrachloride (CCl4) or thioacetamide (TAA).
KEY RESULTS
Azelnidipine inhibited TGF-β1 and angiotensin II (Ang II)-activated α1(I) collagen mRNA expression in HSCs. Furthermore, TGF-β1- and Ang II-induced oxidative stress and TGF-β1-induced p38 and JNK phosphorylation were reduced in HSCs treated with azelnidipine. Azelnidipine significantly decreased inflammatory cell infiltration, pro-fibrotic gene expressions, HSC activation, lipid peroxidation, oxidative DNA damage and fibrosis in the livers of CCl4- or TAA-treated mice. Finally, azelnidipine prevented a decrease in the expression of some antioxidant enzymes and accelerated regression of liver fibrosis in CCl4-treated mice.
CONCLUSIONS AND IMPLICATIONS
Azelnidipine inhibited TGF-β1- and Ang II-induced HSC activation in vitro and attenuated CCl4- and TAA-induced liver fibrosis, and it accelerated regression of CCl4-induced liver fibrosis in mice. The anti-fibrotic mechanism of azelnidipine against CCl4-induced liver fibrosis in mice may have been due an increased level of antioxidant defence. As azelnidipine is widely used in clinical practice without serious adverse effects, it may provide an effective new strategy for anti-fibrotic therapy.
Keywords: reactive oxygen species, LX-2, carbon tetrachloride, thioacetamide, signal transduction
Introduction
Liver fibrosis is characterized by excessive production and deposition of extracellular matrix proteins, especially type I collagen (Bataller and Brenner, 2001). Liver fibrosis is chiefly defined by cellular activation of hepatic stellate cells (HSCs) and elevated activity of TGF-β1 and its downstream cellular mediators (Gressner and Weiskirchen, 2006). HSCs are a major source of type I collagen and remain quiescent in the healthy liver (Kisseleva and Brenner, 2007). In response to liver injury, HSCs become active, developing a myofibroblast-like phenotype linked with increased proliferation and collagen synthesis (Arthur, 2000). Cells other than HSCs are also involved in collagen production (Kisseleva and Brenner, 2007). Activation of HSCs into collagen-producing cells is stimulated by pro-fibrogenic cytokines released from injured hepatocytes, Kupffer cells and inflammatory cells (Kisseleva and Brenner, 2007).
TGF-β1 is the most powerful pro-fibrogenic cytokine (Kisseleva and Brenner, 2007). TGF-β1, which is secreted by activated Kupffer cells and sinusoidal endothelial cells, causes apoptosis of hepatocytes and elicits the activation and recruitment of inflammatory cells into the injured liver and the differentiation of liver resident cells (e.g. fibroblasts, HSCs, epithelial cells) into collagen-producing myofibroblasts (Kisseleva and Brenner, 2007). In turn, activated HSCs themselves can generate TGF-β1, accelerating hepatocyte damage and lymphocyte infiltration (Breitkopf et al., 2005).
Oxidative stress plays a critical role in liver fibrogenesis (Siegmund and Brenner, 2005). Injured hepatocytes, and to some extent Kupffer cells and neutrophils, function as major sources of reactive oxygen species (ROS) during the early stages of liver fibrosis (Kisseleva and Brenner, 2007). ROS stimulate HSCs in a paracrine manner (Bataller and Brenner, 2005). In turn, activated HSCs can also produce ROS (Siegmund and Brenner, 2005). Notably, antioxidants have been reported to have a protective effect against ROS-mediated liver fibrosis. Activated HSCs express L-type voltage-operated calcium channels (Bataller et al., 2001), and the channels are activated or up-regulated by TGF-β1 (Oide and Thurman, 1994), although partially contradictory data (Roth-Eichhorn et al., 1999) have been reported. ROS can also regulate the function of L-type voltage-operated calcium channels (Hool, 2008). In addition, there is reportedly a direct connection between TGF-β-mediated accumulation of H2O2 and up-regulation of the α1(I) collagen (COL1A1) gene in HSCs (García-Trevijano et al., 1999).
Angiotensin II (Ang II), a vasoconstricting agonist implicated in the pathogenesis of liver fibrosis (Kisseleva and Brenner, 2011), plays an important role in ROS formation by activating NADPH oxidase in HSCs (De Minicis and Brenner, 2007). Ang II induces an increase in the intracellular calcium current and accelerates the contraction and growth of HSCs (Baik et al., 2003). Therefore, inhibition of ROS production and calcium influx in HSCs in response to TGF-β1 or Ang II may be a useful strategy for inhibiting HSC activation and liver fibrosis.
Azelnidipine is a novel long-acting dihydropyridine calcium blocker with selectivity for L-type calcium channels, which is used as an antihypertensive drug (Wellington and Scott, 2003). Azelnidipine has been shown to have antioxidative effects in endothelial cells (Yamagishi et al., 2004) and cardiomyocytes (Koyama et al., 2007). Therefore, we evaluated the anti-fibrotic and antioxidative effects of azelnidipine on HSCs activated by TGF-β1 or Ang II, as well as its effects in two well-established experimental animal models of fibrosis, carbon tetrachloride (CCl4)- and thioacetamide (TAA)-induced fibrosis (Tsukamoto et al., 1990).
Methods
Materials
TGF-β1, vitamin E, 3-(4,5-dimethyl-thiazole-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), the MAPK kinase 1 (MEK1) inhibitor, PD98059, carbon tetrachloride, thioacetamide and Ang II were purchased from Sigma-Aldrich, Inc. (Saint Louis, MO, USA). Nitrendipine, nifedipine, diltiazem and ISOGEN RNA extraction reagent were purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). Amlodipine was purchased from LKT laboratories, Inc. (Saint Paul, MN, USA). Transcriptor First strand cDNA Synthesis Kit was purchased from Roche Diagnostics K.K. (Tokyo, Japan). Human Collagen type 1 elisa kit was purchased from Applied Cell Biotechnologies (Kanagawa, Japan). Total Glutathione Quantification Kit and Fura-2 AM was purchased from Dojindo Molecular Technologies, Inc. (Tokyo, Japan). The BCA Protein Assay Kit was purchased from Thermo Fisher Scientific, K.K. (Kanagawa, Japan). Hepatic hydroxyproline assay kit was purchased from BioVision (Mountain View, CA, USA). JNK inhibitor, SP600125 and p38 MAPK inhibitor, SB203580 were purchased from Calbiochem (La Jolla, CA, USA). Dulbecco's modified Eagle's medium, the Invitrogen Reverse Transcription System and fluorescent probe 2′,7′-dichlorodihydrofluorescein diacetate were purchased from Invitrogen (Tokyo, Japan). Phospho-ERK1/2 antibody, ERK1 antibody, phospho-p38 antibody and p38 antibody were purchased from BD Biosciences (San Jose, CA, USA). Phospho-JNK antibody, JNK antibody, phospho-Smad3 antibody and Smad3 antibody were purchased from Cell Signaling Technology, Inc. (Beverly, MA, USA). Vectastain ABC kit was purchased from Vector Laboratories Inc. (Burlingame, CA, USA). Anti 8-hydroxy-2′-deoxyguanosine (8-OHdG) antibody was purchased from NIKKEN SEIL Co., Ltd. (Shizuoka, Japan). α-Smooth muscle actin (α-SMA) antibody and myeloperoxidase (MPO) antibody were purchased from Abcam Inc. (Tokyo, Japan). Direct 8-iso-Prostaglandin F2α Enzyme Immunoassay Kit was purchased from Assay Designs, Inc. (Ann Arbor, MI, USA). Acyl-coenzyme A oxidase 1 (ACOX1) antibody, nonphagocytic oxidase 2 (Nox2) antibody, catalase antibody, glutathione peroxidase-1 (GPx-1) antibody, superoxide dismutase-1 (SOD-1) antibody, superoxide dismutase-2 (SOD-2) antibody and actin antibody were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Cytochrome P450 2E1 (CYP2E1) antibody was purchased from Enzo Life Sciences, Inc. (Farmingdale, NY, USA). CYP2B1 antibody was purchased from Daiichi Pure Chemicals Co., Ltd. (Tokyo, Japan). Azelnidipine (racemate) (R)-(−)-azelnidipine and (S)-(+)-azelnidipine were provided from Daiichi-Sankyo Pharmaceuticals (Tokyo, Japan). In principle, azelnidipine (racemate) is described as azelnidipine.
HSCs and cell cultures
LX-2, an immortalized human HSC line, was kindly provided by Dr SL Friedman, Mount Sinai School of Medicine, NY. LX-2 cells were seeded overnight at 3 × 106 cells per well in six-well plates or 1 × 106 cells per dish in 10 cm dishes in Dulbecco's modified Eagle's medium supplemented with 2% fetal bovine serum, 2 mM l-glutamine, 100 IU penicillin and 100 mg·mL−1 streptomycin. The cells were incubated in a 95% air, 5% CO2–air humidified atmosphere at 37°C and cultured for 1 or 2 days for various assays. At subconfluence, cells were deprived of serum for 36 h by placing them in serum-free Dulbecco's modified Eagle's medium with 0.1% albumin. Then TGF-β1 or Ang II was added, and the cells were incubated for an additional 24 h. Then cells were retrieved and used for RNA expression analysis, type I collagen assays or measurement of glutathione production. The calcium blockers and vitamin E were added 6 h before addition of TGF-β1. The MAPK inhibitors were added 1 h before addition of TGF-β1. Cell cultures for the other analyses are described in detail in the relevant sections below.
Cell viability assays
The cytotoxicity of different concentrations of azelnidipine was evaluated using the MTT assay. The details of the assay are provided in the Supporting methods.
