

Abstract
BACKGROUND AND PURPOSE
Haemorrhagic shock and resuscitation (H/R) induces hepatic injury, strong inflammatory changes and death. Alcohol intoxication is assumed to worsen pathophysiological derangements after H/R. Here, we studied the effects of acute alcohol intoxication on survival, liver injury and inflammation after H/R, in rats.
EXPERIMENTAL APPROACH
Rats were given a single oral dose of ethanol (5 g·kg−1, 30%) or saline (control), 12 h before they were haemorrhaged for 60 min and resuscitated (H/R). Sham groups received the same procedures without H/R. Measurements were made 2, 24 and 72 h after resuscitation. Survival was assessed 72 h after H/R.
KEY RESULTS
Ethanol increased survival after H/R three-fold and also induced fatty changes in the liver. H/R-induced liver injury was amplified by ethanol at 2 h but inhibited 24 h after H/R. Elevated serum IL-6 levels as well as hepatic IL-6 and TNF-α gene expression 2 h after H/R were reduced by ethanol. Ethanol enhanced serum IL-1β at 2 h, but did not affect increased hepatic IL-1β expression at 72 h after H/R. Local inflammatory markers, hepatic infiltration with polymorphonuclear leukocytes and intercellular adhesion molecule 1 expression decreased after ethanol compared with saline, following H/R. Ethanol reduced H/R-induced IκBα activation 2 h after H/R, and NF-κB-dependent gene expression of MMP9.
CONCLUSIONS AND IMPLICATIONS
Ethanol reduced H/R-induced mortality at 72 h, accompanied by a suppression of proinflammatory changes after H/R in ethanol-treated animals. Binge-like ethanol exposure modulated the inflammatory response after H/R, an effect that was associated with NF-κB activity.
Keywords: ethanol, liver, inflammation, survival, in vivo
Introduction
Alcohol consumption is associated with one-third of all traumatic injury deaths each year (Li et al., 1997; Rehm et al., 2003). Almost 50% of trauma victims have positive blood alcohol concentrations (BACs), among them 35% with a BAC greater than 1 mg·mL−1 (Reyna et al., 1985; Rivara et al., 1993; Madan et al., 1999; Hadfield et al., 2001). Alcohol-intoxicated trauma victims have an increased risk for subsequent complications such as pneumonia, sepsis or multiple organ failure (MOF); some studies report a greater morbidity and mortality in these patients (Faunce et al., 1997; Bagby et al., 1998; Ruiz et al., 1999; Boe et al., 2001; 2003; Messingham et al., 2002; Zhang et al., 2002). Other studies report divergent results showing that acute alcohol intoxication does not affect the outcome and is even associated with decreased 24 h mortality after trauma (own unpublished data). The source of complications in trauma victims after alcohol consumption is multifactorial and depends primarily on the injury severity, genetic predisposition of the patient and the history of alcohol abuse and use (chronic vs. acute alcohol abuse respectively) (Madan et al., 1999; von Heymann et al., 2002; Spies et al., 2004; Lau et al., 2009). After H/R, the phagocytic and oxidative burst capacity of polymorphonuclear leukocytes (PMNL) are suppressed in alcohol-intoxicated rodents (Molina et al., 2004). Acute alcohol intoxication suppresses innate immunity and production of inflammatory cytokines, such as TNF-α, IL-1β and IL-6, in human and animals (Gluckman and MacGregor, 1978; Nelson et al., 1989; Verma et al., 1993; Szabo et al., 1999; Boe et al., 2001; 2003). Both chronic and acute alcohol abuses result in significant alterations in haemodynamic stability, metabolic regulation and immune function.
Haemorrhagic shock induces an early proinflammatory response, which may lead to multiple organ dysfunction syndrome (MODS) or MOF that may ultimately lead to a greatly increased mortality rate after trauma (Sauaia et al., 1995; Moore et al., 1996; Baue et al., 1998; Treggiari et al., 2004). Haemorrhage followed by fluid resuscitation (H/R) disturbs the microcirculation and activates immune cells [monocytes and PMNL)] to subsequent excessive responsiveness to inflammatory stimuli (Partrick et al., 1996; Hietbrink et al., 2006). These changes result in damage of cell membranes, DNA and proteins, and subsequent organ damage (Redl et al., 1993; Botha et al., 1995; Partrick et al., 1996; Akgur et al., 2000; Holzer et al., 2005; Passos et al., 2007). The increase of proinflammatory cytokines is strongly associated with an increased mortality in a model of H/R (Cai et al., 2009). Furthermore, elevated cytokine levels contribute to the up-regulation of adhesion molecules, apoptotic and/or necrotic changes of endothelial, parenchymal and immune cells and accompany compromised organ function after H/R, notably in the liver (Yamakawa et al., 2000; Cockerill et al., 2001; Liaw et al., 2005; Shimizu et al., 2007; Lehnert et al., 2008; Relja et al., 2009; 2010). NF-κB is involved in the regulation of inflammatory and immune processes, and is activated in the liver following H/R (Meng et al., 2001). Its activation requires removal and degradation of its endogenous inhibitor IκBα (Zingarelli, 2005). Once activated, NF-κB translocates from the cytosol to the nucleus and regulates gene transcription of several proinflammatory cytokine genes, such as IL-6, IL-1, TNF-α, and intercellular adhesion molecule 1 (ICAM-1) (Tak and Firestein, 2001; nomenclature follows Alexander et al., 2011).
