

Abstract
BACKGROUND AND PURPOSE
5-HT2A receptor antagonists improve antidepressant responses when added to 5-HT-selective reuptake inhibitors (SSRIs) or tricyclic antidepressants. Here, we have studied the involvement of neuroplasticity pathways and/or the 5-hydroxytryptaminergic system in the antidepressant-like effect of this combined treatment, given subchronically.
EXPERIMENTAL APPROACH
Expression of brain-derived neurotrophic factor (BDNF) and its receptor (TrkB), 5-bromo-2′-deoxyuridine (BrdU) incorporation, and β-catenin protein expression in different cellular fractions, as well as 5-HT1A receptor function were measured in the hippocampus of rats treated with fluoxetine, ketanserin and fluoxetine + ketanserin for 7 days, followed by a forced swimming test (FST) to analyse antidepressant efficacy.
KEY RESULTS
mRNA for BDNF was increased in the CA3 field and dentate gyrus of the hippocampus by combined treatment with fluoxetine + ketanserin. Expression of β-catenin was increased in total hippocampal homogenate and in the membrane fraction, but unchanged in the nuclear fraction after combined treatment with fluoxetine + ketanserin. These effects were paralleled by a decreased immobility time in the FST. There were no changes in BrdU incorporation, TrkB expression and 5-HT1A receptor function in any of the groups studied.
CONCLUSIONS AND IMPLICATIONS
The antidepressant-like effect induced by subchronic co-treatment with a SSRI and a 5-HT2A receptor antagonist may mainly be because of modifications in hippocampal neuroplasticity (BDNF and membrane-associated β-catenin), without a significant role for other mechanisms involved in chronic antidepressant response, such as hippocampal neuroproliferation or 5-HT1A receptor desensitization in the dorsal raphe nucleus.
Keywords: 5-HT2A receptor antagonist, SSRI, neuroplasticity, hippocampus, neutrophin, β-catenin, BrdU, behaviour
Introduction
Classically, the pathogenesis of depression has been explained by the monoamine hypothesis, involving a dysfunction of 5-hydroxytryptaminergic, noradrenergic and/or dopaminergic systems (see Ressler and Nemeroff, 2000). More recently, a neurotrophic hypothesis has been proposed, on the basis of the neuroproliferative effects of antidepressants (Duman et al., 1997). It is noteworthy that most antidepressants such as the selective 5-HT reuptake inhibitors (SSRIs) need at least 2–3 weeks to show their therapeutic benefit.
The effects of the neurotransmitter 5-HT are mediated by the 5-HT receptor family, formed by seven different subfamilies (5-HT1 to 5-HT7) and 13 different subtypes (for example, 5-HT2A/B/C; nomenclature follows Alexander et al., 2011). 5-HT2A receptors are present in dendrites and axons of several areas within the rat brain, including the cerebral cortex, septum, hippocampus, basal ganglia, amygdala and brain stem (Pazos et al., 1985). The role of 5-HT2A receptors is especially important in the prefrontal cortex where the activation of this receptor subtype produces an increase of the excitability of pyramidal neurons (Aghajanian and Marek, 2000). Furthermore, 5-HT2A receptors appear to be involved in psychiatric disorders, and antagonism of this receptor subtype is one of the mechanisms of action of atypical antipsychotic drugs (see Schmidt et al., 1995).
The role of 5-HT2A receptors in depression is supported by several studies reporting changes at different levels in tissue samples from suicide victims. A down-regulation of the receptor protein (Rosel et al., 2000), together with an up-regulation in G protein coupling (Rosel et al., 2000) and expression of mRNA for the receptor (Pandey et al., 2002) have been reported in the hippocampus, although other studies have not found changes in this structure (Stockmeier et al., 1997). In contrast, an increase in 5-HT2A receptor density and function has been consistently reported in the frontal cortex (Pandey et al., 2002) and platelets (Serres et al., 1999). In addition, antidepressant treatments have provided conflicting results, as chronic SSRIs produces an up-regulation in 5-HT2A receptors (Massou et al., 1997), while tricyclic and/or monoamine oxidase inhibitors induce a down-regulation of this 5-HT receptor subtype (Attar-Lévy et al., 1999).
Recently, it has been reported that 5-HT2A antagonists produce antidepressant-like effects (Marek et al., 2003; Pandey et al., 2010), acting through the blockade of the postsynaptic 5-HT2A receptors (Rosel et al., 2000). Since the activation of 5-HT2A receptor opposes the therapeutic effects of the SSRIs in major depression (Marek et al., 2003), the antidepressant effect of some SSRIs appears to be potentiated by the co-administration of 5-HT2A subtype antagonists such as risperidone, olanzapine or M100907 (Marek et al., 2003), mainly by increasing 5-HT, dopamine and noradrenaline release in medial prefrontal cortex (Huang et al., 2006). Drugs that mediate both 5-HT reuptake inhibition and 5-HT2A receptor blockade are known as 5-HT2A receptor antagonists/reuptake inhibitors (SARIs) and have been suggested in cases of treatment-resistant depression (Shelton et al., 2001; Marek et al., 2003; Adell et al., 2005).
The neurogenic hypothesis of depression is mainly supported by the fact that chronic antidepressant treatment produces an increase in cell proliferation in the subgranular layer of the dentate gyrus (DG) of the hippocampus (Duman et al., 1997; Malberg et al., 2000; Santarelli et al., 2003), as well as an increase in the expression of brain-derived neurotrophic factor (BDNF) in the hippocampus (Nibuya et al., 1995; Vaidya et al., 1999) and serum (Shimizu et al., 2003). It is noteworthy that BDNF is mainly involved in synaptic plasticity, rather than in neuron growth and survival (see Martinowich and Lu, 2008). The activation of 5-HT2A receptors increases BDNF levels in the prefrontal cortex and decreases BDNF levels in the DG of the hippocampus, effects mediated by glutamatergic and GABAergic neurons, respectively (Vaidya et al., 1999). The decrease in expression of mRNA for BDNF in hippocampus, assessed in a stress model as immobility time, is reversed, at least in part by antagonism of 5-HT2A receptors, thus suggesting the involvement of the 5-HT2A receptor subtype in the increased inhibitory control of the hippocampus and the stress-induced down-regulation of BDNF mRNA (Vaidya et al., 1999).
In the last years, it has been reported that chronic antidepressant treatments modulate the expression of β-catenin, a constituent of the canonical Wnt pathway (Madsen et al., 2003; Mostany et al., 2008), which is accumulated in the cytosol following inhibition of glycogen synthase kinase-3 (GSK-3), and translocates to the nucleus, activating the transcription of genes associated with proliferation (see Wada, 2009). β-Catenin is also associated with N-cadherin and α-catenin in the cell membrane, controlling the size of the reserve vesicle pool in synapse development (Bamji et al., 2003) and providing a link between cadherin-mediated cell–cell adhesion and the F-actin cytoskeleton (Patapoutian and Reichardt, 2000).
In the present experiments, we have analysed the effect of 7 days co-treatment with the SSRI fluoxetine and the 5-HT2A receptor antagonist ketanserin on cell proliferation [5-bromo-2′-deoxyuridine (BrdU) incorporation], expression of proteins involved in neuroplasticity (BDNF expression, β-catenin), and 5-hydroxytryptaminergic markers classically involved in chronic antidepressant responses (5-HT1A receptor function). These studies have been carried out in parallel with the analysis of antidepressant-like behavioural changes.
Methods
Animals
All animal care and experimental procedures complied with Spanish legislation and the European Communities Council Directive on ‘Protection of Animals Used in Experimental and Other Scientific Purposes’ (86/609/EEC). Male Sprague–Dawley rats weighing 270–350 g were group-housed and maintained on 12/12 h light/dark cycle, with access to food and water ad libitum.
Antidepressant treatment and BrdU administration
Rats were divided in four groups, 7–12 rats per group, for each set of experiments, and administered via i.p. vehicle (0.9% NaCl solution), 5 mg·kg−1·day−1 fluoxetine (Fagron Iberica S.A.U., Barcelona, Spain), 0.1 mg·kg−1·day−1 ketanserin (Sigma, Madrid, Spain) or the combination of 5 mg·kg−1·day−1 fluoxetine + 0.1 mg·kg−1·day−1 ketanserin for 7 days or acutely (single treatment). The dose of ketanserin used for this study was based on previous reports using similar i.p. (Andersson et al., 1988) or i.v. doses (Catafau et al., 2009). We have not used a higher dose of ketanserin, as reported by other authors (Jha et al., 2008; Pandey et al., 2010), in an attempt to avoid possible effects on the 5-HT2C receptors.
For immunohistochemical analysis of cell proliferation, animals received BrdU (4 × 75 mg·kg−1 every 2 h, i.p.; Sigma) in sterile 0.9% NaCl solution on the last day of antidepressant treatment and 24 h before killing. All other chemicals used were of analytical grade.