RNA expression analysis
Total RNA extraction from LX-2 cells and the subsequent synthesis of first strand cDNA were performed using ISOGEN RNA extraction reagent and Transcriptor First strand cDNA Synthesis Kit respectively. The details of the assays are provided in the Supporting methods. Specific oligonucleotide primers are given in Table 1.
Table 1.
Specific oligonucleotide primers for real-time PCR
| Species | Genes | Primer sequences |
|---|---|---|
| Mice | Actb | 5′-GTATCCATGAAATAAGTGGTTACAGG-3′, 5′-GCAGTACATAATTTACACAGAAGCAAT-3′ |
| Collagen α1(I) | 5′-CCTCAGGGTATTGCTGGACAAC-3′, 5′-ACCACTTGATCCAGAAGGACCTT-3′ | |
| F4/80 | 5′-CATCTTGCTGGAGACTGT-3′, 5′-CTGCCAAGTTAATGGACTCA-3′ | |
| TIMP-1 | 5′-CATGGAAAGCCTCTGTGGATATG-3′, 5′-GATGTGCAAATTTCCGTTCCTT-3′ | |
| ACOX1 | 5′-TGGTATGGTGTCGTACTTGAATGAC-3′, 5′-AATTTCTACCAATCTGGCTGCAC-3′ | |
| Nox2 | 5′-GAAAACTCCTTGGGTCAGCACT-3′, 5′-ATTTCGACACACTGGCAGCA-3′ | |
| Catalase | 5′-TGAGAAGCCTAAGAACGCAATTC-3′, 5′-CCCTTCGCAGCCATGTG-3′ | |
| GPx-1 | 5′-TTACATTGTTTGAGAAGTGCGA-3′, 5′-CAAAGTTCCAGGCAATGTC-3′ | |
| SOD-1 | 5′-CATTCCATCATTGGCCGT-3′, 5′-TCAGACCACACAGGGAATGTTTA-3′ | |
| SOD-2 | 5′-TGTATATCTCTGGAGAACTGGAC-3′, 5′-GGCCCTCTTGTGACTGTAA-3′ | |
| Human | GADPH | 5′-GAAGGTGAAGGTCGGAGTC-3′, 5′-GAAGATGGTGATGGGATTTC-3′ |
| Collagen α1(I) | 5′-TCCTGGTCCTGCTGGCAAAGAA-3′, 5′-CACGCTGTCCAGCAATACCTTGA-3′ |
Actb, β-actin.
Type I collagen assays
The type I collagen protein concentration in the extracellular matrix of LX-2 cells was quantified by elisa using the Human Collagen type 1 elisa kit according to the manufacturer's instructions.
Measurement of intracellular ROS levels
At subconfluence, LX-2 cells were deprived of serum for 24 h as mentioned above, and then each calcium blocker was added. After 6 h, TGF-β1, Ang II, or H2O2 was added. After 6 h, the medium was changed to 10 µM 2′,7′-dichlorodihydrofluorescein diacetate as the fluorescent probe in PBS, and the cells were incubated for 20 min. The intracellular ROS levels were investigated to detect the liberation of 2′,7′-dichlorofluorescein (DCF). The fluorescence intensity was measured by a spectrophotometer (Flex station, Molecular Devices, Tokyo, Japan) at an excitation of 495 nm and an emission of 525 nm.
Measurement of glutathione production
Glutathione levels in LX-2 cells were determined using the recycling reaction, which improved the sensitivity of total glutathione detection, as follows: 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB) and glutathione react to generate 2-nitro-5-thiobenzoic acid and glutathione disulphide. Glutathione is generated from glutathione disulphide by glutathione reductase and reacts with DTNB again to produce 2-nitro-5-thiobenzoic acid, which is detected by measuring the absorbance at 412 nm.
Measurement of intracellular calcium concentration ([Ca]i)
Changes in [Ca]i were measured in Fura-2 AM-loaded LX-2 cells with a spectrophotometer (Flex station). Briefly, at confluence, LX-2 cells were deprived of serum for 9 h as mentioned before, the culture medium was removed, and the loading buffer, including Fura-2 AM, with or without each calcium blocker, was added and incubated for 1 h. After the cells were washed, recording medium was added. Immediately after TGF-β or Ang II was added, fluorescent calcium ions were detected by monitoring the excitation spectra at 380 nm (calcium-free) and 340 nm (calcium complex) with fixed emission at 510 nm.
Western blotting analysis
At subconfluence, LX-2 cells were deprived of serum for 24 h as mentioned above, and then TGF-β1 was added. At various times after the addition of TGF-β1, the cells were retrieved for time course analysis of the phosphorylation of p38, JNK, ERK and Smad3. Then the TGF-β1 exposure time was optimized for the phosphorylation of each protein. In another series of experiments, 24 h after serum deprivation, azelnidipine or vitamin E was added to the cells. TGF-β1 was added 6 h later, and the cells were retrieved after each optimal exposure time. The details of these assays are provided in the Supporting methods.
Animals, treatment and specimen collection
A total of 104 male C57BL/6 mice were purchased from Charles River Japan Inc. (Kanagawa, Japan). Mice were housed six to eight per cage at room temperature in a 12 h light/dark cycle. A normal chow diet was provided ad libitum 24 h a day, and all mice were given distilled water ad libitum for the duration of the study. All animal care and experimental procedures were performed in accordance with the guidelines for animal care and use established by Gunma University Graduate School of Medicine. Mice were randomly divided into the CCl4 or TAA groups. We used CCl4 (2 mL·kg−1 body weight, dissolved in olive oil [1:9 (v/v)]) or TAA (100 mg·kg−1 body weight, dissolved in water). CCl4, TAA or the corresponding vehicle was i.p. injected into mice three times per week for 6 weeks. Azelnidipine (10 mg·kg−1·day−1) was suspended in 0.5% sodium carboxymethylcellulose and orally given once daily 6 days a week for 6 weeks. The mice were divided into four treatment groups: (1) the control mice, which were not administered CCl4, TAA or azelnidipine but were i.p. injected with the corresponding vehicle; (2) the azelnidipine alone mice, which were injected with vehicle only and orally given azelnidipine; (3) the CCl4 alone or TAA alone mice, which were injected with CCl4 or TAA without azelnidipine treatment; and (4) the CCl4 or TAA plus azelnidipine mice, which were orally given azelnidipine and injected with CCl4 or TAA. As another control, verapamil (10 mg·kg−1·day−1) was administered in the same way as azelnidipine in the CCl4 and TAA groups. Each group comprise six mice. The mice began receiving i.p. injections at 6 weeks of age. Twenty-four hours after the last CCl4 or TAA injection, the mice were deeply anaesthetized with diethyl ether vapour in a chamber. The mice were then exsanguinated via the axillary artery, and the liver was excised. The sera and livers were immediately stored at −80°C until required.
In another set of experiments, mice at 6 weeks of age received CCl4 i.p. injections three times per week for 8 weeks to establish pronounced fibrosis. CCl4 was then stopped, and the peak of fibrosis was calculated to occur 3 days after the last CCl4 injection (day 0 of fibrosis regression). Liver fibrosis regression was then monitored 3 and 7 days after the peak of fibrosis in mice that had received azelnidipine (10 mg·kg−1·day−1) or vehicle (n= 8 mice per group per time point) during fibrosis regression. The method of killing and sample storage were the same as described above.
Analyses of animal specimens
For histological examination, liver tissue specimens embedded in paraffin were stained with haematoxylin–eosin. Immunohistochemical analyses for 8-OHdG, α-SMA and MPO were performed by the avidin–biotin–peroxidase complex method using 8-OHdG antibody, α-SMA antibody or MPO antibody, respectively. The sites of peroxidase binding were determined using the diaminobenzidine method. Sirius red staining was performed according to the usual method. MPO-stained cells and 8-OHdG-stained cells were measured in 10 microscopic fields at a 200-fold magnification by a person unaware of the treatment for each group. The α-SMA-stained area and Sirius red-stained area were measured using the National Institutes of Health (NIH) image software programme in 10 microscopic fields at a 200-fold magnification. Total RNA extraction from the liver was performed using ISOGEN RNA extraction reagent, and the subsequent RNA expression analysis was the same as described above. Specific oligonucleotide primers are shown in Table 1. For Western blotting analysis, liver tissue lysate was used, but microsome extracts were used exclusively for analysis of CYP2E1 and CYP2Bs. Hepatic 8-isoPGF2α concentrations were measured with a Direct 8-iso-Prostaglandin F2α Enzyme Immunoassay Kit. Hepatic hydroxyproline contents were measured using a Hydroxyproline Assay Kit.
Statistical analysis
Data were analysed by one-way anova, except the regression models of CCl4-induced liver fibrosis, which were analysed by Student's unpaired two-sided t-test. Thus, anova was performed unless otherwise indicated. Values are expressed as the means ± SD. Unless indicated, at least three independent experiments were performed.
Results
Effects of azelnidipine on LX-2 cell viability
LX-2 cells could be treated with azelnidipine at concentrations up to 2 µM without an effect on cell viability (Supporting Figure S1A).