Thus, we studied the influence of a binge-like exposure to alcohol (using a single high dose of ethanol) upon mortality, hepatic injury and inflammation after H/R in rats.
Methods
Animals and experimental model
All animal care and experimental protocols were approved by the Veterinary Department of the Regional Council in Darmstadt, Germany. As effects of alcohol exposure vary with gender, and several studies have reported a pronounced susceptibility for alcohol-induced inflammation and organ damage in females, in both humans and animals (Loft et al., 1987; Emanuele et al., 2001; Nanji et al., 2001), we used female rats (180–250 g, Harlan, Borchen, Germany). Also induction of an acute fatty liver after ethanol gavage is highly reliable in Lewis rats, this strain was chosen for our study (Zhong et al., 1999). Twelve hours before H/R, rats were gavaged with a single dose of ethanol (30% solution, equivalent to 5 g·kg−1; … .); referred to as H/R+EtOH (Sigma-Aldrich, Steinheim, Germany) or an equal volume of saline (H/R+Ctrl), before induction of the H/R procedure. Using a 20% solution of ethanol in preliminary studies, we found the risk of aspiration due to high gavage volume was substantial and we reduced the volume administered by increasing the concentration to 30%. Using this concentration, no aspiration occurred, and hepatic fat accumulation and hepatic damage both after sham and H/R procedures were comparable (data not shown; Kozan et al., 2009). The BAC at 12 h after ethanol gavage was measured using an Ethanol Assay Kit (BioVision, Heidelberg, Germany) according to the manufacturer's instructions. After an overnight fast, rats were anaesthetized with isoflurane (1.5%; Baxter, Unterschleißheim, Germany), and the right carotid artery, the right femoral artery and the left jugular vein were cannulated with polyethylene tubing. Then, haemorrhage was induced over 5 min by withdrawing blood from the right carotid artery into a heparinized syringe (10 U) to a mean blood pressure of 30 ± 2 mmHg. Systemic blood pressure was monitored in the right femoral artery using a blood pressure analyzer (BPA 400, Digi-Med, Louisville, KY, USA). Constant pressure was maintained by further withdrawal of small volumes of blood as necessary for 60 min. Then, rats were resuscitated by transfusion of 60% of the shed blood plus a volume of lactated Ringer's solution corresponding to 50% of the shed blood volume with a syringe pump over 30 min via the left jugular vein (Relja et al., 2009). After the end of resuscitation, catheters were removed, the vessels were occluded and the wounds were closed. Sham groups underwent surgical procedures without H/R (sham+Ctrl and sham+EtOH groups). At euthanasia, the vena cava was punctured, blood was collected and tissue samples were collected. For each rat, the two right dorsal liver lobes were snap-frozen in liquid nitrogen. The remaining liver was flushed with normal saline, perfused and fixed with 10% buffered formalin through the portal vein, embedded in paraffin and subsequently sectioned and stained with haematoxylin–eosin. Body temperature was measured in the colon and maintained at 37°C throughout the experiment with a heating pad. Thirty-two animals were observed for 72 h to assess survival.
Group allocation
Survival study
Thirty-two female Lewis rats (180–250 g) were randomly allocated to receive either saline gavage (Ctrl), or ethanol gavage before either sham or the H/R procedure (n= 7 and n= 9 per group, respectively). Survival was assessed at 72 h.
Organ injury and inflammatory changes
Twenty-eight animals were randomly allocated to four groups and treated in the same way as described above (n= 7 per each group) and tissues samples were collected at 2 h after resuscitation. Twelve animals, also randomly allocated to four groups (n= 3 per each group) were killed at 24 h after resuscitation. The animals from the survival group and additional animals in the H/R+Ctrl group were killed at 72 h after resuscitation (n= 7).
Examination of ethanol-induced hepatic fat accumulation
Hepatic 5 µm frozen sections were fixed with 10% buffered formalin. Lipids were stained with Oil red O, counterstained with haematoxylin, and photographs were taken.
Examination of tissue injury
Sera were stored at −80°C for later analysis of alanine aminotransferase (ALT) and LDH using the Vitros 250 device (Ortho-Clinical Diagnostics, Neckargemünd, Germany). Determination of histological damage was performed by an independent examiner who allocated the haematoxylin–eosin-stained liver sections to the various experimental groups, without knowledge of the treatments, as described by Meng et al., (2001).
Detection of PMNL
Analysis of the hepatic infiltration with PMNL was performed by chloroacetate esterase staining (4% pararosanilin, 4% sodium nitrite and naphthol solution) for 30 min at room temperature. Sections were counterstained with haematoxylin. PMNL were determined by counting the number of chloroacetate esterase-positive cells in a total of 25 high-power (400×) fields per liver section per rat, without knowledge of the treatments. Data from each tissue section were pooled to determine means.