Forced swimming test (FST)
Rats were placed in swimming tanks 18 cm in diameter and 40 cm tall. The tank was filled with enough water at 25°C, so that the rat could not touch the bottom. The rat was placed in the swimming tank for a single 5 min session approximately 24 h after the last treatment in the subchronic set of animals or after 30 min to 1 h, following an acute (single) administration of ketanserin and/or fluoxetine. The time spent in immobility, swimming and climbing behaviours were scored by an observer unaware of the treatments given (Cryan et al., 2005a). The behavioural effect is stronger in FST performed within the first 5 min of the test, without performing pre-test induction (Cryan et al., 2005a). Antidepressant effect was defined as a decrease in immobility time.
In situ hybridization
Rats were killed by decapitation, and the brains were quickly removed, frozen in dry ice and stored at −80°C until sectioning. Cryostat sections (20 µm) were thaw-mounted onto slides and pre-treated for in situ hybridization using a standard protocol previously published (Zetterström et al., 1998). Briefly, the tissues were fixed with 4% paraformaldehide in PBS for 5 min, acetylated with 0.25% acetic anhydride in 0.1 M triethanolamine buffer for 10 min, dehydrated in a series of ethanol washes (70, 80, 95 and 100%), incubated in chloroform for 10 min, and finally rehydrated with 100 and 95% ethanol. The sections were air-dried and stored at −20°C until use.
Oligonucleotides complementary to mRNAs for BDNF (5′ GGT CTC GTA GAA ATA TTG GTT CAG TTG GCC TTT TGA TAC CGG GAC 3′, Zetterström et al., 1998) were 3′ end-labelled with [35S]dATP (PerkinElmer Inc., Waltham, MA, USA) using terminal deoxynucleotide transferase (TdT). The labelled oligonucleotide probe was purified and added to each section (250 000 cpm per section) in the hybridization buffer [50% formamide (v/v), 4× saline sodium citrate buffer (SSC), 10 mM sodium phosphate, pH 7.0, 1 mM sodium pyrophosphate, 5× Denhardt's Solution, 0.2 mg·mL−1 salmon sperm DNA, 10% (w/v) dextran sulphate, 0.1 mg·mL−1 polyadenylic acid, 0.12 mg·mL−1 heparin and 20 mM dithiothreitol (DTT) added freshly]. After incubation at 42°C in humidity chambers for 16–20 h, the slides were washed twice in 2× SSC containing 4 mM DTT, at 50°C followed by 5 min washes in 1× SSC, 0.1× SSC, ethanol (80%) and 1 min ethanol (96%) at room temperature. Sections were then air-dried and exposed to films (Biomax MR, Kodak, Madrid, Spain) together with 14C microscales (Amersham, Buckinghamshire, UK) at 4°C for 3 weeks. Controls included hybridization of sections with an excess of unlabelled probe (200×). The abundance of mRNA in hippocampus was analysed and quantified using a computerized image analysis system (Scion Image, Scion Corporation, Maryland, MD, USA). Optical density values were calibrated to 35S tissue equivalents using 14C microscales (Amersham). The data are presented as a percentage of the mean of the saline group (set to 100%).
Immunohistochemistry
Twenty-four hours after the last BrdU injection, rats were anaesthetized with sodium pentobarbital (50 mg·kg−1, i.p.; Sigma) and transcardially perfused with 4% paraformaldehyde in PBS. Brains were post-fixed and cryoprotected with 30% sucrose. Serial coronal sections (40 µm) of the brains were obtained through the entire hippocampus. BrdU inmunohistochemistry was performed as previously described (Mostany et al., 2008); sections were incubated for 2 h in 50% formamide/2× SSC at 65°C, followed by incubation in 2 N HCl for 30 min. Then sections were incubated for 10 min in 0.1 M borate buffer. After washing in PBS, sections were incubated in 1% H2O2 for 30 min, blocked with 5% goat serum in PBS (PBS-TS) for 30 min and then incubated with monoclonal mouse anti-BrdU (1:600; Roche Diagnostics, Barcelona, Spain) overnight at 4°C. Sections were washed in PBS-TS, and incubated with a biotinylated donkey anti-mouse IgG secondary antibody (1:200; Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA) and amplified with avidin–biotin complex (Vector Laboratories, Burlingame, CA, USA). BrdU-positive cells were labeled using diaminobenzidine (DAB) + Ni as chromogen (Vector Laboratories).
For β-catenin immunohistochemistry, sections were boiled at 90°C in 10 mM citric acid, pH 6.0, for 20 min, blocked with 5% donkey normal serum and then incubated overnight at 4°C with an anti-β-catenin monoclonal IgG (1:500; Santa Cruz Biotechnology, Inc., Heidelberg, Germany) and subsequently with a biotinylated donkey anti-mouse IgG (1:200; Jackson ImmunoResearch Laboratories, Inc.) followed by ABC Vectastain Kit (Vector Laboratories). Finally, they were developed with DAB (Invitrogen, Barcelona, Spain).
For quantification of BrdU-positive cells and β-catenin accumulation, every sixth section corresponding to interaural stereotaxic coordinates ranging from 4.48 to 5.70 mm (Paxinos and Watson, 1998) throughout the hippocampus was processed and counted under a light microscope (Carl Zeiss Axioskop 2 Plus) at 40× and 100× magnification. The total number of BrdU-positive cells or β-catenin positive aggregates per section were determined and multiplied by 6 to obtain the total number of BrdU-positive cells or β-catenin-positive aggregates per hippocampus.
Western blot
For Western blot analysis, animals were killed by decapitation, their brains removed and the hippocampi dissected and stored at −80°C. Each sample was homogenized and processed to obtain the total cell lysate (TCL), and membrane, cytosol and nuclear fractions as described by Mostany et al. (2008). Every sample was homogenized (1:15, 500 µL approx.) using a Potter homogenizer in homogenization buffer (10 mM HEPES, pH 7.9, 1.5 mM MgCl2, 10 mM KCl) containing protease and phosphatase inhibitors (PPI; 1 mM PMSF, 10 µL·mL−1 aprotinin, 10 µg·mL−1 leupetin, 10 µg·mL−1 pepstatin A, 10 µg·mL−1 antipain, 10 µg·mL−1 chymostatin, 5 µg·mL−1 trypsin inhibitor, 1 mM NaV, 1 mM NaF, 1 mM cantharidin and 10 µM E-64). After homogenization, 250 µL of homogenate was lysed in lysis buffer (HB containing 1% Igepal, 0.1% sodium deoxycholate, 0.2% SDS and 0.1% Triton X-100) 30 min on ice for the TCL, and centrifuged at 14 000×g 10 min at 4°C. Aliquots of the supernatant (TCL) were stored at −20°C. The remaining homogenate (250 µL) for subcellular fractionation was centrifuged at 1000×g for 10 min at 4°C, and the resulting supernatant (S1) and pellet were (P1) separated. The S1 fraction was centrifuged at 100 000×g for 15 min at 4°C, and the resulting pellet P2 membrane fraction was resuspended in buffer containing detergents and PPI, incubated 30 min at 4°C, centrifuged for 10 min at 14 000×g. Aliquots of this supernatant were stored as the membrane fraction (M). Nuclear proteins (N) were isolated by high salt extraction from the P1 fraction. The P1 fraction was homogenized in 20 mM HEPES pH 7.9, 0.45 M NaCl, 1 mM EDTA containing PPI and incubated in ice for 30 min. Solubilized proteins were recovered in the supernatant after centrifugation at 14 000×g for 10 min at 4°C. Protein was quantified by the Lowry method.
About 30–50 µg of protein was resolved on 12.5 or 15% SDS-PAGE and transferred to PVDF membranes. These membranes were incubated in mouse anti-β-catenin (1:1000), mouse anti-N-cadherin (1:1000), rabbit anti-BDNF (1:300), mouse anti-GAPDH (1:2000) and mouse anti-α-tubulin (1:20 000) primary antibodies,(Santa Cruz Biotechnology, Inc.) overnight. After extensive washings in 0.05% Tween 20 in Tris buffered saline, membranes were incubated with horseradish peroxidase-conjugated secondary antibodies. Secondary antibodies were detected with ECL Advance kit (GE Healthcare Europe GmbH, Munich, Germany). Blot quantitations were performed by densitometric scanning using Scion Image Software (Scion Corporation). The densitometry values were normalized with respect to GAPDH values for TCL and M fractions, and with respect to β-tubulin for the N fraction. Data for every sample was the mean of at least two independent experiments.
Autoradiography for 5-HT1A receptor function
Experiments were performed following modifications to a previously described protocol (Sim et al., 1997). Sections were pre-incubated for 30 min at 25°C in a buffer containing 50 mM Tris-HCl, 0.2 mM EGTA, 3 mM MgCl2, 100 mM NaCl, 1 mM DTT and 2 mM GDP (pH 7.7), and subsequently incubated for 2 h at 25°C in the same buffer containing 3 mU·mL−1 adenosine deaminase and 0.04 nM [35S]guanosine-5-O-(3-thio) triphosphate (GTPγS; PerkinElmer Inc.). Consecutive sections were incubated with 10 µM (±)-8-hydroxy-N,N-dipropyl-2-aminotetralin (8-OH-DPAT; Sigma) alone or in the presence of 10 µM WAY100635. Non-specific binding was determined in the presence of 10 µM GTPγS. After the incubation, the sections were washed twice for 15 min in 50 mM Tris-HCl buffer (pH 7.4) at 4°C, rinsed in distilled cold water and cold air dried. Sections were exposed to radiation-sensitive films (Hyperfilm™-βmax, Amersham) together with 14C-polymer standards (Amersham) for 2 days at 4°C.