Azelnidipine reduces TGF-β1- or Ang II-induced expression of LX-2 COL1A1 mRNA and collagen I protein
Treatment of LX-2 cells with TGF-β1 led to a significant increase in COL1A1 gene expression in a concentration-dependent manner (Figure 1A, a). Treatment of LX-2 cells with 100 nM azelnidipine significantly reduced the up-regulation of COL1A1 mRNA expression that was induced by treatment with 5 ng·mL−1 TGF-β1 alone. Similar results were observed in cells treated with 50 µg·mL−1 of the antioxidant vitamin E (Figure 1A, b). Moreover, azelnidipine reduced the induction of COL1A1 mRNA expression by 10 ng·mL−1 TGF-β1 in a concentration-dependent manner (Figure 1A, c). TGF-β1-induced collagen I protein expression was significantly reduced in LX-2 cells treated with either azelnidipine or vitamin E (Figure 1A, d). Furthermore, pretreatment of LX-2 cells with vitamin E before azelnidipine treatment blocked further azelnidipine-induced inhibition of COL1A1 mRNA expression (data not shown). There were no significant differences in the inhibitory effects on TGF-β1-induced expression of COL1A1 mRNA among cells treated with azelnidipine (racemate), (R)-(−)-azelnidipine or (S)-(+)-azelnidipine (Figure 1B), although the L-type voltage-operated calcium channel blocking activity of (R)-(−)-enantiomer was more potent than that of the (S)-(+)-enantiomer (Koike et al., 2002). TGF-β1-induced expression of COL1A1 mRNA was not significantly changed in LX-2 cells treated with four different calcium blockers, including amlodipine, diltiazem, nifedipine or nitrendipine, at 100 nM (Supporting Figure S1B). TGF-β1-induced expression of COL1A1 mRNA in HSCs was not reduced with 1 or 10 µM nitrendipine or verapamil (data not shown).
Figure 1.
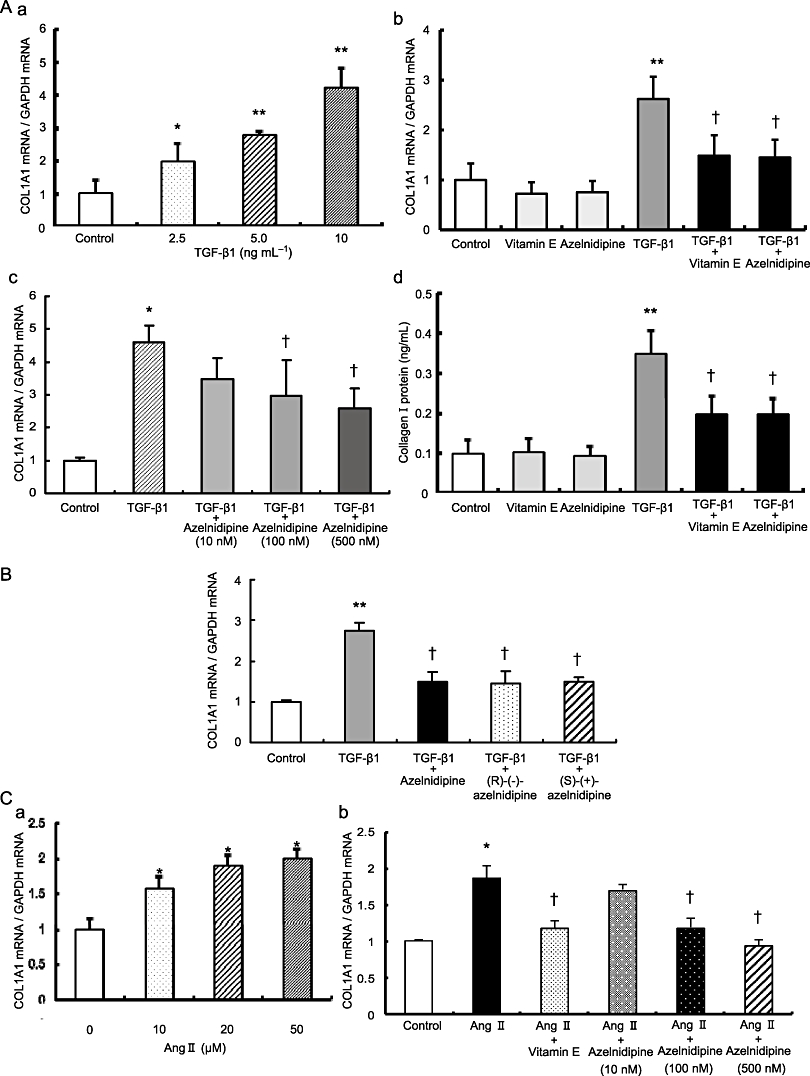
The effects of calcium blockers on TGF-β1 or Ang II-induced stimulation of α1(I) collagen (COL1A1) mRNA and collagen I protein expression in LX-2 cells. (A) TGF-β1-induced stimulation of COL1A1 mRNA and collagen I protein expression and its inhibition by azelnidipine or vitamin E. (a) Concentration-dependent increase in COL1A1 mRNA expression stimulated by the indicated concentrations of TGF-β1. (b) The effects of 100 nM azelnidipine or 50 µg·mL−1 vitamin E on COL1A1 mRNA expression with or without 5 ng·mL−1 TGF-β1. (c) Concentration-dependent reduction of TGF-β1-induced COL1A1 mRNA expression by the indicated concentrations of azelnidipine. The concentration of TGF-β1 was 10 ng·mL−1. (d) The effects of 100 nM azelnidipine or 50 µg·mL−1 vitamin E on collagen I protein expression with or without 5 ng·mL−1 TGF-β1. (B) The effects of azelnidipine (racemate), (R)-(−)-azelnidipine and (S)-(+)-azelnidipine on TGF-β1-induced stimulation of COL1A1 mRNA expression in LX-2 cells. The concentration of each chemical was 100 nM, and the concentration of TGF-β1 was 5 ng·mL−1. (C) Ang II-induced stimulation of COL1A1 mRNA and its inhibition by azelnidipine or vitamin E. (a) Concentration-dependent increase in COL1A1 mRNA expression stimulated by the indicated concentrations of Ang II. (b) Concentration-dependent reduction of Ang II-induced COL1A1 mRNA expression by the indicated concentrations of azelnidipine. The concentration of Ang II was 20 µM. *P < 0.05 and **P < 0.01 versus control, and †P < 0.05 versus corresponding TGF-β1 or Ang II.
Treatment of LX-2 cells with Ang II led to a significant increase in COL1A1 gene expression in a concentration-dependent manner (Figure 1C, a). Treatment of LX-2 cells with azelnidipine significantly reduced the elevated COL1A1 mRNA expression induced by 20 µM Ang II in a concentration-dependent manner, and similar results were observed in cells treated with vitamin E (Figure 1C, b).
Azelnidipine reduces TGF-β1- or Ang II-induced increase in intracellular ROS levels and reverses the TGF-β1-induced decrease in glutathione levels in LX-2 cells
Treatment with 5 ng·mL−1 TGF-β1 or 20 µM Ang II led to a significant increase in DCF fluorescence in LX-2 cells compared with LX-2 cells incubated without TGF-β1 or Ang II, respectively, which is similar to treatment with 100 µM H2O2 as an oxidant (Figure 2A). Intracellular ROS levels induced by TGF-β1, Ang II or H2O2 were significantly reduced in LX-2 cells treated with 100 nM azelnidipine (Figure 2A). However, treatment with amlodipine, diltiazem, nifedipine or nitrendipine did not affect intracellular ROS levels in LX-2 cells (Supporting Figure S1C). Treatment with TGF-β1 led to a significant decrease in glutathione levels in LX-2 cells compared with LX-2 cells incubated without TGF-β1 (Figure 2B). The reduction in glutathione levels induced by TGF-β1 was significantly reversed in LX-2 cells treated with azelnidipine, which was similar to the effect of 50 µg·mL−1 vitamin E (Figure 2B).
Figure 2.
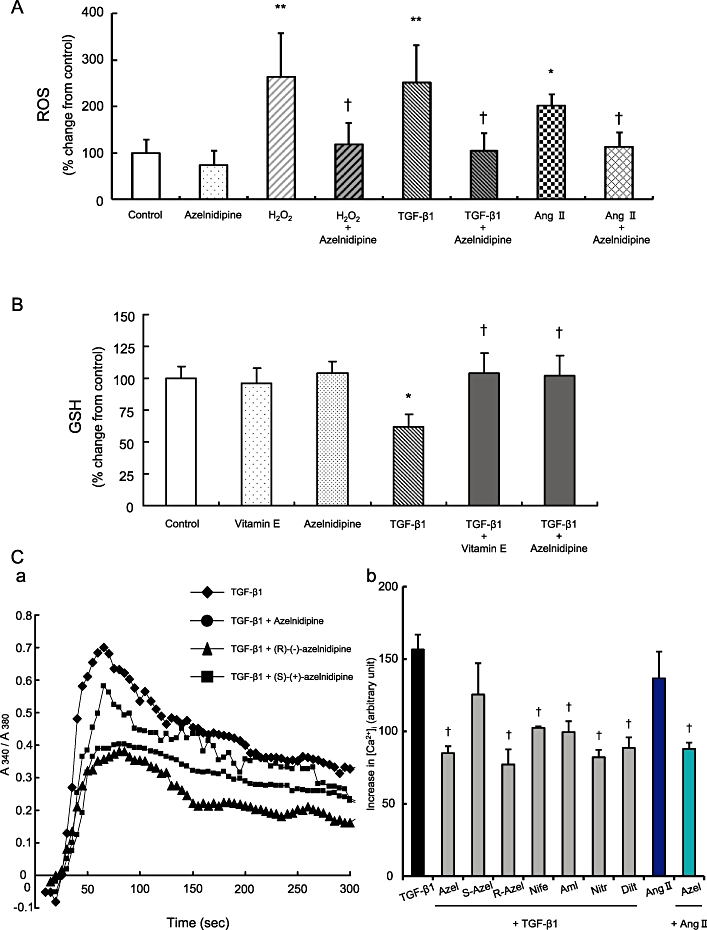
The effects of azelnidipine or vitamin E on stimulant-induced oxidative stress and the effects of various calcium blockers on the stimulant-induced increase in intracellular calcium concentration ([Ca]i) in LX-2 cells. The concentration of each calcium blocker was 100 nM, and TGF-β1, Ang II or H2O2 were applied at 10 ng·mL−1, 50 µM and 50 µM respectively. (A) TGF-β1-, Ang II-, or H2O2-induced increase of intracellular ROS and its inhibition by azelnidipine. (B) TGF-β1-induced decrease in glutathione levels and its inhibition by azelnidipine or vitamin E. (C) TGF-β1 or Ang II-induced increase in [Ca]i in LX-2 cells and its inhibition by various calcium blockers. Representative changes of [Ca]i (a) and the area under the curve (AUC) of [Ca]i with the indicated chemicals in LX-2 cells (b). *P < 0.05 versus control, **P < 0.01 versus control, and †P < 0.05 versus corresponding H2O2, TGF-β1 or Ang II. Azel, azelnidipine; S-Azel, (S)-(+)-azelnidipine; R-Azel, (R)-(+)-azelnidipine; Nife, nifedipine; Aml, amlodipine; Nitr, nitrendipine; Dilt, diltiazem.