Quantification of cytokine levels
Concentrations of serum IL-6, TNF-α and IL-1β level were determined using a Quantikine Rat IL-6, Rat TNF-α and Rat IL-1β/IL-1F2 elisa kit of R&D Systems (Wiesbaden-Nordenstadt, Germany) according to the manufacturer's instuctions. elisa was performed using Infinite M200 microplate reader (Tecan, Männedorf, Switzerland).
Western blotting for intracellular signalling
Liver tissue was homogenized in lysis buffer at 4°C, followed by centrifugation for 30 min at 4°C at 20 000×g. Supernatants were stored at −80°C for later analysis. Lysates (50 µg protein) were separated by electrophoresis on 12% polyacrylamide SDS gels and transferred to nitrocellulose membranes (Amersham-Buchler, Braunschweig, Germany). Phosphorylated IκBα (phospho-IκBα) was detected using rabbit polyclonal phospho-IκBα (phospho-Ser32+ Ser36) antibody, and IκBα using rabbit polyclonal IκBα antibody (Abcam, Cambridge, UK). Determination of β-actin with anti-β-actin antibody (Sigma, Taufkirchen, Germany) served as a loading control. Blots were blocked (10% non-fat dry milk in 1 mM Tris, 150 mM NaCl, pH 7.4) for 1 h, incubated for 1 h at room temperature with primary antibody (diluted according to manufacturer's instructions in blocking buffer with 0.5% Tween 20 and 0.5% BSA) and then incubated for 1 h with horseradish peroxidase-conjugated secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) diluted 1:1000 in blocking buffer with 0.5% Tween 20 and 0.5% BSA at room temperature. Proteins were detected with ECL™ Western blot detection reagents (GE Healthcare, Munich, Germany). Films were digitized, and the integrated density of individual bands was determined using the software Multianalyst (Biorad, Munich, Germany). By densitometric measurements using the same software, the amount of protein expression was normalized to β-actin. The activation state of IκBα was calculated as the ratio of phosphorylated and total (phosphorylated plus non-phosphorylated) protein values of densitometry results in per cent (phospho/total*100).
Staining of ICAM-1
Cryosectioned liver samples (5 µm) were air dried for 10 min, fixed in acetone 10 min at room temperature and air dried for 60 min at room temperature. Sections were washed with PBS and water. Then, endogenous peroxidase activity was blocked with hydrogen peroxide (2% for 10 min, room temperature). Hepatic sections were washed and incubated with blocking solution (2% BSA in PBS) for 1 h at room temperature. Mouse anti-rat CD54 monoclonal antibody (BD Pharmingen, Heidelberg, Germany) diluted 1:150 in PBS (pH 7.4) containing 1% BSA was used as primary antibody (overnight incubation, 4°C). Anti-mouse horseradish peroxidase-linked secondary antibody (30 min, room temperature) and diaminobenzidine (Peroxidase EnVision Kit, DakoCytomation, Hamburg, Germany) were used to detect specific binding. Sections were counterstained with haematoxylin. The immunostained tissue sections were captured at 400× and analysed without knowledge of the treatments. The extent of labelling in the liver lobule was defined as the percentage of the field area within a preset colour range determined by the software (Adobe Photoshop 7.0, Adobe Systems Inc., San Jose, CA, USA). Data from each tissue section (10 fields per section) were pooled to determine mean values.
RNA isolation, quantitative reverse transcription (RT)-PCR
Total RNA of the snap-frozen liver lobes was isolated using the RNeasy-system (Qiagen, Hilden, Germany) according to the manufacturer's instructions. The residual amounts of DNA remaining were removed using the RNase-Free DNase Set according to the manufacturer's instructions (Qiagen). The RNA was stored immediately at −80°C. Quality and amount of the RNA were determined photometrically using the NanoDrop ND-1000 device (NanoDrop Technologies, Wilmington, DE, USA).
RNA was subsequently used for quantitative RT-PCR. In brief, 100 ng of total hepatic RNA was reversely transcribed using the Affinity script QPCR-cDNA synthesis kit (Stratagene, La Jolla, CA, USA) following the manufacturer's instructions. To determine the mRNA expression of IL-6, TNF-α, IL-1β and MMP9, qRT-PCR was carried out on a Stratagene MX3005p QPCR system (Stratagene) using gene-specific primers for rat Il6 (NM_012589, UniGene#: Rn.9873, Cat#: PPR06483A), rat Il1b (NM_031512, UniGene#: Rn.9869, Cat#: PPR06480A), rat Tnf (NM_012675, UniGene#: Rn.2275, Cat#: PPR06411E) and rat Mmp9 (NM_031055, UniGene#: Rn.10209, Cat#: PPR44728B) purchased from SABiosciences (SuperArray, Frederick, MD, USA). As reference gene, the expression of GAPDH with rat Gapdh (NM_017008, UniGene#: Rn.91450, Cat#: PPR06557A, SABiosciences, SuperArray) was measured. Sequences of these primers are not available. PCR reaction was set up with 1× RT2 SYBR Green/Rox qPCR Master mix (SABiosciences) in a 25 µL volume according to the manufacturer's instructions. A two-step amplification protocol consisting of initial denaturation at 95°C for 10 min followed by 40 cycles with 15 s denaturation at 95°C and 60 s annealing/extension at 60°C was chosen. A melting-curve analysis was applied to control the specificity of amplification products.