Data analysis
Data were expressed as percentage of values from vehicle-treated rats, with these values set to 100%. Results shown are means ± SEM. Data were analysed using a two-way anova followed by a Bonferroni post hoc test to analyze the possible interaction between the antidepressant (fluoxetine) administration and the 5-HT2A receptor blockade (administration of ketanserin) in the results obtained. Statistical significance was set at P < 0.05.
Results
Effect of fluoxetine, ketanserin and their co-treatment on the FST
In the FST the immobility times observed in the different treatment groups after subchronic (7 days administration) are shown in Figure 1A. A two-way anova analysis showed a significant interaction in the immobility time between treatment with fluoxetine and 5-HT2A receptor antagonist [F(1,35) = 56.23, P < 0.001], and a significant main effect for the fluoxetine [F(1,35) = 204.9, P < 0.001], while the group treated with ketanserin alone did not show statistical changes [F(1,35) = 1.37, P= 0.25]. In the swimming time, two-way anova analysis presented a significant interaction between 5-HT2A blockade and fluoxetine treatment [F(1,35) = 17.33, P < 0.001], a significant main effect of fluoxetine treatment [F(1,35) = 23.16, P < 0.001], and a significant main effect of 5-HT2A blockade [F(1,35) = 26.70, P < 0.001] (Figure 1B). Regarding climbing time, no significant interaction was observed but there was a significant main effect of fluoxetine treatment [F(1,35) = 6.74, P < 0.05] (Figure 1C).
Figure 1.
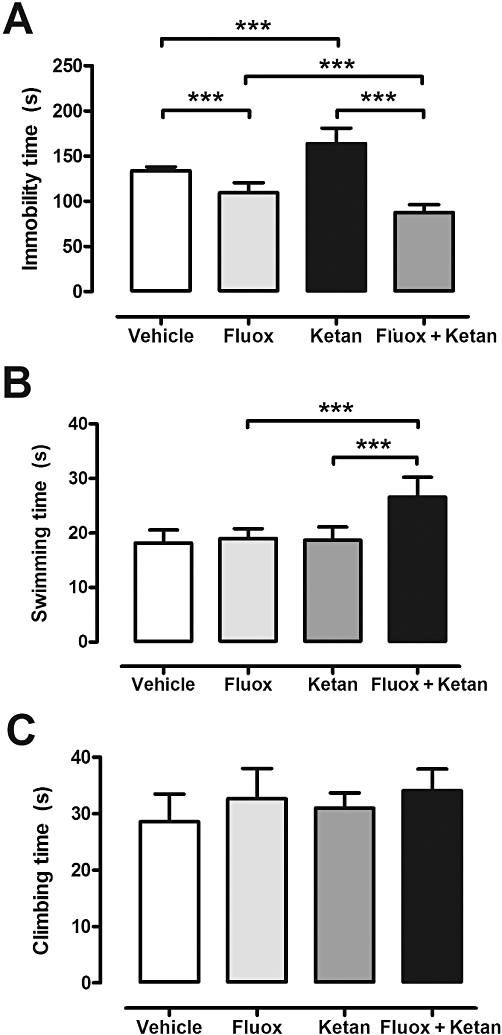
Graphs showing immobility time (A), swimming time (B) and climbing time (C) in the FST for vehicle, fluoxetine (Fluox), ketanserin (Ketan) and fluoxetine + ketanserin (Fluox + Ketan) treatment groups. Data are expressed in seconds (s) (mean ± SEM, n= 8–10). Note that a two-way anova shows significant changes in the immobility time: fluoxetine × ketanserin interaction [F(1,35) = 56.23, P < 0.001]; and in the swimming time: fluoxetine × ketanserin interaction [F(1,35) = 17.33, P < 0.001]. ***P < 0.001 for the Bonferroni post hoc test.
After an acute treatment, there were no significant changes between the different groups analysed for the immobility time: 118 ± 9 s vehicle group, 112 ± 17 s fluoxetine group, 147 ± 17 s ketanserin group and 121 ± 7 s co-treatment group.
Effect of fluoxetine, ketanserin and their co-treatment on BDNF and TrkB expression
Subchronic fluoxetine treatment showed a trend towards increased expression of BDNF mRNA compared with the vehicle group in some regions such as the CA3 field (CA3) and the DG of the hippocampus, but this was not significant. After treatment with ketanserin alone, BDNF mRNA expression in the CA3 and DG regions, were also not different from those of the vehicle group. However, co-treatment with fluoxetine + ketanserin for 7 days increased BDNF mRNA expression in several brain areas, including CA3 and DG (Figure 2F,G). A two-way anova revealed a non-significant interaction in the CA3 region of the hippocampus between the antidepressant treatment with fluoxetine and the 5-HT2A receptor antagonist group [F(1,44) = 0.20, P= 0.66], and the 5-HT2A receptor antagonist alone [F(1,44) = 0.81, P= 0.37], while the antidepressant effect was significant [F(1,44) = 13.4, P < 0.001] (Figure 2F). The DG of the hippocampus showed an interaction between the two variables studied [F(1,44) = 4.45, P < 0.05], the blockade of the 5-HT2A receptor [F(1,44) = 4.77, P < 0.05] and the antidepressant effect [F(1,44) = 17.66, P < 0.001], using a two-way anova (Figure 2G). Other brain areas showing a tendency towards increased in BDNF mRNA expression in the co-treatment group included the amygdala (147 ± 19%) and piriform cortex (151 ± 21%). Areas such as the CA1 field of the hippocampus and medial prefrontal cortex did not show significant changes using the two-way anova following any drug treatment assayed (data not shown).
Figure 2.
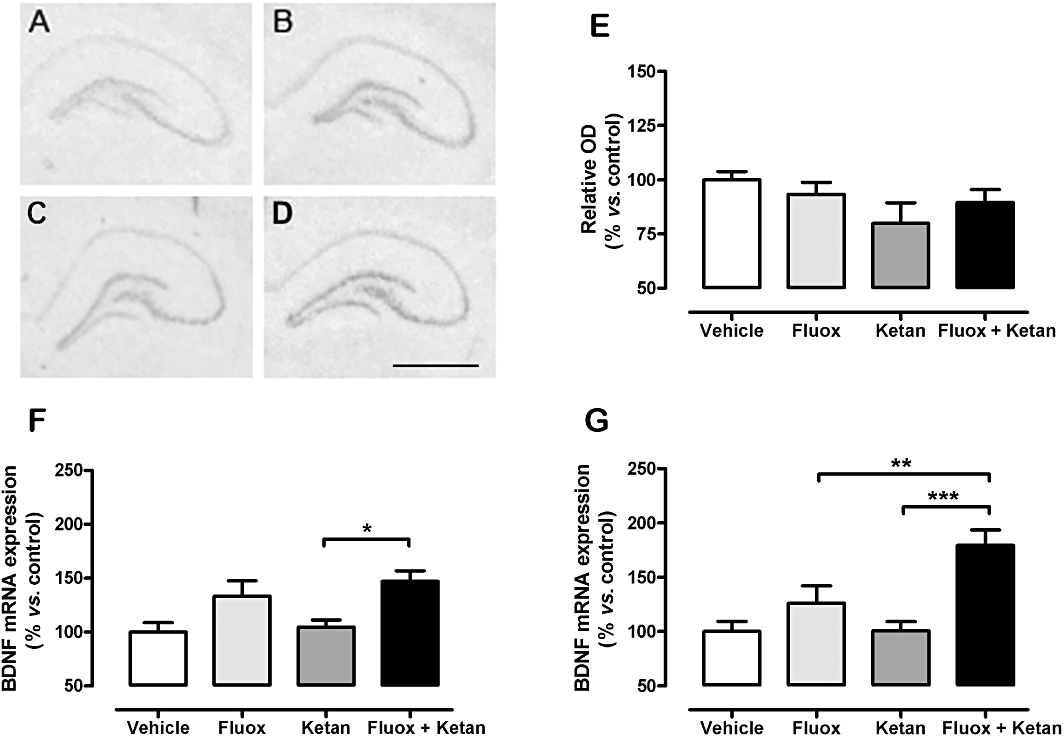
Autoradiograms illustrating BDNF in situ hybridization of vehicle (A), fluoxetine (Fluox; B), ketanserin (Ketan; C), and fluoxetine + ketanserin (Fluox + Ketan; D) treatments (upper images). Graphs show BDNF protein expression in hippocampus TCL (E), and mRNA expression in both CA3 subfield (F) and DG (G) of the hippocampus. Data expressed as percentage of vehicle-treated animals (100%). Data are expressed as mean ± SEM; n= 10–12. Two-way anova for BDNF mRNA in DG: fluoxetine × ketanserin interaction [F(1,44) = 4.45, P < 0.05]. *P < 0.05, **P < 0.01, ***P < 0.001 in the Bonferroni post hoc test. Bar: 2 mm. OD, optic density.