Azelnidipine reduces the TGF-β1- or Ang II-induced increase of [Ca]i in LX-2 cells
Treatment with 10 ng·mL−1 TGF-β1 or 50 µM Ang II increased [Ca]i, and all calcium blockers at 100 nM, with the exception of (S)-(+)-azelnidipine, significantly reduced the TGF-β1-induced increase in [Ca]i. Azelnidipine also significantly reduced the Ang II-induced increase in [Ca]i (Figure 2C).
The JNK inhibitor SP600125 and the p38 inhibitor SB203580, but not the MEK1 inhibitor PD98059, inhibit TGF-β1-induced COL1A1 mRNA expression in LX-2 cells
TGF-β1-induced increased expression of COL1A1 mRNA was significantly reduced in LX-2 cells treated with SP600125 or SB203580, but not with PD98059 (Figure 3A).
Figure 3.
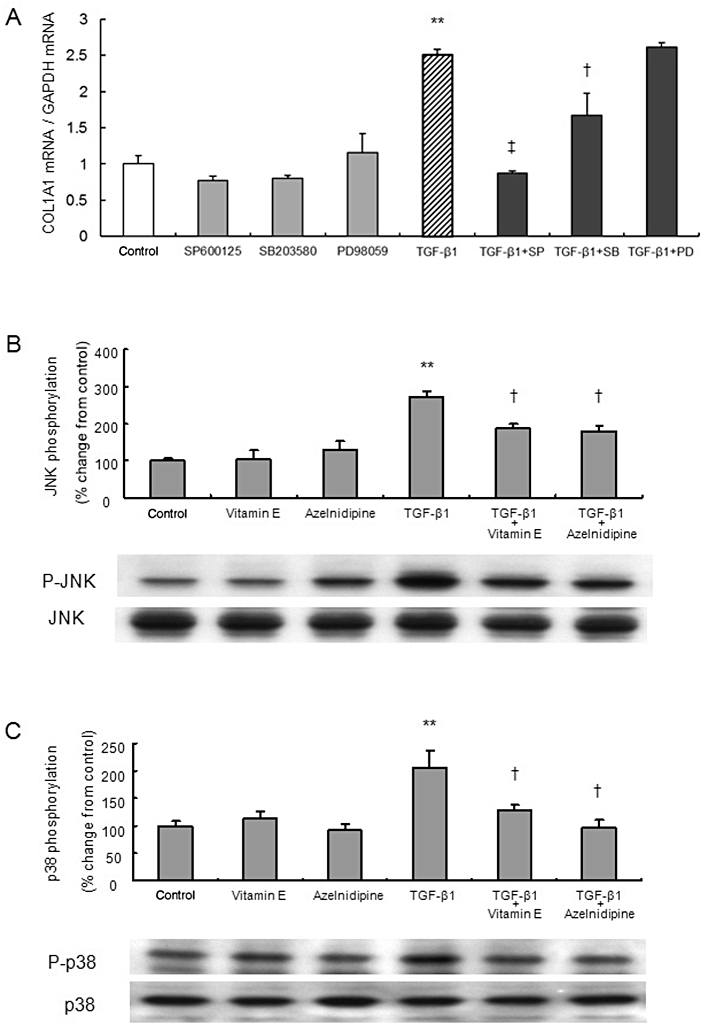
The roles of MAPK signal transduction pathways and the effects of azelnidipine on TGF-β1-induced activation of LX-2 cells. The concentration of azelnidipine was 100 nM, and TGF-β1 and vitamin E were applied at 5 ng·mL−1 and 50 µg·mL−1, respectively. (A) The effects of MAPK inhibitors on TGF-β1-induced COL1A1 mRNA expression. The effect of 10 µM SP600125 (SP), a JNK inhibitor, 10 µM SB203580 (SB), a p38 inhibitor, or 30 µM of PD98059 (PD), a MEK1 inhibitor on COL1A1 mRNA expression with or without stimulation by TGF-β1. (B, C) TGF-β1-induced phosphorylation of JNK or p38 and its inhibition by azelnidipine or vitamin E are indicated. **P < 0.01 versus control, †P < 0.05 and ‡P < 0.01 versus corresponding TGF-β1.
Treatment with TGF-β1 increases phosphorylation of JNK and p38 but not ERK1/2 in LX-2 cells
TGF-β1 induced phosphorylation of JNK and p38. ERK phosphorylation was not affected by TGF-β1 signalling (Supporting Figure S2A).
Azelnidipine inhibits TGF-β1-induced phosphorylation of p38 and JNK in LX-2 cells
TGF-β1-induced phosphorylation of JNK was significantly reduced in LX-2 cells that had been treated with azelnidipine or vitamin E (Figure 3B). TGF-β1-induced phosphorylation of p38 was significantly reduced in LX-2 cells treated with azelnidipine, and the effect was greater than or comparable with that of vitamin E (Figure 3C). Treatment of LX-2 cells with azelnidipine alone did not significantly affect phosphorylation of JNK (Figure 3B), p38 (Figure 3C) or ERK1/2 (Supporting Figure S2B).
TGF-β1-induced phosphorylation of Smad3 is not inhibited by azelnidipine in LX-2 cells
TGF-β1 increased phosphorylation of Smad3, but the increase was not inhibited by azelnidipine in LX-2 cells (Supporting Figure S2C and D).
Azelnidipine reduces macrophage and neutrophil infiltration in CCl4- or TAA-treated mice
F4/80 mRNA expression levels in liver tissue in the control mice were similar to those in mice treated with azelnidipine alone (Figure 4A, a). F4/80 mRNA expression levels in mice treated with CCl4 plus azelnidipine were significantly lower than those in mice treated with CCl4 alone (Figure 4A, a). The same was true for F4/80 mRNA expression levels in the TAA-treated mice (Figure 4A, b). There were no MPO-stained cells in the samples from control mice or the mice treated with azelnidipine alone (Figure 4B, a). The number of MPO-stained cells in the mice treated with CCl4 plus azelnidipine was significantly lower than that in mice treated with CCl4 alone (Figure 4B, a). The same was true for the number of MPO-stained cells in the TAA-treated mice (Figure 4B, b).
Figure 4.
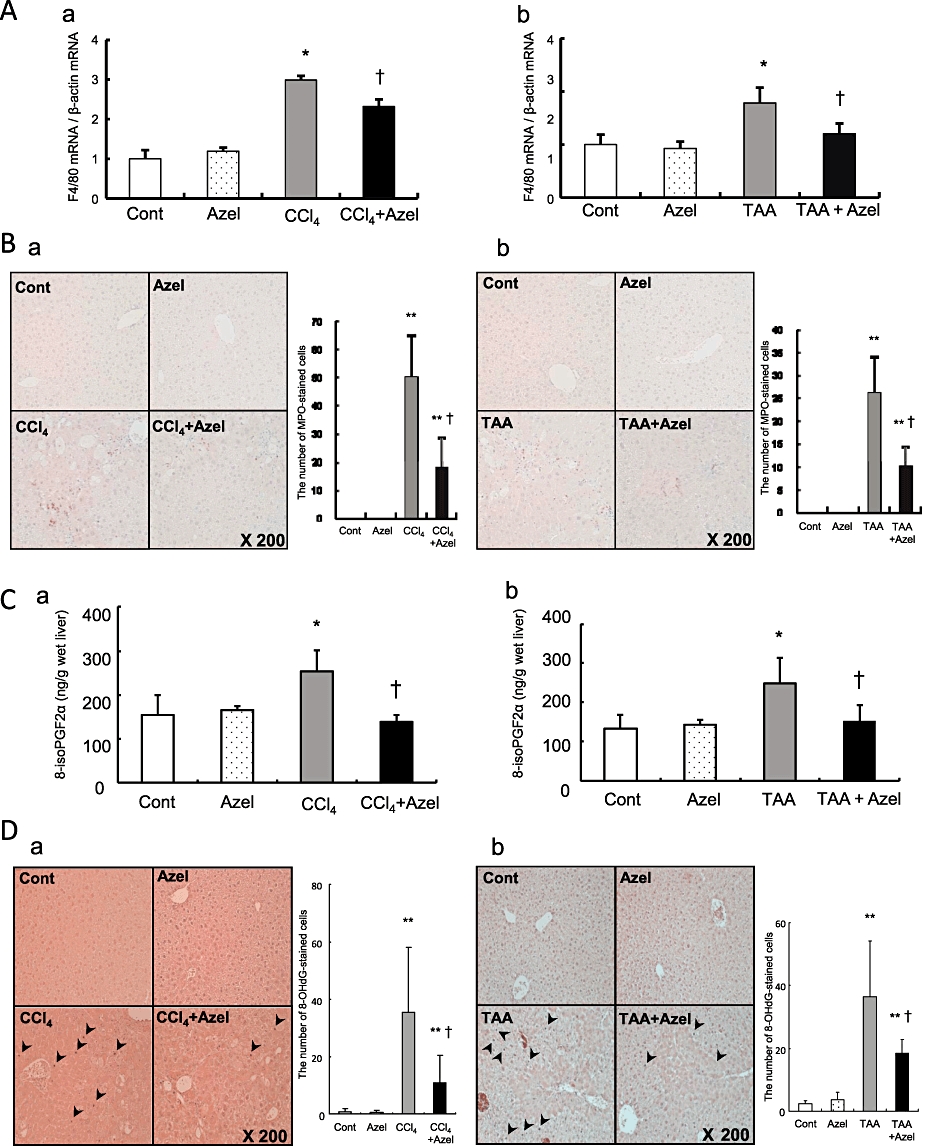
The effects of azelnidipine on carbon tetrachloride (CCl4)- or thioacetamide (TAA)-induced liver injury in mice. Mice in CCl4 groups are grouped as follows: Cont, control (no CCl4, no azelnidipine); Azel, azelnidipine alone (with azelnidipine, no CCl4); CCl4, CCl4 alone (with CCl4, no azelnidipine); CCl4+ Azel, CCl4 plus azelnidipine (with CCl4 and azelnidipine). Mice in the TAA groups are shown as with the CCl4 groups except using TAA instead of CCl4. F4/80 mRNA expression (A), myeloperoxidase (MPO) staining (B), hepatic 8-isoPGF2α concentrations (C) and 8-hydroxy-2′-deoxyguanosine (8-OHdG) staining (D) in the CCl4 groups (a) or the TAA groups (b) are shown. MPO-stained cells and 8-OHdG-stained cells (arrowhead) were quantified in ten microscopic fields at a 200-fold magnification. *P < 0.05 and **P < 0.01 versus Cont, †P < 0.05 versus CCl4 alone.