Relative expression of each target genes, mRNA level was then calculated using the comparative threshold-cycle (CT) method (2_ΔΔCT method). In brief, the amount of target mRNA in each sample was first normalized to the amount of GAPDH mRNA, to give ΔCT and then to a calibrator consisting of samples obtained from the sham+Ctrl group. The relative mRNA expression of target genes is presented as fold increase calculated in relation to sham+Ctrl after normalization to GAPDH.
Statistical analysis
Differences between groups were determined by one-way anova using a multiple comparison procedure (Student–Newman–Keuls post hoc). Changes in target gene expression were analysed by Wilcoxon matched-pair analysis followed by Bonferroni correction. A P-value of less than 0.05 was considered significant. Data are given as mean ± SEM. Kaplan–Meier test followed by log-rank test was used for analysis of survival data. All statistical analyses were performed employing GraphPad Prism 5 (Graphpad Software, Inc., San Diego, CA).
Results
Lipid deposition in the liver after ethanol administration
Lipid deposition representing ethanol-induced fatty liver was demonstrated by staining of lipid accumulation with Oil red O, 12 h after ethanol or saline gavage. Two representative histological pictures demonstrate ethanol-induced accumulation of lipid droplets in both periportal and pericentral zones of liver lobules (Figure 1B). In livers of saline-gavaged rats, no lipid deposition was detected (Figure 1A). However, the BAC at 12 h after ethanol gavage was almost not detectable (10–15 mM, data not shown).
Figure 1.
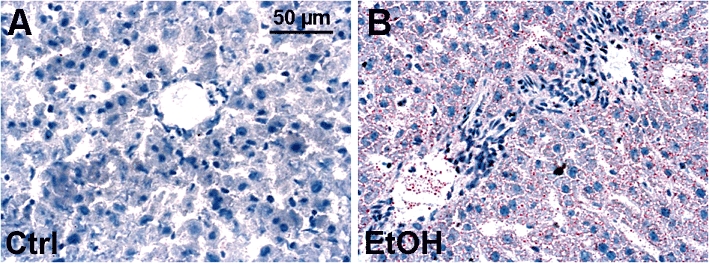
Oil red O staining of lipid droplets after acute alcohol exposure. Lipid deposition after ethanol gavage was analyzed by Oil red O staining. Left figure (A) depicts livers of saline-gavaged rats (ctrl), right figure (B) depicts livers of ethanol-gavaged rats (EtOH). Tissues were collected 12 h after saline or ethanol gavage. Representative Oil red O stained liver sections are shown; bar is 50 µm.
Survival rates of ethanol-intoxicated rats after H/R
In order to reveal the effect of acute ethanol intoxication on survival after H/R, rat survival at 72 h after H/R was evaluated. In both sham-operated groups, no deaths occurred. Haemorrhagic shock followed by resuscitation induced a mortality of 80% at 72 h after H/R in our model. Acute ethanol gavage before H/R strikingly attenuated the mortality to 19% after H/R (P < 0.001, Figure 2).
Figure 2.
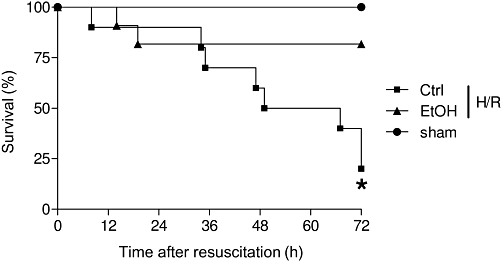
Acute ethanol (EtOH) ingestion reduces mortality after H/R. Mortality is shown as survival (% group size) after 72 h. Group sizes were: sham+Ctrl and and sham+EtOH, each n= 7; H/R+EtOH, n= 9; H/R+Ctrl, n= 9. *P < 0.001 vs. other groups; Kaplan–Meier test followed by log-rank test.
Histopathological changes in hepatic tissue after H/R
Liver sections from saline-pretreated (Ctrl) rats (Figure 3, left row) revealed areas of coagulative necrosis at 2 h after H/R (Figure 3B). H/R-induced necrosis was increased 24 h after resuscitation as indicated by cellular enlargement and nuclear dissolution in predominantly midzonal and pericentral areas of liver lobules from ctrl rats (Figure 3C). Necrotic areas were reduced 72 h after H/R, in H/R+Ctrl rats (Figure 3D). In ethanol-intoxicated rats (Figure 3, right row), hepatocytes with cell enlargement and nuclear dissolution were most pronounced at 2 h after resuscitation, with a marked reduction of necrosis at 24 h and 72 h after H/R respectively (Figure 3F–H). These changes were not detected after sham operation (Figure 3A and E).
Figure 3.