The analysis of BDNF protein levels in hippocampus after 7 days of treatment did not reveal significant changes in any of the experimental groups studied: vehicle 100 ± 4%, fluoxetine 93 ± 6%, ketanserin 80 ± 10% and fluoxetine + ketanserin 90 ± 6%. Acute treatment with fluoxetine, ketanserin or the combination of both drugs also showed no changes: 100 ± 8%, vehicle; 110 ± 8%, fluoxetine; 102 ± 9%, ketanserin; and 100 ± 4%, co-treatment group.
TrkB receptor expression was also measured by in situ hybridization. No modification was found for any of the experimental groups analysed after 7 days of treatment (Supporting Information Figure S1).
Lack of effect of fluoxetine, ketanserin and the co-treatment on BrdU incorporation
The effects of the treatments (fluoxetine, ketanserin and fluoxetine + ketanserin) on the BrdU-positive cells are shown in Figure 3. Statistical analysis of the data using a two-way anova showed no interaction between fluoxetine treatment and 5-HT2A receptor blockade but a significant main effect of ketanserin treatment [F(1,24) = 34.60, P < 0.001].
Figure 3.
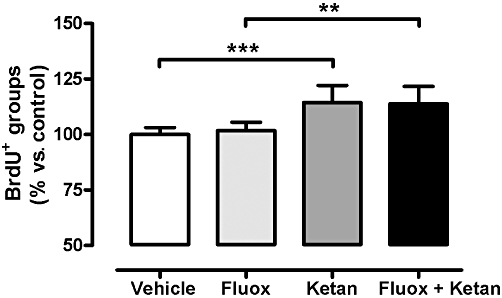
Graph showing BrdU immunolabelling (BrdU+) in the SGZ of the DG of the hippocampus for vehicle, fluoxetine (Fluox), ketanserin (Ketan), and fluoxetine + ketanserin (Fluox + Ketan) treatment groups. Data are presented as percentage of vehicle group (100%). Data are expressed as mean ± SEM, n= 7. **P < 0.01, ***P < 0.001 using a two-way anova analysis followed by a Bonferroni post hoc test.
Effect of fluoxetine, ketanserin and the co-treatment on β-catenin protein expression
Western blot analysis of the TCL showed no changes in β-catenin protein levels in the fluoxetine alone and ketanserin alone groups, with a significant increase in the fluoxetine + ketanserin group (Figure 4A). The interaction between antidepressant administration and 5-HT2A receptor blockade was statistically significant using a two-way anova[F(1,44) = 9.14, P < 0.01], and there was a significant main effect of fluoxetine treatment [F(1,44) = 15.55, P < 0.001] and for 5-HT2A receptor blockade [F(1,44) = 6.77, P < 0.05]. In the membrane fraction, there were no changes in β-catenin in the fluoxetine and ketanserin groups, while there was a significant increase of β-catenin labelling in the fluoxetine + ketanserin group (Figure 4B,D). Two-way anova showed a significant interaction of fluoxetine treatment and 5-HT2A blockade [F(1,44) = 4.65, P < 0.05], a significant main effect of the fluoxetine treatment [F(1,44) = 7.71, P < 0.01] and of the 5-HT2A receptor antagonist [F(1,44) = 6.23, P < 0.05]. No significant modifications in β-catenin in the nuclear fraction were observed in any of the experimental groups (Figure 4C,D), using a two-way anova[F(1,44) = 0.061, P= 0.806] for the interaction between both drugs.
Figure 4.
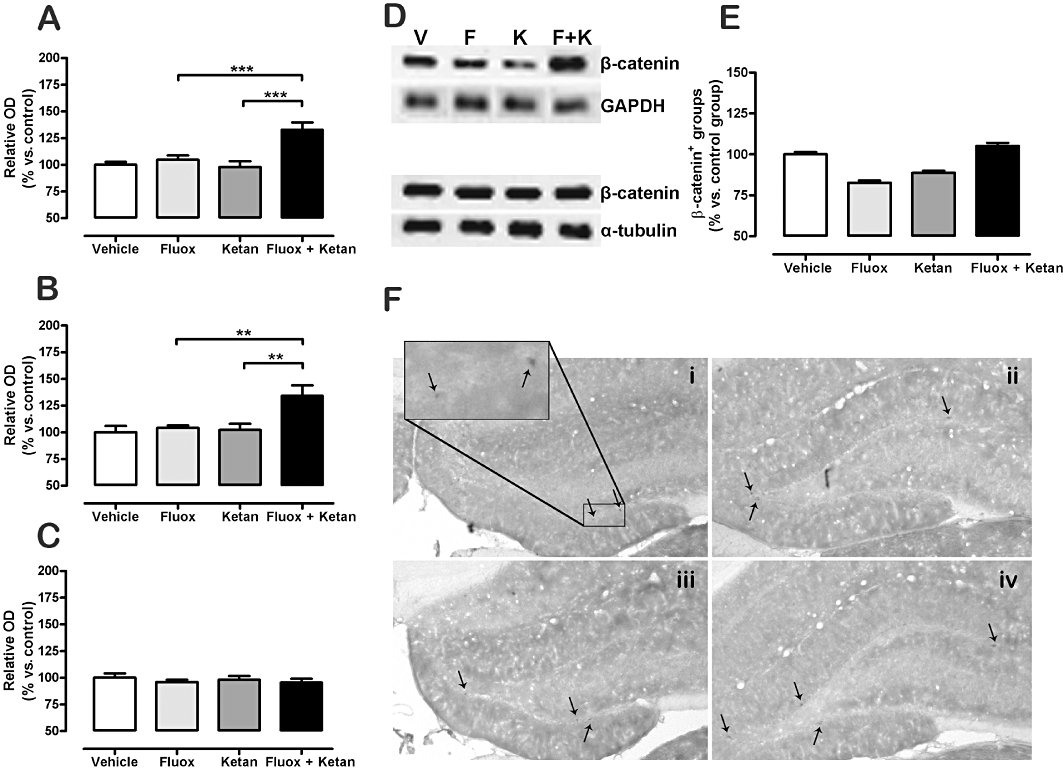
Effect of fluoxetine (Fluox), ketanserin (Ketan), and fluoxetine + ketanserin (Fluox + Ketan) treatments on β-catenin level in rat hippocampus TCL (A), membrane fraction (B) and nuclear fraction (C). Representative Western blots of β-catenin protein expression in membrane and nuclear fraction from hippocampus [vehicle (V), fluoxetine (F), ketanserin (K) and fluoxetine + ketanserin (F + K)] (D). β-catenin immunohistochemical positive clusters in the SGZ of the hippocampus (E) and representative images showing β-catenin immunolabelling in SGZ of the hippocampus (F): (i) vehicle, (ii) fluoxetine, (iii) ketanserin and (iv) fluoxetine + ketanserin. Western blot data are expressed as percentage of the relative optical density (OD) of the control, n= 10–12, and data for immunolabelling are presented as β-catenin immunopositive groups SGZ, n= 7 (vs. vehicle-treated animals). Data are presented as mean ± SEM Two-way anova for the antidepressant fluoxetine × ketanserin interaction in TCL [F(1,44) = 9.14, P < 0.01], membrane fraction [F(1,44) = 4.65, P < 0.05] and β-catenin immunolabelling [F(1,24) = 36.28, P < 0.001]. **P < 0.01, ***P < 0.001 in the Bonferroni post hoc test.
Acute drug administration did not change β-catenin protein levels in hippocampal total homogenate, membrane or nuclear fraction in any of the different treatment groups (data not shown).
The β-catenin immunopositive cells in the different treatment groups (Figure 4E,F) showed an interaction between fluoxetine administration and the 5-HT2A receptor antagonist, [F(1,24) = 36.28, P < 0.001] and a significant main effect of the fluoxetine treatment [F(1,24) = 5.59, P < 0.05] using a two-way anova.
Effect of fluoxetine, ketanserin and the co-treatment on N-cadherin protein expression
The analysis by Western blot of the hippocampal cell lysate (TCL) of the different experimental groups revealed a significant increase in N-cadherin protein levels in the co-treatment group over those in the fluoxetine alone and ketanserin alone groups (Bonferroni post hoc test). Two-way anova showed significant fluoxetine and ketanserin interaction [F(1,44) = 5.95, P < 0.05], a significant main effect of the fluoxetine treatment [F(1,44) = 10.62, P < 0.01] and a significant main effect of the ketanserin treatment [F(1,44) = 6.19, P < 0.05] (Figure 5A). N-cadherin protein levels in the membrane fraction (Figure 5B) were increased both in the fluoxetine and in the co-treatment groups. A significant interaction between antidepressant treatment and 5-HT2A receptor antagonism was observed [F(1,44) = 4.79, P < 0.05], as was a significant main effect of the fluoxetine treatment [F(1,44) = 42.64, P < 0.001], following two-way anova (Figure 5B). The analysis of N-cadherin protein levels in the nuclear fraction (Figure 5C) showed a significant main effect of the ketanserin treatment [F(1,44) = 4.15, P < 0.05], but not significant changes following a Bonferroni post hoc test.