Azelnidipine reduces lipid peroxidation and oxidative DNA damage in livers of CCl4- and TAA-treated mice
Hepatic 8-isoPGF2α concentrations and the number of 8-OHdG-stained cells in the control mice were similar to those in mice treated with azelnidipine alone (Figure 4C, a and D, a). Hepatic 8-isoPGF2α concentrations and the number of 8-OHdG-stained cells in mice treated with CCl4 plus azelnidipine were significantly lower than those in mice treated with CCl4 alone (Figure 4C, a and D, a). The same was true for hepatic 8-isoPGF2α concentrations and the number of 8-OHdG-stained cells in the TAA-treated mice (Figure 4C, b and D, b).
Azelnidipine reduces hepatic pro-fibrotic gene expression, liver fibrosis and HSC activation in CCl4- and TAA-treated mice
The mRNA expression levels of COL1A1 and TIMP-1 in the control mice were similar to those in mice treated with azelnidipine alone (Figure 5A, a and B, a). The mRNA expression levels of COL1A1 and TIMP-1 in mice treated with CCl4 plus azelnidipine were significantly lower than those in mice treated with CCl4 alone (Figure 5A, a and B, a). The same was true for the mRNA expression levels of COL1A1 and TIMP-1 in the TAA-treated mice (Figure 5A, b and B, b). Hepatic hydroxyproline contents and the Sirius red-stained area in the control mice were similar to those in mice treated with azelnidipine alone (Figure 5C, a and D, a). Hepatic hydroxyproline contents and the Sirius red-stained area in mice treated with CCl4 plus azelnidipine were significantly lower than those in mice treated with CCl4 alone (Figure 5C, a and D, a). The same was true for hepatic hydroxyproline contents and the Sirius red-stained area in the TAA-treated mice (Figure 5C, b and D, b). No expression of α-SMA was observed in either the control mice or the mice treated with azelnidipine alone (Figure 5E, a). The expression levels of α-SMA in mice treated with CCl4 plus azelnidipine were significantly lower than those in mice treated with CCl4 alone (Figure 5E, a). The same was true for the expression levels of α-SMA in the TAA-treated mice (Figure 5E, b).
Figure 5.
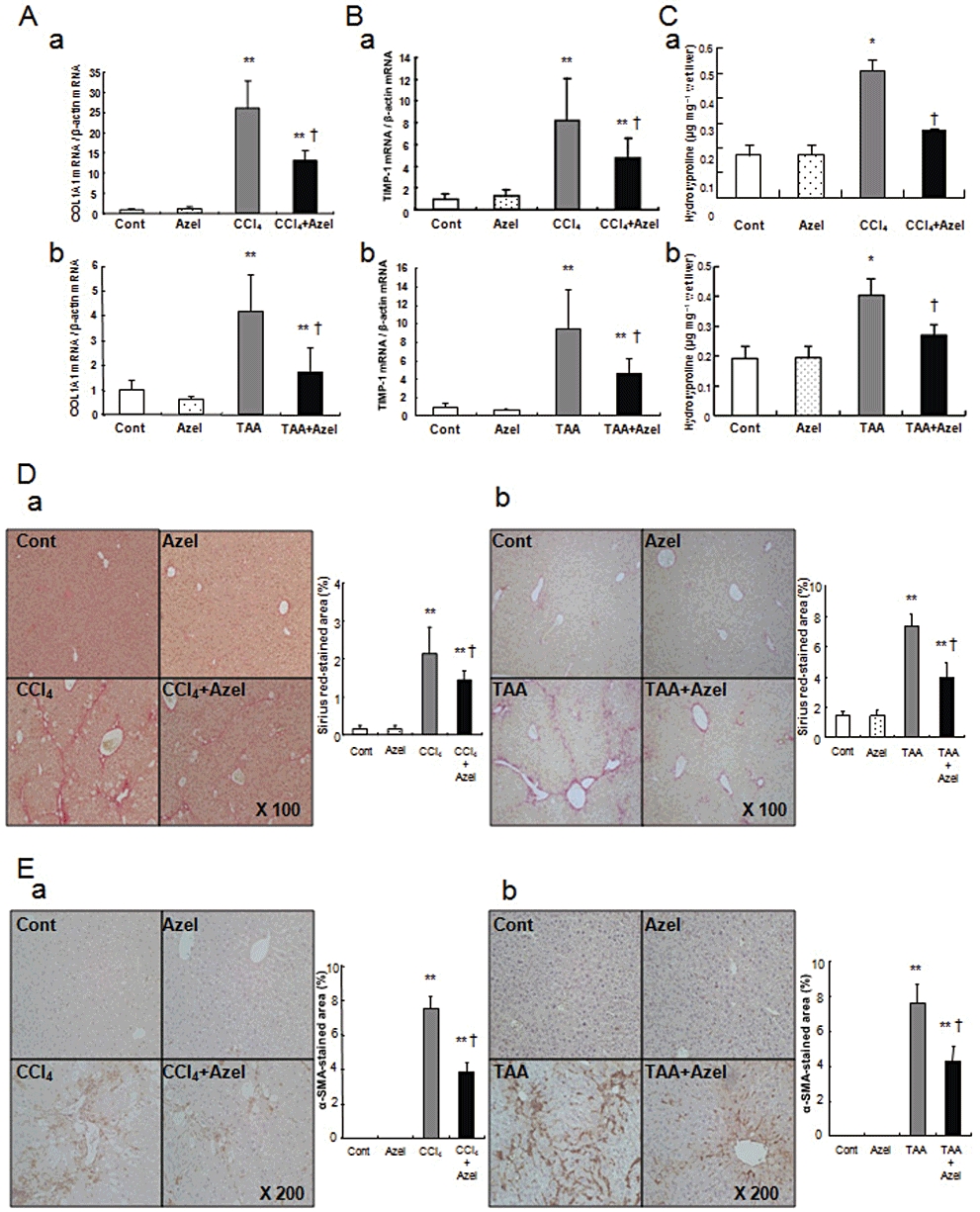
The effects of azelnidipine on CCl4- or TAA-induced fibrogenesis in mice. The groups are the same as those in Figure 4. Mice in the TAA groups are shown as with the CCl4 groups except TAA was used instead of CCl4. COL1A1 mRNA expression (A), TIMP-1 mRNA expression (B), hepatic hydroxyproline contents (C), Sirius red staining (D) and α-SMA staining (E) in the CCl4 groups (a) or the TAA groups (b) are shown. *P < 0.05 and **P < 0.01 versus Cont, †P < 0.05 versus CCl4 alone. Quantification of the Sirius red-stained area or α-SMA-stained area was performed using the NIH image software programme.
Verapamil did not affect liver fibrosis in CCl4-treated mice
Verapamil did not affect the Sirius red-stained area or the hepatic hydroxyproline contents in CCl4-treated mice (Supporting Figure S3).
Azelnidipine prevents the decrease in the expression levels of ROS-eliminating enzymes in CCl4-treated mice
Azelnidipine partially reversed the inhibition of ACOX1 protein expression caused by CCl4 (Figure 6B). By contrast, azelnidipine did not influence the protein expression of Nox2 (Figure 6B). On the other hand, azelnidipine partially reversed inhibition of the protein expression of ROS-eliminating enzymes caused by CCl4 (Figure 6D). The same was true for changes in their mRNA levels (Figure 6A and C).
Figure 6.