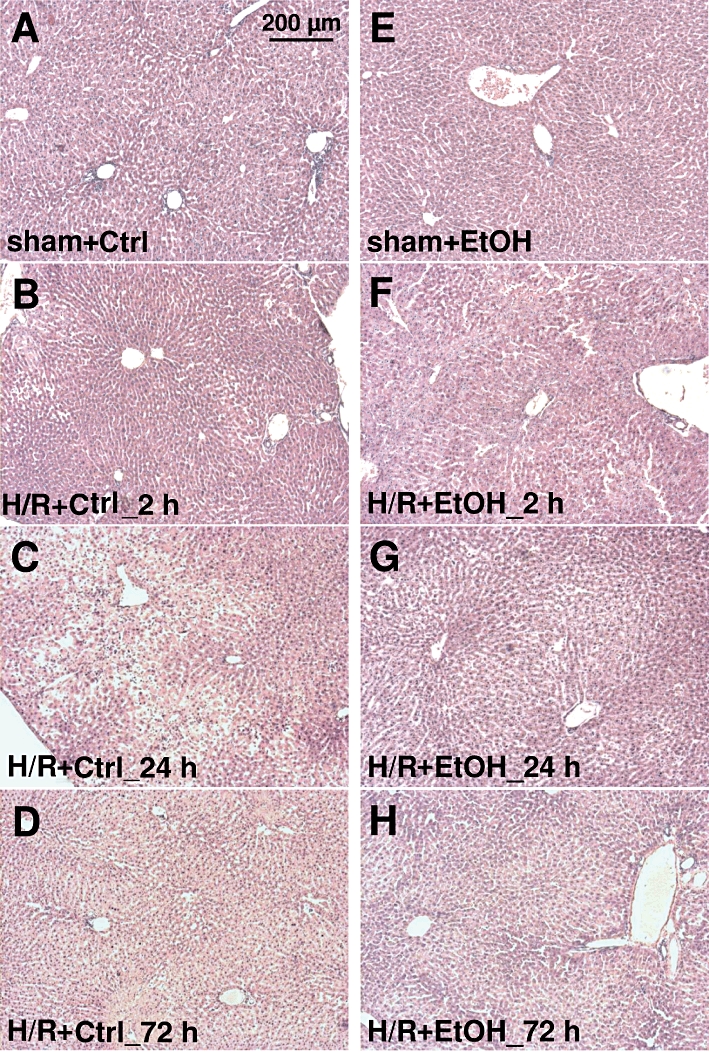
Histological liver necrosis after acute ethanol ingestion after at 2 h, 24 h and 72 h after H/R. Rats were gavaged with saline (Ctrl) or ethanol (5 g·kg−1, 30% EtOH), 12 h before H/R. Sham-operated animals underwent the same surgical procedures but H/R was not carried out. Representative haematoxylin- and eosin- stained liver lobes from saline-treated (Ctrl) (A: after sham operation, B, C and D, 2, 24 and 72 h after H/R, respectively) or ethanol-treated (E: after sham operation, F, G and H, 2, 24 and 72 h after H/R) rats are shown. Bar is 200 µm.
Cell damage after H/R
H/R induced an increase of serum ALT, a marker of hepatocellular damage at 2 h after H/R, (compare H/R+Ctrl group value with the corresponding sham value (131 ± 16 IU·L−1; P < 0.01, Figure 4A). The sham values did not vary between 2, 24 and 72h and are shown as a single point in Figure 4A. Twenty-four hours after H/R, ALT increased further (P < 0.05, Figure 4A) and then fell by 72 h after resuscitation. Pre-treatment with ethanol enhanced ALT release at 2 h after H/R (compare H/R+EtOH values with H/R+Ctrl) (P < 0.05, Figure 4A). In contrast to the saline-gavaged rats, ALT decreased in rats that received an ethanol gavage before H/R at 24 and 72 h.
Figure 4.
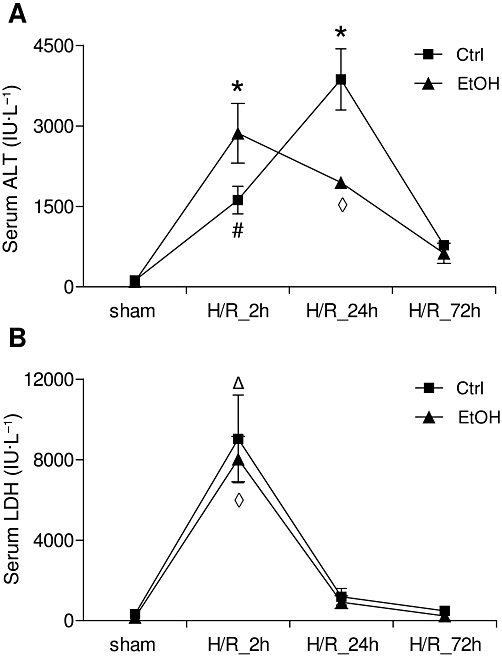
Serum ALT and LDH levels after acute ethanol ingestion and H/R in rats. 12 h before H/R, rats were gavaged with saline (Ctrl) or ethanol (EtOH).Blood samples were collected at 2, 24 and 72 h after H/R for measurement of ALT (A) and LDH (B) enzyme activity. *P < 0.05 vs. all, #P < 0.05 vs. all except H/R+EtOH 24 h group, ◊P < 0.001 vs. all except H/R+Ctrl 2 h, ΔP < 0.001 vs. all except H/R+EtOH 2 h, n= 3 for 24 h groups, and n= 7 for other groups.