Figure 5.
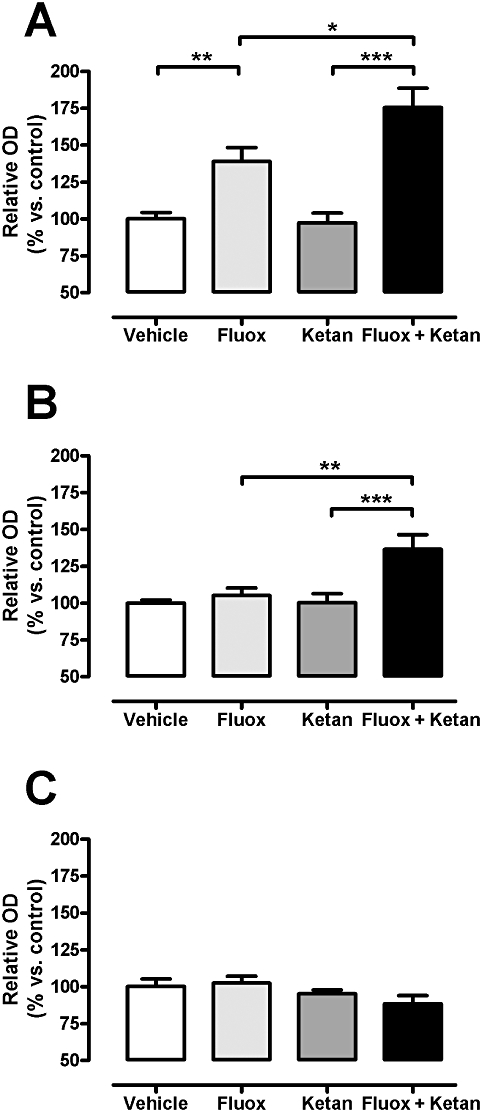
Parallel effect of fluoxetine (Fluox), ketanserin (Ketan), and fluoxetine + ketanserin (Fluox + Ketan) treatments on N-cadherin protein levels in rat hippocampus total cell lysate (A), membrane fraction (B) and nuclear fraction (C). Western blot data are expressed as percentage of the relative optical density (OD) in the vehicle-treated animals (100%). Data are presented as mean ± SEM, n= 10–12. Two-way anova analysis for fluoxetine × ketanserin interaction in total cell lysate: [F(1,44) = 5.95, P < 0.05] and in membrane fraction: [F(1,44) = 4.79, P < 0.05]. *P < 0.05, **P < 0.01, ***P < 0.001 using a Bonferroni post hoc test.
The acute administration of fluoxetine or ketanserin alone or combined did not change the N-cadherin protein levels in hippocampal total homogenate (data not shown).
Lack of effect of fluoxetine, ketanserin and the co-treatment on 5-HT1A receptor function
Basal [35S]GTPγS binding and 8-OH-DPAT-mediated [35S]GTPγS stimulation were not significantly modified in the different brain structures studied in fluoxetine, ketanserin and fluoxetine + ketanserin treatment groups (Table 1).
Table 1.
Effect of 7 day treatment with fluoxetine, ketanserin or fluoxetine + ketanserin on basal [35S]GTPγS binding and (±)-8-OH-DPAT-stimulated [35S]GTPγS binding in some rat brain areas, reflecting 5-HT1A function
| Vehicle | Fluoxetine | Ketanserin | Fluox + ketan | |
|---|---|---|---|---|
| CA1 | 126 ± 6 | 122 ± 4 | 135 ± 8 | 138 ± 6 |
| CA3 | 122 ± 8 | 124 ± 6 | 132 ± 7 | 137 ± 6 |
| DG | 173 ± 15 | 170 ± 5 | 192 ± 13 | 183 ± 13 |
| DRN | 154 ± 8 | 177 ± 8 | 168 ± 11 | 176 ± 11 |
Values are presented as percentage of basal values (100%) (mean ± SEM) for 8-OH-DPAT-stimulated [35S]GTPγS binding. Two-way anova showed no statistical differences.
CA1, CA1 field of hippocampus; CA3, CA3 field of hippocampus; DG, dentate gyrus of hippocampus; DRN, dorsal raphe nucleus.
Discussion and conclusions
Current antidepressant treatments require several weeks to achieve therapeutic efficacy. Different strategies based on the pharmacological manipulation of monoaminergic systems have been developed in an effort to accelerate the onset of antidepressant action, including the combination of 5-HT reuptake blockers with antagonists of serotonin receptors (Adell et al., 2005). Recent studies have reported a potentiation of the antidepressant effects by co-treatment with drugs with a 5-HT2A receptor antagonist profile (Shelton et al., 2001; Marek et al., 2003; Celada et al., 2004; Pandey et al., 2010). The aim of this study was to evaluate the effect of subchronic co-treatment with the SSRI fluoxetine and the 5-HT2A antagonist ketanserin on hippocampal neurogenic markers, along with behavioural findings. The dose of ketanserin used (0.1 mg·kg−1·day−1) should result in a selective blockade of the 5-HT2A subtype (Hoyer et al., 2002). Our results show that the administration for 7 days of either fluoxetine or ketanserin did not significantly modify BDNF, β-catenin expression or depression-related behaviour. In contrast, co-treatment with fluoxetine and ketanserin significantly increased those neurogenic markers, inducing an antidepressant-like response in the FST.
To date, some reports indicate that the combination of 5-HT2A receptor antagonists and tricyclic antidepressants (Pandey et al., 2010) or SSRIs (Marek et al., 2003) produces antidepressant-like effects in animals. More interestingly, this combination also increases antidepressant responses in humans, even in treatment-resistant cases (Shelton et al., 2001; Marek et al., 2003). Furthermore, the acute administration of 5-HT2A receptor antagonists decreases immobility and increases the swimming behaviour in the FST (Patel et al., 2004). In line with this, stimulation of 5-HT2A receptors results in depressogenic-like behaviour (Rajkumar et al., 2009). Our results in the co-treatment group after 7 days of treatment show a decrease in immobility in the FST, a response previously reported to appear after 14 days of SSRI treatment (Cryan et al., 2005a), and an increase of the swimming time, which shows statistical significance for the interaction of the fluoxetine treatment and 5-HT2A receptor blockade, which reflects an involvement of the 5-hydroxytryptaminergic system in this antidepressant-like response (Cryan et al., 2005a,b). In addition, the lack of changes in climbing time excludes a possible role of noradrenergic/dopaminergic mechanisms (Cryan et al., 2005b). Acute administration of the different drugs alone or combined did not produce any change compared with vehicle. In this regard, our data are in accordance with others' in which low doses of fluoxetine (8–20 mg·kg−1) (Castagnéet al., 2006; Ulak et al., 2010), or the acute administration of ketanserin alone (Campos et al., 2005) or in combination with some antidepressant drugs (Campos et al., 2005) did not produce any significant change in immobility time in the FST.
Furthermore, we have not found any change associated to the function of the 5-HT1A receptor subtype, although a significant reduction of the basal values was observed in the basal values in the dorsal raphe nucleus (DRN) between the fluoxetine and the co-treatment groups, suggesting that the duration of the treatment (7 days) is not enough to desensitize 5-HT1A receptors in the DRN, as occurs with chronic (14 days) treatments (Castro et al., 2003). Thus, an increased 5-hydroxytryptaminergic activity might be a consequence of the influence of other regulatory mechanisms on DRN activity, such as GABAergic neurons (containing 5-HT2A receptors) and/or glutamate neurotransmission (see Celada et al., 2004), or other early neuroplastic changes, as discussed later.
BDNF expression is reduced in some brain areas (Duman et al., 1997) and serum (Shimizu et al., 2003) of depressed patients, as well as in the hippocampus of some animal models of depression (Elfving et al., 2009). The expression of this protein is increased after chronic antidepressant treatment in rat hippocampus (Nibuya et al., 1995; Larsen et al., 2007), human brain (Duman et al., 1997) and human serum (Shimizu et al., 2003). In the present work, no changes were observed in BDNF mRNA expression in the hippocampus following subchronic treatment with fluoxetine or ketanserin alone, in agreement with previous work (Larsen et al., 2007). However, after subchronic co-treatment with fluoxetine and ketanserin, we found an increased expression of BDNF mRNA in CA3, reaching statistical significance in the DG, an area clearly related to the pathogenesis of depression (see Lucassen et al., 2006). This increase in BDNF mRNA expression was not accompanied by an increase in the expression of the protein. At least 3 weeks are required to show a significant increase in the hippocampus (De Foubert et al., 2004).