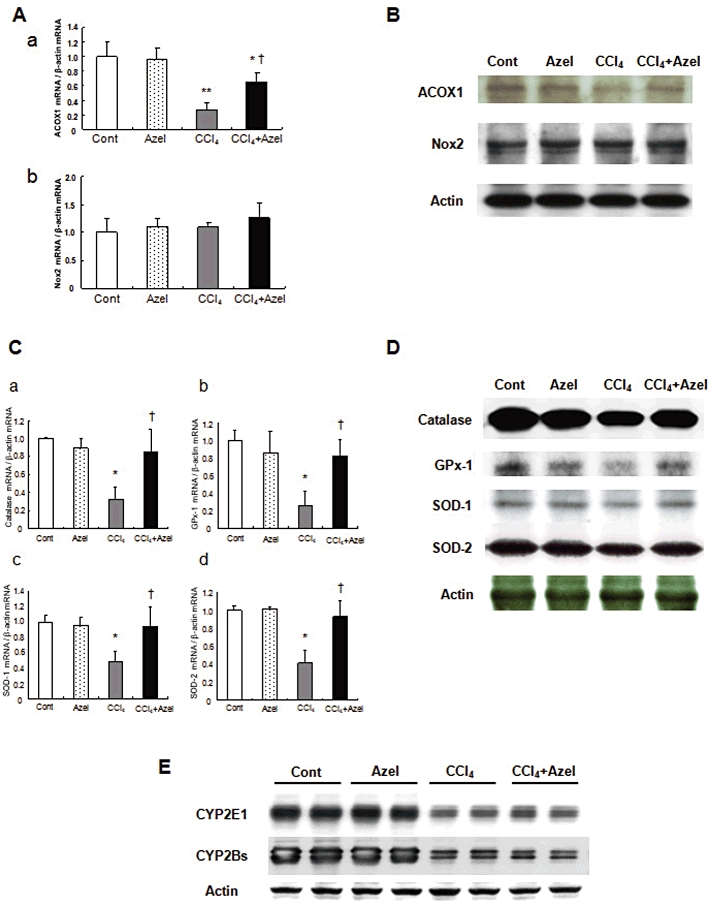
The mechanisms of the antioxidant effect of azelnidipine (Azel) and the effects of Azel on CYP metabolism and fibrogenesis in mice with CCl4-induced liver injury. The groups are the same as the CCl4 groups shown in Figure 4. mRNA (A) and protein (B) expression of ROS-generating enzymes, ACOX1, Nox2. mRNA (C) and protein (D) expression of ROS-eliminating enzymes, catalase, GPx-1, SOD-1, SOD-2 and (E) CYP2E1 and CYP2Bs protein expression. *P < 0.05 and **P < 0.01 versus Cont, †P < 0.05 versus CCl4 alone.
CYP2E1 and CYP2Bs protein levels are not affected by azelnidipine in CCl4-treated mice
CYP2E1 and CYP2Bs protein levels in the control mice were similar to those in the mice treated with azelnidipine alone (Figure 6E). Although CCl4 administration significantly reduced those protein levels, the levels were similar between mice treated with CCl4 plus azelnidipine and mice treated with azelnidipine alone (Figure 6E).
Azelnidipine accelerates fibrosis regression in CCl4-treated mice
Azelnidipine resulted in accelerated resolution of liver scarring compared with mice treated with vehicle. This difference was histologically evident after 3 and 7 days (Figure 7A); this finding was confirmed by a reduction in the Sirius red-stained area and hepatic hydroxyproline levels in the azelnidipine-treated group (Figure 7B). These results were analysed using Student's unpaired two-sided t-test.
Figure 7.
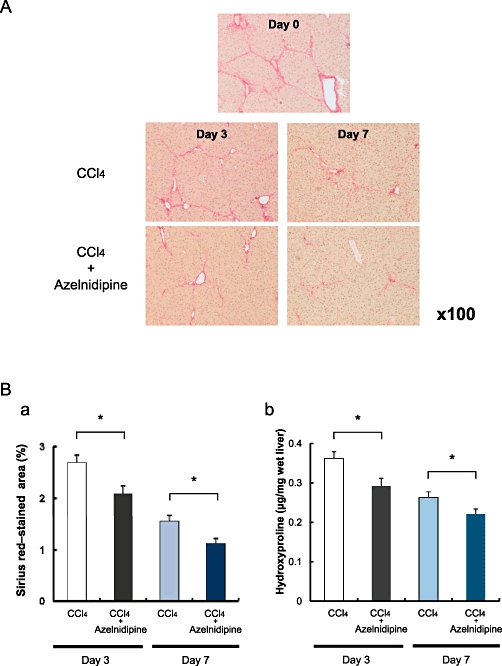
The effects of azelnidipine on the regression of CCl4-induced liver fibrosis in mice. C57BL/6 mice were challenged with CCl4 for 8 weeks to establish advanced liver scarring. (A) Three days after the last CCl4 injection (at the peak of fibrosis, day 0), mice received either azelnidipine (CCl4+ Azel) or vehicle (CCl4 alone) and were assessed for fibrosis regression based on histology at day 3 and day 7. (B) Sirius red-stained area (a) and hepatic hydroxyproline contents (b) were evaluated at day 3 and day 7. *P < 0.05 between CCl4 versus CCl4+ Azel. Quantification of the Sirius red-stained area was performed using the NIH image software programme.
Discussion and conclusions
The major finding of our study was that azelnidipine inhibited TGF-β1- or Ang II-induced HSC activation in vitro and attenuated liver fibrosis induced by chronic CCl4 or TAA administration in vivo. Moreover, azelnidipine could accelerate regression of liver fibrosis in CCl4-treated mice. The use of a calcium blocker as an anti-fibrotic agent in animals has not been reported, with the exception of verapamil (Xu et al., 2007) and tetrandrine (Hsu et al., 2007; Yin et al., 2007), which have some obstacles for clinical application as discussed below. This is the first study to show that regression of liver fibrosis could be achieved by a pure calcium blocker administered at an appropriate pharmacological concentration. Because azelnidipine is safe and currently available for clinical use in Japan, our study has a particularly practical value. In addition, our study is the first in-depth evaluation of the inhibitory effect of a calcium blocker on HSC activation, including activation of intracellular signalling pathways, although inhibitory effects of calcium blockade on HSC contraction (Bataller et al., 1998) and calcium influx (Oide and Thurman, 1994) have been reported previously.
The hepatotoxicity of CCl4 (Kim et al., 2000; Basu, 2003) or TAA (Kang et al., 2005; 2008) has been proposed to be increased by oxidative stress, supporting our findings that the anti-fibrotic effect of azelnidipine on CCl4- or TAA-induced fibrosis was possibly due to its antioxidant effect. This is in line with the anti-fibrotic effect of curcumin (a polyphenolic antioxidant) on CCl4- or TAA-induced injury (Bruck et al., 2007; Fu et al., 2008). Neither improved blood lipid profiles nor calcium channel-blocking effects were likely to be involved in the anti-fibrotic effects of azelnidipine because the 10 mg·kg−1·day−1 dosage of azelnidipine, corresponding to a pharmacological dose in humans (Kuramoto et al., 2003; Nakano et al., 2006), had no effect on haemodynamic parameters, including systemic arterial blood pressure and blood lipid profiles, in mice (Nakano et al., 2008).
Furthermore, the mechanism of the antioxidant effect of azelnidipine may be related to prevention of the decrease in the expression of antioxidant enzymes in CCl4-treated mice. This conclusion is based on the following results: (i) the ROS-generating enzyme ACOX1 was down-regulated, while Nox2 expression was unchanged in CCl4-treated mice; (ii) azelnidipine partially reversed ACOX1 expression and had no effects on Nox2 expression; (iii) ROS-eliminating enzymes were all down-regulated in CCl4-treated mice, and azelnidipine partially reversed inhibition of expression of ROS-eliminating enzymes; (iv) azelnidipine did not affect CYP metabolism in mice with CCl4-induced liver injury. CCl4 is transformed by CYP to its active metabolite, which readily interacts with oxygen to form free radicals. An excessive formation of superoxide anions may inactivate SOD, thus resulting in an inactivation of the H2O2 scavenging enzymes, catalase and GPx-1. The DPPH (1,1-diphenyl-2-picrylhydrazyl) free-radical scavenging ability of azelnidipine (Fujiwara and Konno, 2004) may prevent the decrease in the expression of ROS-eliminating enzymes.
The inhibitory effect of azelnidipine on HSCs stimulated by TGF-β1 or Ang II may have been due to the antioxidative effect of azelnidipine. This conclusion is supported by the following results: (i) azelnidipine (racemate), (R)-(−)-azelnidipine or (S)-(+)-azelnidipine, which have similar antioxidant effects but different calcium channel blocking activity, inhibited TGF-β1-induced expression of COL1A1 mRNA in LX-2 cells; (ii) azelnidipine, but not the other equimolar calcium blockers, inhibited TGF-β1-induced intracellular ROS levels and reversed the TGF-β1-induced decrease in glutathione levels in LX-2 cells; (iii) azelnidipine inhibited both Ang II-induced expression of COL1A1 mRNA and Ang II-induced intracellular ROS levels; (iv) pretreatment of LX-2 cells with vitamin E before azelnidipine blocked further azelnidipine-induced inhibition of expression of COL1A1 mRNA; (v) azelnidipine exerted its inhibitory effects on TGF-β1-induced COL1A1 expression in HSCs through the stress-activated protein kinases, p38 and JNK pathways, which are involved in oxidative stress (Reeves et al., 2000; Vayalil et al., 2007).
Previous studies showed that activated HSCs are associated with an up-regulation of the L-type voltage-operated calcium channels that mediate calcium influx and cell contraction (Bataller et al., 2001), and that the channels are activated or up-regulated by TGF-β1 (Oide and Thurman, 1994). However, the long-term treatment of HSCs with TGF-β1 (5 days) influences depolarization-dependent calcium signals in HSCs, but the short-term treatment (24 h) did not (Roth-Eichhorn et al., 1999). The results reported by Roth-Eichhorn et al. are partially contradictory to the results reported by Oide and Thurman, but they support our finding that inhibition of the expression of COL1A1 mRNA in HSCs was not due to the calcium channel blocking effect. In summary, 24 h treatment of HSCs with TGF-β1, which was also performed in our study, may influence calcium influx, but the level of calcium influx may not be enough to induce the expression of COL1A1 mRNA. Thus, the exact role of L-type voltage-operated calcium channels in HSCs stimulated by TGF-β1 remains to be elucidated.