LDH, an indicator of general cell damage, was clearly raised at 2 h in the H/R+Ctrl group compared with the sham+Ctrl rats (320 ± 120 IU·L−1, P < 0.001, Figure 4B); the sham+Ctrl values did not vary between 2, 24 and 72 h and are shown as a single point in Figure 4B. The raised LDH in the H/R+Ctrl groups decreased at 24 h and 72 h after resuscitation to levels comparable with the corresponding sham+Ctrl rats. In ethanol-intoxicated rats, LDH release increased at 2 h after resuscitation, compared with sham+EtOH (163 ± 16 IU·L−1, P < 0.001, Figure 4B; one point is shown). LDH decreased in ethanol-intoxicated rats 24 and 72 h after resuscitation. Ethanol gavage itself did not increase ALT or LDH values after sham operation compared with saline-gavaged rats (Figure 4A and B).
Systemic proinflammatory changes after H/R – serum IL-6, IL-1β and TNF-α levels
Haemorrhage followed by resuscitation induces a systemic immune response, which was assessed at 2, 24 or 72 h after H/R by serum IL-6, IL-1β and TNF-α levels. Compared with the sham+Ctrl group, serum IL-6 levels increased in the H/R+Ctrl group, 2 h after resuscitation (P < 0.05, Figure 5A). IL-6 levels in ethanol-gavaged rats, at 2 h after resuscitation, were markedly reduced (P < 0.05, Figure 5A), comparing H/R+EtOH with H/R+Ctrl groups. After sham operation, serum IL-6 levels did not differ between sham+Ctrl and sham+EtOH groups (Figure 5A). 24 h after resuscitation, IL-6 decreased further in both H/R groups without significant differences, followed by a decrease to basal and partly undetectable levels at 72 h in all groups (Figure 5A).
Figure 5.
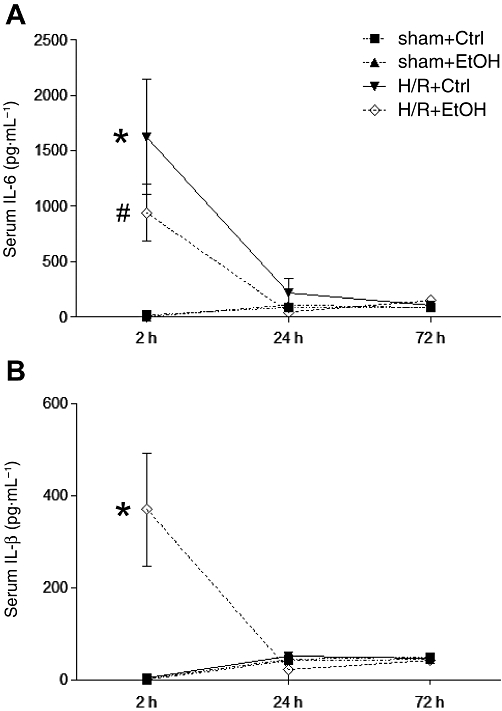
Acute ethanol ingestion affects serum IL-6 and IL-1β after H/R. 12 h before H/R, rats were gavaged with saline (Ctrl) or ethanol (EtOH). Blood samples were collected at 2, 24 and 72 after resuscitation. Serum concentrations of IL-6 (A) and IL-1β (B) were determined by elisa. Sham-operated animals underwent the same surgical procedures as the H/R groups but H/R was not carried out (*P < 0.05 vs. all, #P < 0.05 vs. H/R+Ctrl group 2 h group, n= 7 for 2 h group, n= 3 for 24 h group, n= 3 for 72 h group).
Serum IL-1β levels (Figure 5B) were not increased after H/R in the H/R+Ctrl group at 2 h, compared with both sham groups (4 ± 2 and 0 pg mL−1). IL-1β levels in ethanol-treated rats increased markedly after H/R, compared with all other groups (P < 0.05, Figure 5B). At 24 and 72 h, the initial systemic increase in IL-1β was lost and levels were comparable to the results obtained for sham groups at these time points (Figure 5B).
Serum TNF-α concentrations were not detectable at any time (2, 24 and 72 h) in any group.
Hepatic gene expression of local proinflammatory markers after H/R – IL-6, IL-1β and TNF-α
The real-time PCR showed an increase of IL-6 and TNF-α gene expression at 2 h after resuscitation in liver samples obtained from saline treated (H/R+Ctrl) animals, compared with all other groups (P < 0.05, Figure 6A and C). The expression of IL-1β was initially increased at 72 h after H/R in both saline and ethanol pre-treated groups (P < 0.05, Figure 6B).
Figure 6.