A relationship between 5-HT2A receptors and BDNF has been suggested by studies where activation of 5-HT2A receptors decreased hippocampal BDNF (Vaidya et al., 1999). Furthermore, studies in mice with genetically low levels of BDNF (BDNF+/−), which present a depression-like profile, show higher hippocampal 5-HT2A receptor levels, which are down-regulated in hippocampal primary cultures and organotypic hippocampal slices after 7 days of incubation with BDNF (Trajkovska et al., 2009). It is noteworthy that antidepressant-like effects have been described 3–10 days after acute bilateral infusion of BDNF in CA3 and DG (Shirayama et al., 2002).
In contrast to our data on BDNF mRNA expression, TrkB mRNA expression was not modified after the 7 day treatment with fluoxetine, ketanserin or the co-treatment. An increase of TrkB mRNA expression after chronic antidepressant treatment (Nibuya et al., 1995) has been previously reported, although 21 days of treatment appear to be necessary before an up-regulation of TrkB gene is achieved. The possibility exists that the regulation of TrkB receptor expression is slower than that of BDNF or that an increase of BDNF mRNA could lead to the activation of another signalling pathway, such as the p75 neurotrophin receptor (Rantamäki et al., 2007).
The 5-HT2A receptor subtype is located postsynaptically (Peddie et al., 2008) in the pyramidal and granular cell layers within the hippocampus, and in GABAergic interneurons (Peddie et al., 2008), involved in the down-regulation of BDNF expression (Zafra et al., 1991). Furthermore, specific 5-HT2A receptor antagonists facilitate the induction of long-term potentiation in the hippocampus (Wang and Arvanov, 1998). The increase in BDNF expression in CA3 and DG after subchronic treatment with fluoxetine and ketanserin may be partially mediated by 5-HT2A receptor blockade, inhibiting GABAergic interneurons and producing an increase in the activation of pyramidal neurons in the hippocampus (Vaidya et al., 1999), thus modulating dendritic activation and synaptic plasticity (Gulyás et al., 1999).
The increase in β-catenin accumulation found in total hippocampal homogenates after subchronic co-treatment with fluoxetine and ketanserin was comparable with that reported following electroconvulsive seizures (Madsen et al., 2003) and chronic antidepressant treatment (Mostany et al., 2008), not observing any changes after acute administration. However, this increase in β-catenin was associated with the membrane fraction, not the nuclear fraction, in contrast to the results reported after chronic antidepressants (Mostany et al., 2008). This is further supported by the results from immunohistochemical labelling in the subgranular zone (SGZ) of the hippocampus where those immunopositive cells are co-localized with progenitor cells (Mostany et al., 2008). β-Catenin is an important regulator of synaptic plasticity and long-term memory formation (Maguschak and Ressler, 2008). The cadherin–β-catenin complex localized in symmetrical synaptic junctions (Uchida et al., 1996) is involved in the recruitment and control of the size of the reserve vesicle pool (Bamji et al., 2003), as well as in synaptic targeting (Nishimura et al., 2002). The relevance of β-catenin as a part of the N-cadherin/β-catenin complex, in contrast to its role in the Wnt/β-catenin pathway, is supported by the parallel increase that we observed in N-cadherin protein attached to the membrane fraction as a result of the co-treatment with the antidepressant fluoxetine and the 5-HT2A receptor antagonist ketanserin. The intracellular levels of cadherin/catenin complex are a limiting factor in dendritic morphogenesis. Its overexpression increases dendritic branching (Yu and Malenka, 2003), and the association with cadherins effectively sequesters β-catenin from the cytoplasmic pool (Patapoutian and Reichardt, 2000). Regarding the factors regulating the interaction between Wnt/β-catenin signalling and cadherin-mediated adhesion, an increase in N-cadherin would result in the inhibition of Wnt (see Nelson and Nusse, 2004), thus favouring the cell–cell adhesion role of β-catenin. In experiments deleting β-catenin in hippocampal pyramidal neurons, a reduction in the number of reserve pool vesicles per synapse and an impaired response to prolonged repetitive stimulation was observed (Bamji et al., 2003). The increase in synaptic activity is also associated with the redistribution of β-catenin into synapses, which depends on differential phosphorylation of this protein (see Nelson and Nusse, 2004), mediated by neurotrophic Trk receptors (David et al., 2008). Some authors indicate that β-catenin located in adherent junctions can also be released and translocated to the nucleus (Kam and Quaranta, 2009). Thus, the co-treatment with fluoxetine and ketanserin could result in an increase of the role of β-catenin in the facilitation of synaptic plasticity.
We have not observed a significant interaction between both drug treatments in the modulation of hippocampal cell proliferation, although an increase was found following either ketanserin or fluoxetine + ketanserin administration. An increase in cell proliferation in the DG of the hippocampus has been related to chronic antidepressant treatments, but it has not been reported following subchronic treatment with fluoxetine (Malberg et al., 2000). The effects of 5-HT2A receptor antagonists, given alone, depend on the duration of the treatment as a reduction in proliferation after an acute treatment (Jha et al., 2008) and an increase after 7 days administration (Jha et al., 2008) have both been reported.
In summary, the subchronic (1 week) co-treatment with an SSRI and a 5-HT2A antagonist was sufficient to increase some neuroplastic markers previously associated with chronic (2–3 weeks) antidepressant treatment, such as BDNF and β-catenin. In addition, this treatment did not induce the well characterized changes in monoaminergic neurotransmission following chronic antidepressants, such as 5-HT1A receptor desensitization in the DRN. However, this combined treatment was enough to promote antidepressant-like behavioural changes with a shorter onset of action. Thus, we propose that the modifications in synaptic plasticity induced by this drug combination are enough to produce an antidepressant-like response, preceding the appearance of changes in cell proliferation and 5-hydroxytryptaminergic markers. Although further experiments are needed to fully clarify the role of 5-HT2A receptors in this early antidepressant response, our results strongly support the pharmacological blockade of this receptor subtype as a promising target for the treatment of depression.
Acknowledgments
We wish to thank Drs Jesús Pascual-Brazo, Alvaro Díaz, Elena Castro and Elsa Valdizán for their scientific advice, and Ms Lourdes Lanza, Rebeca Madureira, Isabel Ruiz, Alicia Martin and Helena Blanco for their technical assistance. This work has been supported by Instituto de Salud Carlos III (Red Investigación de Enfermedades Mentales, REMTAP), Ministerio de Ciencia e Innovación SAF07-61862 and Fundación Alicia Koplowitz.
Glossary
- 8-OH-DPAT
8-hydroxy-N,N-dipropyl-2-aminotetralin
- BDNF
brain-derived neurotrophic factor
- BrdU
5-bromo-2′-deoxyuridine
- CA1 and CA3
CA1 and CA3 subfields of the hippocampus
- DG
dentate gyrus of the hippocampus
- DRN
dorsal raphe nucleus
- FST
forced swimming test
- GSK
glycogen synthase kinase-3
- [35S]GTPγS
[35S]guanosine 5′-O-[γ–thio]triphosphate
- SARI
5-HT2A receptor antagonists/reuptake inhibitor
- SGZ
subgranular zone
- SSRI
5-HT-selective reuptake inhibitor
Conflicts of interest
The authors declare that AP has received support for research from Faes Farma, SA, FP-C and RV report no biomedical financial interests or potential conflicts of interest. The present study is not related to any of these professional or collaborative relationships.
Supporting information
Figure S1 Representative autoradiograms of TrkBin situ hybridization in vehicle (A), fluoxetine (B),ketanserin (C) and fluoxetine + ketanserine (D) treatment groups.Graphs showing TrkB mRNA expression in the different groups studiedin two representative regions, as CA3 subfield (E) and dentategyrus (DG) (F) of the hippocampus. TrkB expression is representedas percentage versus vehicle TrkB mRNA expression for each region.Data expressed as percentage of vehicle group. Data are expressedas mean ± SEM; n = 10–12. Two-way ANOVAfollowed by Bonferroni post hoc test showed no interactionbetween fluoxetine treatment and 5-HT2A receptor blockade. Bar: 2 mm.