Verapamil was reported to suppress liver fibrosis induced by multiple hepatotoxic factors, including CCl4, ethanol and high cholesterol, in rats (Xu et al., 2007). However, doses of 20, 40 and 80 mg·kg−1·day−1 were suggested to be inappropriately high doses for the following reasons. The maximum plasma concentration after oral administration of verapamil at doses of 80–120 mg·day−1 in humans is lower than or at least equal to that after oral administration of 10 mg·kg−1 verapamil in rats (Echizen and Eichelbaum, 1986; Bhatti and Foster, 1997). Moreover, 2 mg·kg−1 of verapamil significantly decreased both arterial pressure and cardiac output in rats (Lay et al., 2006). In addition, verapamil shows a markedly lower systemic and/or oral clearance and significant prolongation of elimination half-life in patients with liver cirrhosis (Klotz, 2007). This differs from azelnidipine, which has pharmacokinetic properties that are not changed in the plasma of patients with liver cirrhosis (Konno and Takagi, 2004). In fact, 10 mg·kg−1·day−1 of verapamil did not affect liver fibrosis in CCl4-treated mice in our study. Furthermore, TGF-β1-induced expression of COL1A1 mRNA in HSCs was not reduced with 1 or 10 µM verapamil. Taken together, these indicate that the clinical application of verapamil for liver fibrosis is disputable.
Tetrandrine, an alkaloid used in Chinese medicine, ameliorated liver fibrosis in animal models (Hsu et al., 2007; Yin et al., 2007). However, tetrandrine has multiple pharmacological effects, including a natrium channel-blocking effect (Chen et al., 2009) and anti-proliferative effects on several cancer cell lines (Dong et al., 1997; Yoo et al., 2002). Thus, tetrandrine is not merely a non-selective calcium blocker. Additionally, tetrandrine is a potential hepatotoxin that induces multiple apoptogenic signals (Yan et al., 2006). The pharmacokinetic and toxicokinetic characteristics of tetrandrine have not been well established (Song et al., 2008), and further research is necessary before it can be considered for clinical use.
In summary, we revealed that azelnidipine inhibited TGF-β1- or Ang II-induced HSC activation in vitro, attenuated CCl4- or TAA-induced liver fibrosis, and accelerated the regression of CCl4-induced liver fibrosis when administered at a clinically relevant dose. Furthermore, we have elucidated the mechanism of the anti-fibrotic effect of azelnidipine. Because azelnidipine is widely used in clinical practice without serious adverse effects, it may provide an effective new strategy for anti-fibrotic therapy.
Acknowledgments
We are very grateful to Prof Scott L Friedman, Division of Liver Diseases, Mount Sinai School of Medicine, New York, New York 10029-6574, USA, for generous provision of the human HSC cell line, LX-2. We appreciate Daiichi Sankyo Co., Ltd, Tokyo, Japan, for providing azelnidipine (racemate), (R)-(−)-azelnidipine and (S)-(+)-azelnidipine.
Glossary
- 8-OHdG
8-hydroxy-2′-deoxyguanosine
- α-SMA
α-smooth muscle actin
- ACOX1
acyl-coenzyme A oxidase 1
- ALT
alanine aminotransferase
- Ang
II, angiotensin II
- AUC
area under the curve
- [Ca]i
intracellular calcium concentration
- COL1A1
α1(I) collagen
- CCl4
carbon tetrachloride
- CYP
cytochrome P450
- DCF
2′,7′-dichlorofluorescein fluorescence
- DPPH
1,1-diphenyl-2-picrylhydrazyl
- DTNB
5,5′-dithiobis(2-nitrobenzoic acid)
- GPx-1
glutathione peroxidase-1
- HSC
hepatic stellate cell
- MEK1
MAPK kinase 1
- MPO
myeloperoxidase
- MTT
3-(4,5-dimethyl-thiazole-2-yl)-2,5-diphenyl tetrazolium bromide
- NIH
National Institutes of Health
- Nox
nonphagocytic oxidase
- ROS
reactive oxygen species
- SOD
superoxide dismutase
- TAA
thioacetamide
- TIMP-1
tissue inhibitor of metalloproteinases-1
Conflicts of interest
None.
Supporting information
Figure S1 (A) The effects of azelnidipine onLX-2 cell viability. The cells were incubated with azelnidipine atvarious concentrations for 24 hours. (B) The effects of eachcalcium blocker on the TGF-β1-induced stimulation of COL1A1mRNA expression in LX-2 cells. (C) The effects of each calciumblocker on the TGF-β1-induced increase of intracellular ROS inLX-2 cells. The concentration of each calcium blocker andTGF-β1 were 100 nM and 5 ng·mL−1,respectively. **P < 0.01 versus control.
Figure S2 (A) The roles of MAPK signaltransduction pathways in TGF-β1-induced activation of LX-2cells. Time courses of TGF-β1-induced phosphorylation of MAPKsignal transduction molecules. The effects of TGF-β1 onphosphorylation of JNK (a) p38 (b) or ERK1/2 (c) at different timepoints after adding TGF-β1. LX-2 cells activated byTGF-α were used as a positive control. (B) The effect of 100nM azelnidipine without TGF-β1 on phosphorylation of ERK1/2 inLX-2 cells. Time course of phosphorylation of ERK1/2 at differenttime points after adding azelnidipine. (C) Time course ofTGF-β1-induced phosphorylation of Smad3. (D) The effect ofazelnidipine (Azel) on TGF-β1-induced phosphorylation of Smad3in LX-2 cells. The concentration of TGF-β1 was 5ng·mL−1.
Figure S3 The effects of verapamil onCCl4-induced fibrogenesis in mice. The groups are thesame as those in Figure 4 except verapamil was used instead ofazelnidipine. **P < 0.01 versus Control. Sirius red staining (A) and hepatic hydroxyproline contents (B) are indicated. Quantification of the Sirius red-stained area was performed using the NIH image software programme.
References
- Arthur MJ. Fibrogenesis II. Metalloproteinases and their inhibitors in liver fibrosis. Am J Physiol Gastrointest Liver Physiol. 2000;279:G245–G249. doi: 10.1152/ajpgi.2000.279.2.G245. [DOI] [PubMed] [Google Scholar]
- Baik SK, Jo HS, Suk KT, Kim JM, Lee BJ, Choi YJ, et al. Inhibitory effect of angiotensin II receptor antagonist on the contraction and growth of hepatic stellate cells. Korean J Gastroenterol. 2003;42:134–141. [PubMed] [Google Scholar]
- Basu S. Carbon tetrachloride-induced lipid peroxidation: eicosanoid formation and their regulation by antioxidant nutrients. Toxicology. 2003;189:113–127. doi: 10.1016/s0300-483x(03)00157-4. [DOI] [PubMed] [Google Scholar]
- Bataller R, Brenner DA. Hepatic stellate cells as a target for the treatment of liver fibrosis. Semin Liver Dis. 2001;21:437–451. doi: 10.1055/s-2001-17558. [DOI] [PubMed] [Google Scholar]
- Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bataller R, Nicolas JM, Ginees P, Görbig MN, Garcia-Ramallo E, Lario S, et al. Contraction of human hepatic stellate cells activated in culture: a role for voltage-operated calcium channels. J Hepatol. 1998;29:398–408. doi: 10.1016/s0168-8278(98)80057-3. [DOI] [PubMed] [Google Scholar]
- Bataller R, Gasull X, Gines P, Hellemans K, Görbig MN, Nicolás JM, et al. In vitro and in vivo activation of rat hepatic stellate cells results in de novo expression of L-type voltage-operated calcium channels. Hepatology. 2001;33:956–962. doi: 10.1053/jhep.2001.23500. [DOI] [PubMed] [Google Scholar]
- Bhatti MM, Foster RT. Pharmacokinetics of the enantiomers of verapamil after intravenous and oral administration of racemic verapamil in a rat model. Biopharm Drug Dispos. 1997;18:387–396. doi: 10.1002/(sici)1099-081x(199707)18:5<387::aid-bdd26>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Breitkopf K, Haas S, Wiercinska E, Singer MV, Dooley S. Anti-TGF-beta strategies for the treatment of chronic liver disease. Alcohol Clin Exp Res. 2005;29:121S–131S. doi: 10.1097/01.alc.0000189284.98684.22. [DOI] [PubMed] [Google Scholar]
- Bruck R, Ashkenazi M, Weiss S, Goldiner I, Shapiro H, Aeed H, et al. Prevention of liver cirrhosis in rats by curcumin. Liver Int. 2007;27:373–383. doi: 10.1111/j.1478-3231.2007.01453.x. [DOI] [PubMed] [Google Scholar]
- Chen L, Li QY, Yang Y, Li ZW, Zeng XR. Inhibitory effects of tetrandrine on the Na(+) channel of human atrial fibrillation myocardium. Acta Pharmacol Sin. 2009;30:166–174. doi: 10.1038/aps.2008.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Minicis S, Brenner DA. NOX in liver fibrosis. Arch Biochem Biophys. 2007;462:266–272. doi: 10.1016/j.abb.2007.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Yang MM, Kwan CY. In vitro inhibition of proliferation of HL-60 cells by tetrandrine and coriolus versicolor peptide derived from Chinese medicinal herbs. Life Sci. 1997;60:PL135–PL140. doi: 10.1016/s0024-3205(96)00695-9. [DOI] [PubMed] [Google Scholar]