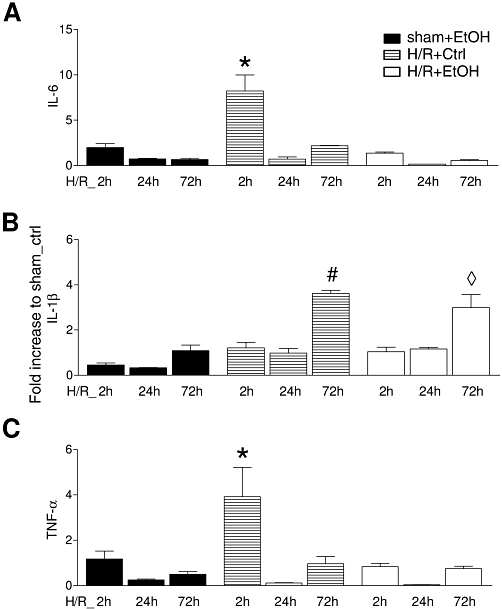
Acute ethanol ingestion modifies gene expression of cytokines (IL-6, TNF-α and IL-1β) in liver at 2 h after H/R. 12 h before H/R, rats were gavaged with saline (Ctrl) or ethanol (EtOH). Hepatic gene expression of IL-6 (A), IL-1β (B) and TNF-α (C) was analysed, at 2, 24 and 72 h after H/R, Sham-operated animals underwent the same surgical procedures but H/R was not carried out. After normalization to GAPDH expression, gene expression was measured as fold increase compared with the sham+Ctrl group (*P < 0.05 vs. all, #P < 0.01 vs. all except H/R+EtOH 72 h group, ◊P < 0.05 vs. all except H/R+Ctrl 72 h, n= 3 for 24 h groups, and n= 7 for other groups).
Local proinflammatory changes after haemorrhage and resuscitation – ICAM-1 expression and hepatic neutrophil accumulation
The expression of ICAM-1, as revealed by immunohistology, increased markedly 2 h after resuscitation as compared with sham-operated ctrl rats in the saline-gavaged group (Figure 7A and B). A typical sinusoidal lining pattern was present. ICAM-1 was strongly expressed in livers of ctrl animals also at 24 h after H/R with a decline of ICAM-1 expression at 72 h after H/R (Figure 7C and D). Hepatic ICAM-1 expression was markedly reduced in the group of ethanol-gavaged animals at 2 h after resuscitation, as compared with H/R+Ctrl_2 h group, and declined further at 24 h after H/R (Figure 7B, F and G). At 72 h after H/R, in the ethanol group, hepatic ICAM-1 expression was comparable with both sham groups (Figure 7H, A and B).
Figure 7.
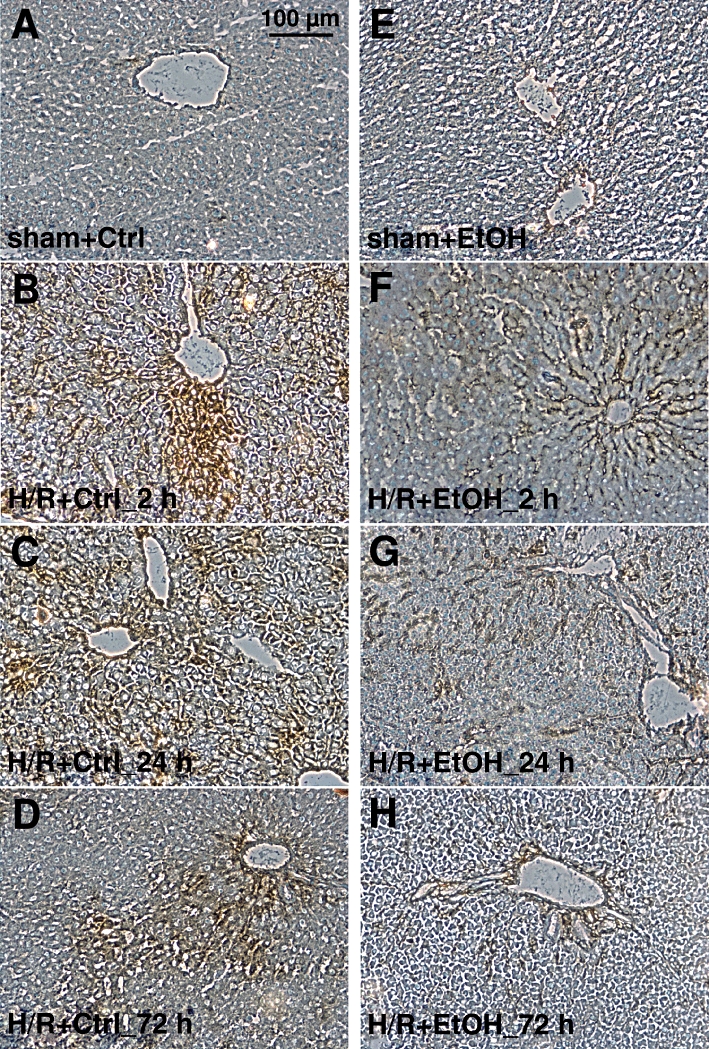
Acute ethanol ingestion decreases ICAM-1 expression after H/R. Hepatic ICAM-1 expression was assessed by immunhistological staining with anti-CD54 antibody (brown). 12 h before H/R, rats were gavaged with saline (Ctrl) or ethanol (EtOH). Sham-operated animals underwent the same surgical procedures without blood withdrawal. Representative liver sections from Ctrl groups (A: after sham operation, B, C and D, 2, 24 and 72 h after H/R, respectively) and EtOH groups (E: after sham operation, F, G and H, 2, 24 and 72 h after H/R) rats are shown. Bar is 100 µm.