References
- Adell A, Castro E, Celada P, Bortolozzi A, Pazos A, Artigas F. Strategies for producing faster acting antidepressants. Drug Discov Today. 2005;10:578–585. doi: 10.1016/S1359-6446(05)03398-2. [DOI] [PubMed] [Google Scholar]
- Aghajanian GK, Marek GJ. Serotonin model of schizophrenia: emerging role of glutamate mechanisms. Brain Res Brain Res Rev. 2000;31:302–312. doi: 10.1016/s0165-0173(99)00046-6. [DOI] [PubMed] [Google Scholar]
- Alexander SP, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 5th edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson K, Fuxe K, Eneroth P, Härfstrand A, Agnati LF. Involvement of D1 dopamine receptors in the nicotine-induced neuro-endocrine effects and depletion of diencephalic catecholamine stores in the male rat. Neuroendocrinology. 1988;48:188–200. doi: 10.1159/000125007. [DOI] [PubMed] [Google Scholar]
- Attar-Lévy D, Martinot JL, Blin J, Dao-Castellana MH, Crouzel C, Mazoyer B, et al. The cortical serotonin2 receptors studied with positron-emission tomography and [18F]-setoperone during depressive illness and antidepressant treatment with clomipramine. Biol Psychiatry. 1999;45:180–186. doi: 10.1016/s0006-3223(98)00007-9. [DOI] [PubMed] [Google Scholar]
- Bamji SX, Shimazu K, Kimes N, Huelsken J, Birchmeier W, Lu B, et al. Role of beta-catenin in synaptic vesicle localization and presynaptic assembly. Neuron. 2003;40:719–731. doi: 10.1016/s0896-6273(03)00718-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos MM, Fernandes ES, Ferreira J, Santos AR, Calixto JB. Antidepressant-like effects of Trichilia catigua (Catuaba) extract: evidence for dopaminergic-mediated mechanisms. Psychopharmacology (Berl) 2005;182:45–53. doi: 10.1007/s00213-005-0052-1. [DOI] [PubMed] [Google Scholar]
- Castagné V, Porstol RD, Moser P. Early behavioral screening for antidepressants and anxiolytics. Drug Dev Res. 2006;67:729–742. [Google Scholar]
- Castro ME, Diaz A, del Olmo E, Pazos A. Chronic fluoxetine induces opposite changes in G protein coupling at pre and postsynaptic 5-HT1A receptors in rat brain. Neuropharmacology. 2003;44:93–101. doi: 10.1016/s0028-3908(02)00340-4. [DOI] [PubMed] [Google Scholar]
- Catafau AM, Searle GE, Bullich S, Gunn RN, Rabiner EA, Herance R, et al. Imaging cortical dopamine D(1) receptors using [(11)C]NNC112 and ketanserin blockade of the 5-HT(2A) receptors. J Cereb Blood Flow Metab. 2009;30:985–993. doi: 10.1038/jcbfm.2009.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celada P, Puig M, Amargós-Bosch M, Adell A, Artigas F. The therapeutic role of 5-HT1A and 5-HT2A receptors in depression. J Psychiatry Neurosci. 2004;29:252–265. [PMC free article] [PubMed] [Google Scholar]
- Cryan JF, Page ME, Lucki I. Differential behavioral effects of the antidepressants reboxetine, fluoxetine, and moclobemide in a modified forced swim test following chronic treatment. Psychopharmacology (Berl) 2005a;182:335–344. doi: 10.1007/s00213-005-0093-5. [DOI] [PubMed] [Google Scholar]
- Cryan JF, Valentino RJ, Lucki I. Assessing substrates underlying the behavioral effects of antidepressants using the modified rat forced swimming test. Neurosci Biobehav Rev. 2005b;29:547–569. doi: 10.1016/j.neubiorev.2005.03.008. [DOI] [PubMed] [Google Scholar]
- David MD, Yeramian A, Duñach M, Llovera M, Cantí C, de Herreros AG, et al. Signalling by neurotrophins and hepatocyte growth factor regulates axon morphogenesis by differential beta-catenin phosphorylation. J Cell Sci. 2008;121:2718–2730. doi: 10.1242/jcs.029660. [DOI] [PubMed] [Google Scholar]
- De Foubert G, Carney SL, Robinson CS, Destexhe EJ, Tomlinson R, Hichs CA, et al. Fluoxetine-induced change in rat brain expression of brain-derived neurotrophic factor varies depending on length of treatment. Neuroscience. 2004;128:597–604. doi: 10.1016/j.neuroscience.2004.06.054. [DOI] [PubMed] [Google Scholar]
- Duman RS, Heninger GR, Nestler EJ. A molecular and cellular theory of depression. Arch Gen Psychiatry. 1997;54:597–606. doi: 10.1001/archpsyc.1997.01830190015002. [DOI] [PubMed] [Google Scholar]
- Elfving B, Plougmann PH, Müller HK, Mathé AA, Rosenberg R, Wegener G. Inverse correlation of brain and blood BDNF levels in a genetic rat model of depression. Int J Neuropsychopharmacol. 2009;2:1–10. doi: 10.1017/S1461145709990721. [DOI] [PubMed] [Google Scholar]
- Gulyás AI, Acsády L, Freund TF. Structural basis of the cholinergic and serotonergic modulation of GABAergic neurons in the hippocampus. Neurochem Int. 1999;34:359–372. doi: 10.1016/s0197-0186(99)00041-8. [DOI] [PubMed] [Google Scholar]
- Hoyer D, Hannon JP, Martin GR. Molecular, pharmacological and functional diversity of 5-HT receptors. Pharmacol Biochem Behav. 2002;71:533–554. doi: 10.1016/s0091-3057(01)00746-8. [DOI] [PubMed] [Google Scholar]
- Huang M, Ichiwaka J, Li Z, Dai J, Meltzer HY. Augmentation by citalopram of risperidone-induced monoamine release in rat prefrontal cortex. Psychopharmacology (Berl) 2006;185:274–281. doi: 10.1007/s00213-005-0206-1. [DOI] [PubMed] [Google Scholar]
- Jha S, Rajendran R, Fernandes KA, Vaidya VA. 5-HT2A/2C receptor blockade regulates progenitor cell proliferation in the adult rat hippocampus. Neurosci Lett. 2008;441:210–214. doi: 10.1016/j.neulet.2008.06.028. [DOI] [PubMed] [Google Scholar]
- Kam Y, Quaranta V. Cadherin-bound beta-catenin feeds into the Wnt pathway upon adherens junctions dissociation: evidence for an intersection between beta-catenin pools. PLoS ONE. 2009;4:e4580. doi: 10.1371/journal.pone.0004580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen MH, Rosenbrock H, Sams-Dodd F, Mikkelsen JD. Expression of brain derived neurotrophic factor, activity-regulated cytoskeleton protein mRNA, and enhancement of adult hippocampal neurogenesis in rats after sub-chronic and chronic treatment with the triple monoamine re-uptake inhibitor tesofensine. Eur J Pharmacol. 2007;555:115–121. doi: 10.1016/j.ejphar.2006.10.029. [DOI] [PubMed] [Google Scholar]
- Lucassen PJ, Heine VM, Muller MB, van der Beek EM, Wiegant VM, De Kloet ER, et al. Stress, depression and hippocampal apoptosis. CNS Neurol Disord Drug Targets. 2006;5:531–546. doi: 10.2174/187152706778559273. [DOI] [PubMed] [Google Scholar]
- Madsen TM, Newton SS, Eaton ME, Russell DS, Duman RS. Chronic electroconvulsive seizure up-regulates β-catenin expression in rat hippocampus: role in adult neurogenesis. Biol Psychiatry. 2003;54:1006–1014. doi: 10.1016/s0006-3223(03)00700-5. [DOI] [PubMed] [Google Scholar]
- Maguschak KA, Ressler KJ. Beta-catenin is required for memory consolidation. Nat Neurosci. 2008;11:1319–1326. doi: 10.1038/nn.2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malberg JE, Eisch AJ, Nestler EJ, Duman RS. Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J Neurosci. 2000;20:9104–9110. doi: 10.1523/JNEUROSCI.20-24-09104.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marek GJ, Carpenter LL, McDougle CJ, Price LH. Synergistic action of 5-HT2A antagonists and selective serotonin reuptake inhibitors in neuropsychiatric disorders. Neuropsychopharmacology. 2003;28:402–412. doi: 10.1038/sj.npp.1300057. [DOI] [PubMed] [Google Scholar]
- Martinowich K, Lu B. Interaction between BDNF and serotonin: role in mood disorders. Neuropsychopharmacology. 2008;33:73–83. doi: 10.1038/sj.npp.1301571. [DOI] [PubMed] [Google Scholar]
- Massou JM, Trichard C, Attar-Levy D, Feline A, Corruble E, Beaufils B, et al. Frontal 5-HT2A receptors studied in depressive patients during chronic treatment by selective serotonin reuptake inhibitors. Psychopharmacology (Berl) 1997;133:99–101. doi: 10.1007/s002130050377. [DOI] [PubMed] [Google Scholar]
- Mostany R, Valdizán EM, Pazos A. A role for nuclear beta-catenin in SNRI antidepressant-induced hippocampal cell proliferation. Neuropharmacology. 2008;55:18–26. doi: 10.1016/j.neuropharm.2008.04.012. [DOI] [PubMed] [Google Scholar]