- Echizen H, Eichelbaum M. Clinical pharmacokinetics of verapamil, nifedipine and diltiazem. Clin Pharmacokinet. 1986;11:425–449. doi: 10.2165/00003088-198611060-00002. [DOI] [PubMed] [Google Scholar]
- Fu Y, Zheng S, Lin J, Ryerse J, Chen A. Curcumin protects the rat liver from CCl4-caused injury and fibrogenesis by attenuating oxidative stress and suppressing inflammation. Mol Pharmacol. 2008;73:399–409. doi: 10.1124/mol.107.039818. [DOI] [PubMed] [Google Scholar]
- Fujiwara H, Konno Y. Discovery and characterization of azelnidipine: aiming at an ultimate calcium channel blocker. Prog Med. 2004;24:2631–2639. in Japanese with English Abstract. [Google Scholar]
- García-Trevijano ER, Iraburu MJ, Fontana L, Domínguez-Rosales JA, Auster A, Covarrubias-Pinedo A, et al. Transforming growth factor beta1 induces the expression of alpha1(I) procollagen mRNA by a hydrogen peroxide-C/EBPbeta-dependent mechanism in rat hepatic stellate cells. Hepatology. 1999;29:960–970. doi: 10.1002/hep.510290346. [DOI] [PubMed] [Google Scholar]
- Gressner AM, Weiskirchen R. Modern pathogenetic concepts of liver fibrosis suggest stellate cells and TGF-beta as major players and therapeutic targets. J Cell Mol Med. 2006;10:76–99. doi: 10.1111/j.1582-4934.2006.tb00292.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hool LC. Evidence for the regulation of L-type Ca2+ channels in the heart by reactive oxygen species: mechanism for mediating pathology. Clin Exp Pharmacol Physiol. 2008;35:229–234. doi: 10.1111/j.1440-1681.2007.04727.x. [DOI] [PubMed] [Google Scholar]
- Hsu YC, Chiu YT, Cheng CC, Wu CF, Lin YL, Huang YT. Antifibrotic effects of tetrandrine on hepatic stellate cells and rats with liver fibrosis. J Gastroenterol Hepatol. 2007;22:99–111. doi: 10.1111/j.1440-1746.2006.04361.x. [DOI] [PubMed] [Google Scholar]
- Kang JS, Morimura K, Salim EI, Wanibuchi H, Yamaguchi S, Fukushima S, et al. Persistence of liver cirrhosis in association with proliferation of nonparenchymal cells and altered location of alpha-smooth muscle actin-positive cells. Toxicol Pathol. 2005;33:329–335. doi: 10.1080/01926230590922901. [DOI] [PubMed] [Google Scholar]
- Kang JS, Wanibuchi H, Morimura K, Wongpoomchai R, Chusiri Y, Gonzalez FJ, et al. Role of CYP2E1 in thioacetamide-induced mouse hepatotoxicity. Toxicol Appl Pharmacol. 2008;228:295–300. doi: 10.1016/j.taap.2007.11.010. [DOI] [PubMed] [Google Scholar]
- Kim KY, Choi I, Kim SS. Progression of hepatic stellate cell activation is associated with the level of oxidative stress rather than cytokines during CCl4-induced fibrogenesis. Mol Cells. 2000;10:289–300. [PubMed] [Google Scholar]
- Kisseleva T, Brenner DA. Role of hepatic stellate cells in fibrogenesis and the reversal of fibrosis. J Gastroenterol Hepatol. 2007;22(Suppl. 1):S73–S78. doi: 10.1111/j.1440-1746.2006.04658.x. [DOI] [PubMed] [Google Scholar]
- Kisseleva T, Brenner DA. Anti-fibrogenic strategies and the regression of fibrosis. Best Pract Res Clin Gastroenterol. 2011;25:305–317. doi: 10.1016/j.bpg.2011.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klotz U. Antiarrhythmics: elimination and dosage considerations in hepatic impairment. Clin Pharmacokinet. 2007;46:985–996. doi: 10.2165/00003088-200746120-00002. [DOI] [PubMed] [Google Scholar]
- Koike H, Kimura T, Kawasaki T, Sada T, Ikeda T, Sanbuissho A, et al. Azelnidipine, a long-acting calcium channel blocker with slow onset and high vascular affinity. Annu Rep Sankyo Res Lab. 2002;54:1–64. [Google Scholar]
- Konno Y, Takagi H. Azelnidipine. Med Drug J. 2004;40:284–288. in Japanese. [Google Scholar]
- Koyama Y, Takeishi Y, Takahashi H, Shishido T, Arimoto T, Niizeki T, et al. Azelnidipine inhibits H2O2-induced cell death in neonatal rat cardiomyocytes. Cardiovasc Drugs Ther. 2007;21:69–72. doi: 10.1007/s10557-007-6008-4. [DOI] [PubMed] [Google Scholar]
- Kuramoto K, Ichikawa S, Hirai A, Kanada S, Nakachi T, Ogihara T. Azelnidipine and amlodipine: a comparison of their pharmacokinetics and effects on ambulatory blood pressure. Hypertens Res. 2003;26:201–208. doi: 10.1291/hypres.26.201. [DOI] [PubMed] [Google Scholar]
- Lay CS, May CM, Lee FY, Tsai YT, Lee SD, Chien S, et al. Effect of verapamil on nitric oxide synthase in a portal vein-ligated rat model: role of prostaglandin. World J Gastroenterol. 2006;12:2351–2356. doi: 10.3748/wjg.v12.i15.2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano K, Egashira K, Tada H, Kohjimoto Y, Hirouchi Y, Kitajima S. A third-generation, long-acting, dihydropyridine calcium antagonist, azelnidipine, attenuates stent-associated neointimal formation in non-human primates. J Hypertens. 2006;24:1881–1889. doi: 10.1097/01.hjh.0000242414.96277.95. [DOI] [PubMed] [Google Scholar]
- Nakano K, Egashira K, Ohtani K, Gang Z, Iwata E, Miyagawa M. Azelnidipine has anti-atherosclerotic effects independent of its blood pressure-lowering actions in monkeys and mice. Atherosclerosis. 2008;196:172–179. doi: 10.1016/j.atherosclerosis.2007.03.036. [DOI] [PubMed] [Google Scholar]
- Oide H, Thurman RG. Hepatic Ito cells contain calcium channels: increases with transforming growth factor-beta 1. Hepatology. 1994;20:1009–1014. doi: 10.1002/hep.1840200433. [DOI] [PubMed] [Google Scholar]
- Reeves HL, Dack CL, Peak M, Burt AD, Day CP. Stress-activated protein kinases in the activation of rat hepatic stellate cells in culture. J Hepatol. 2000;32:465–472. doi: 10.1016/s0168-8278(00)80398-0. [DOI] [PubMed] [Google Scholar]
- Roth-Eichhorn S, Eberheim A, Bode HP, Gressner AM. Transformation-dependent calcium influx by voltage-operated calcium channels in stellate cells of rat liver. J Hepatol. 1999;30:612–620. doi: 10.1016/s0168-8278(99)80191-3. [DOI] [PubMed] [Google Scholar]
- Siegmund SV, Brenner DA. Molecular pathogenesis of alcohol-induced hepatic fibrosis. Alcohol Clin Exp Res. 2005;29:102S–109S. doi: 10.1097/01.alc.0000189275.97419.58. [DOI] [PubMed] [Google Scholar]
- Song N, Zhang S, Li Q, Liu C. Establishment of a liquid chromatographic/mass spectrometry method for quantification of tetrandrine in rat plasma and its application to pharmacokinetic study. J Pharm Biomed Anal. 2008;48:974–979. doi: 10.1016/j.jpba.2008.06.002. [DOI] [PubMed] [Google Scholar]
- Tsukamoto H, Matsuoka M, French SW. Experimental models of hepatic fibrosis: a review. Semin Liver Dis. 1990;10:56–65. doi: 10.1055/s-2008-1040457. [DOI] [PubMed] [Google Scholar]
- Vayalil PK, Iles KE, Choi J, Yi AK, Postlethwait EM, Liu RM. Glutathione suppresses TGF-beta-induced PAI-1 expression by inhibiting p38 and JNK MAPK and the binding of AP-1, SP-1, and Smad to the PAI-1 promoter. Am J Physiol Lung Cell Mol Physiol. 2007;293:L1281–L1292. doi: 10.1152/ajplung.00128.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellington K, Scott LJ. Azelnidipine. Drugs. 2003;63:2613–2621. doi: 10.2165/00003495-200363230-00004. discussion 2623–2624. [DOI] [PubMed] [Google Scholar]
- Xu D, Wu Y, Liao ZX, Wang H. Protective effect of verapamil on multiple hepatotoxic factors-induced liver fibrosis in rats. Pharmacol Res. 2007;55:280–286. doi: 10.1016/j.phrs.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Yamagishi S, Inagaki Y, Nakamura K, Imaizumi T. Azelnidipine, a newly developed long-acting calcium antagonist, inhibits tumor necrosis factor-alpha-induced interleukin-8 expression in endothelial cells through its anti-oxidative properties. J Cardiovasc Pharmacol. 2004;43:724–730. doi: 10.1097/00005344-200405000-00016. [DOI] [PubMed] [Google Scholar]
- Yan C, Xin-Ming Q, Li-Kun G, Lin-Lin L, Fang-Ping C, Ying X. Tetrandrine-induced apoptosis in rat primary hepatocytes is initiated from mitochondria: caspases and endonuclease G (Endo G) pathway. Toxicology. 2006;218:1–12. doi: 10.1016/j.tox.2005.08.024. [DOI] [PubMed] [Google Scholar]
- Yin MF, Lian LH, Piao DM, Nan JX. Tetrandrine stimulates the apoptosis of hepatic stellate cells and ameliorates development of fibrosis in a thioacetamide rat model. World J Gastroenterol. 2007;13:1214–1220. doi: 10.3748/wjg.v13.i8.1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo SM, Oh SH, Lee SJ, Lee BW, Ko WG Moon CK. Inhibition of proliferation and induction of apoptosis by tetrandrine in HepG2 cells. J Ethnopharmacol. 2002;81:225–229. doi: 10.1016/s0378-8741(02)00082-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.