- Nelson WJ, Nusse R. Convergence of Wnt, beta-catenin, and cadherin pathways. Science. 2004;303:1483–1487. doi: 10.1126/science.1094291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nibuya M, Morinobu S, Duman RS. Regulation of BDNF and TrkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J Neurosci. 1995;15:7539–7547. doi: 10.1523/JNEUROSCI.15-11-07539.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura W, Yao I, Iida J, Tanaka N, Hata Y. Interaction of synaptic scaffolding molecule and Beta-catenin. J Neurosci. 2002;22:757–765. doi: 10.1523/JNEUROSCI.22-03-00757.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey DK, Mahesh R, Kumar AA, Rao VS, Arjun M, Rajkumar R. A novel 5-HT(2A) receptor antagonist exhibits antidepressant-like effects in a battery of rodent behavioural assays: approaching early-onset antidepressants. Pharmacol Biochem Behav. 2010;94:363–373. doi: 10.1016/j.pbb.2009.09.018. [DOI] [PubMed] [Google Scholar]
- Pandey GN, Dwivedi Y, Rizavi HS, Ren X, Pandey SC, Pesold C, et al. Higher expression of serotonin 5-HT(2A) receptors in the postmortem brains of teenage suicide victims. Am J Psychiatry. 2002;159:419–429. doi: 10.1176/appi.ajp.159.3.419. [DOI] [PubMed] [Google Scholar]
- Patapoutian A, Reichardt LF. Roles of Wnt proteins in neural development and maintenance. Curr Opin Neurobiol. 2000;10:392–399. doi: 10.1016/s0959-4388(00)00100-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel JG, Bartoszyk GD, Edwards E, Ashby CR., Jr The highly selective 5-hydroxytryptamine (5-HT)2A receptor antagonist, EMD 281014, significantly increases swimming and decreases immobility in male congenital learned helpless rats in the forced swim test. Synapse. 2004;52:73–75. doi: 10.1002/syn.10308. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. 4th. Sydney, Australia: Academic Press Inc; 1998. [Google Scholar]
- Pazos A, Cortés R, Palacios JM. Quantitative autoradiographic mapping of serotonin receptors in the rat brain. II. Serotonin-2 receptors. Brain Res. 1985;346:231–249. doi: 10.1016/0006-8993(85)90857-1. [DOI] [PubMed] [Google Scholar]
- Peddie CJ, Davies HA, Colyer FM, Stewart MG, Rodríguez JJ. Colocalisation of serotonin2A receptors with the glutamate receptor subunits NR1 and GluR2 in the dentate gyrus: an ultrastructural study of a modulatory role. Exp Neurol. 2008;211:561–573. doi: 10.1016/j.expneurol.2008.03.003. [DOI] [PubMed] [Google Scholar]
- Rajkumar R, Pandey DK, Mahesh R, Radha R. 1-(m-Chlorophenyl)piperazine induces depressogenic-like behaviour in rodents by stimulating the neuronal 5-HT(2A) receptors: proposal of a modified rodent antidepressant assay. Eur J Pharmacol. 2009;608:32–41. doi: 10.1016/j.ejphar.2009.02.041. [DOI] [PubMed] [Google Scholar]
- Rantamäki T, Hendolin P, Kankaanpää A, Mijatovic J, Piepponen P, Domenici E, et al. Pharmacologically Diverse Antidepressants Rapidly Activate Brain-Derived Neurotrophic Factor Receptor TrkB and Induce Phospholipase-Cγ Signaling Pathways in Mouse Brain. Neuropsychopharmacology. 2007;32:2152–2162. doi: 10.1038/sj.npp.1301345. [DOI] [PubMed] [Google Scholar]
- Ressler KJ, Nemeroff CB. Role of serotonergic and noradrenergic systems in the pathophysiology of depression and anxiety disorders. Depress Anxiety. 2000;12(Suppl. 1):2–19. doi: 10.1002/1520-6394(2000)12:1+<2::AID-DA2>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Rosel P, Arranz B, San L, Vallejo J, Crespo JM, Urretavizcaya M, et al. Altered 5-HT(2A) binding sites and second messenger inositol trisphosphate (IP(3)) levels in hippocampus but not in frontal cortex from depressed suicide victims. Psychiatry Res. 2000;99:173–181. doi: 10.1016/s0925-4927(00)00076-7. [DOI] [PubMed] [Google Scholar]
- Santarelli L, Saxe M, Gross C, Surget A, Battaglia F, Dulawa S, et al. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science. 2003;301:805–809. doi: 10.1126/science.1083328. [DOI] [PubMed] [Google Scholar]
- Schmidt CJ, Sorensen SM, Kehne JH, Carr AA, Palfreyman MG. The role of 5-HT2A receptors in antipsychotic activity. Life Sci. 1995;56:2209–2222. doi: 10.1016/0024-3205(95)00210-w. [DOI] [PubMed] [Google Scholar]
- Serres F, Azorin JM, Valli M, Jeanningros R. Evidence for an increase in functional platelet 5-HT2A receptors in depressed patients using the new ligand [125I]-DOI. Eur Psychiatry. 1999;14:451–457. doi: 10.1016/s0924-9338(99)00222-9. [DOI] [PubMed] [Google Scholar]
- Shelton RC, Tollefson GD, Tohen M, Stahl S, Gannon KS, Jacobs TG, et al. A novel augmentation strategy for treating resistant major depression. Am J Psychiatry. 2001;158:131–134. doi: 10.1176/appi.ajp.158.1.131. [DOI] [PubMed] [Google Scholar]
- Shimizu E, Hashimoto K, Okamura N, Koike K, Komatsu N, Kumakiri C, et al. Alterations of serum levels of brain-derived neurotrophic factor (BDNF) in depressed patients with or without antidepressants. Biol Psychiatry. 2003;54:70–75. doi: 10.1016/s0006-3223(03)00181-1. [DOI] [PubMed] [Google Scholar]
- Shirayama Y, Chen AC, Nakagawa S, Russell DS, Duman RS. Brain-derived neurotrophic factor produces antidepressant effects in behavioral models of depression. J Neurosci. 2002;22:3251–3261. doi: 10.1523/JNEUROSCI.22-08-03251.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim LJ, Xiao R, Childers SR. In vitro autoradiographic localization of 5-HT1A receptor-activated G-proteins in the rat brain. Brain Res Bull. 1997;44:39–45. doi: 10.1016/s0361-9230(97)00061-0. [DOI] [PubMed] [Google Scholar]
- Stockmeier CA, Dilley GE, Shapiro LA, Overholser JC, Thompson PA, Meltzer HY. Serotonin receptors in suicide victims with major depression. Neuropsychopharmacology. 1997;16:162–173. doi: 10.1016/S0893-133X(96)00170-4. [DOI] [PubMed] [Google Scholar]
- Trajkovska V, Santini MA, Marcussen AB, Thomsen MS, Hansen HH, Mikkelsen JD, et al. BDNF downregulates 5-HT(2A) receptor protein levels in hippocampal cultures. Neurochem Int. 2009;55:697–702. doi: 10.1016/j.neuint.2009.06.013. [DOI] [PubMed] [Google Scholar]
- Uchida N, Honjo Y, Johnson KR, Wheelock MJ, Takeichi M. The catenin/cadherin adhesion system is localized in synaptic junctions bordering transmitter release zones. J Cell Biol. 1996;135:767–779. doi: 10.1083/jcb.135.3.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulak G, Mutlu O, Tanyeri P, Komsuoglu FI, Akar FY, Erden BF. Involvement of serotonin receptor subtypes in the antidepressant-like effect of TRIM in the rat forced swimming test. Pharmacol Biochem Behav. 2010;95:308–314. doi: 10.1016/j.pbb.2010.02.006. [DOI] [PubMed] [Google Scholar]
- Vaidya VA, Terwilliger RM, Duman RS. Role of 5-HT2A receptors in the stress-induced down-regulation of brain-derived neurotrophic factor expression in rat hippocampus. Neurosci Lett. 1999;262:1–4. doi: 10.1016/s0304-3940(99)00006-3. [DOI] [PubMed] [Google Scholar]
- Wada A. Lithium and neuropsychiatric therapeutics: neuroplasticity via glycogen synthase kinase-3beta, beta-catenin, and neurotrophin cascades. J Pharmacol Sci. 2009;110:14–28. doi: 10.1254/jphs.09r02cr. [DOI] [PubMed] [Google Scholar]
- Wang RY, Arvanov VL. M100907, a highly selective 5-HT2A receptor antagonist and a potential atypical antipsychotic drug, facilitates induction of long-term potentiation in area CA1 of the rat hippocampal slice. Brain Res. 1998;779:309–313. doi: 10.1016/s0006-8993(97)01174-8. [DOI] [PubMed] [Google Scholar]
- Yu X, Malenka RC. Beta-catenin is critical for dendritic morphogenesis. Nat Neurosci. 2003;6:1169–1177. doi: 10.1038/nn1132. [DOI] [PubMed] [Google Scholar]
- Zafra F, Castren E, Thoenen H, Lindholm D. Interplay between glutamate and g-aminobutyric acid transmitter systems in the physiological regulation of brain-derived neurotrophic factor and nerve growth factor synthesis in hippocampal neurons. Proc Natl Acad Sci U S A. 1991;88:10037–10041. doi: 10.1073/pnas.88.22.10037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetterström TS, Pei Q, Grahame-Smith DG. Repeated electroconvulsive shock extends the duration of enhanced gene expression for BDNF in rat brain compared with a single administration. Brain Res Mol Brain Res. 1998;57:106–110. doi: 10.1016/s0169-328x(98)00077-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.