Hepatic neutrophil infiltration increased at 2 and 24 h after resuscitation, respectively, compared with values after sham operation (1 ± 1 cells per high-power field, P < 0.05, Figure 8). The sham values did not vary between 2, 24 and 72 h and are shown as a single point. By 72 h after H/R, PMNL infiltration was markedly reduced (Figure 8). Ethanol markedly diminished hepatic neutrophil infiltration after H/R (Figure 8) compared with the corresponding saline-gavaged group (H/R+Ctrl) to levels not different from those in the sham+EtOH animals (2 ± 1 cells per high-power field, Figure 8; sham values did not vary between 2, 24 and 72 h and are shown as a single point). Ethanol gavage did not increase ICAM-1 expression or PMNL infiltration after sham operation compared with saline-gavaged rats.
Figure 8.
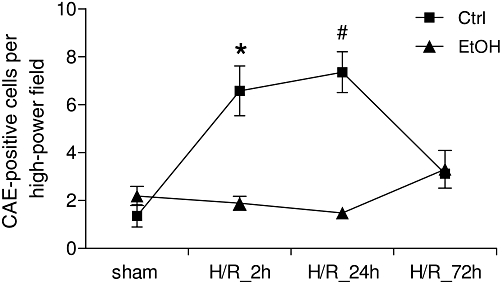
Acute ethanol (EtOH) ingestion reduces hepatic neutrophil infiltration after H/R. Saline (Ctrl) or EtOH-gavaged rats were subjected to H/R or sham operation. Liver samples were collected at 2, 24 and 72 h after H/R and neutrophils in liver sections were identified by chloroacetate esterase (CAE) cytochemistry and counted in 25 high-power fields as described previously (Lehnert et al., 2008). *P < 0.05 vs. all except H/R+Ctrl 24 h, #P < 0.05 vs. all except H/R+Ctrl 2 h group, n= 3 for 24 h groups, and n= 7 for other groups.
Analysis of the NF-κB inhibitor IκBα and NF-κB-dependent expression of MMP9 after H/R
To analyze how ethanol attenuates inflammation after H/R, phosphorylated and non-phosphorylated IκBα was determined by Western blots in liver homogenates collected at 2 h after resuscitation. Densitometric analysis of relative protein expression related to β-actin and subsequent ratio of phosphorylated to total protein indicated an increase in IκBα phosphorylation after H/R, compared with the ratio in sham-operated rats (P < 0.05, Figure 9A and B). Pre-treatment with ethanol prevented the increase in the amount of phosphorylated IκBα protein after H/R (P < 0.05, Figure 9A and B).
Figure 9.
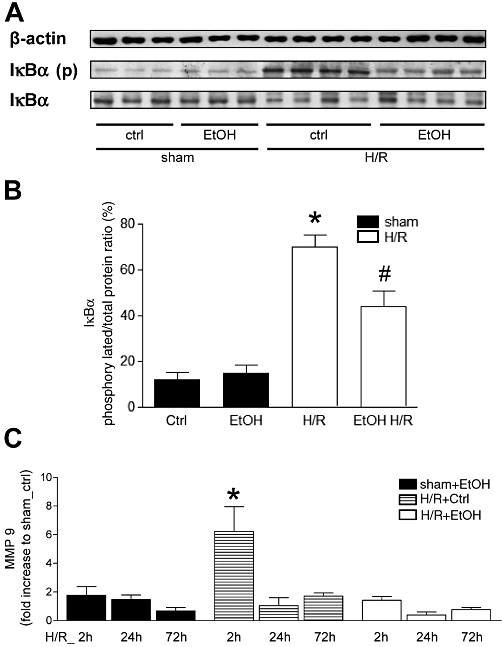
Acute ethanol (EtOH) ingestion reduces hepatic IκBα phosphorylation and MMP9 gene expression at 2 h after H/R. Saline- (Ctrl) or EtOH-gavaged rats were subjected to H/R or sham operation. Two hours after the end of resuscitation, liver tissue was collected and Western blot for IκBα, IκBα-phospho and β-actin was performed. A: 1–6 were liver protein extracts from rats after sham operation (sham, lane 1–3: Ctrl, lane 4–6: EtOH gavage) or H/R (lanes 7–10: Ctrl, lane 11–14: EtOH gavage). In B, ratio of phosphorylated IκBα and total protein after densitometric measurements and normalization to β-actin are represented (*P < 0.05 vs. all, #P < 0.05 vs. H/R+Ctrl, n= 6, representative gel from 3 experiments is shown). At 2, 24 and 72 h after resuscitation, hepatic gene expression of MMP (C) was analysed. After normalization to GAPDH expression, gene expression was measured as fold increase compared with sham+Ctrl group (*P < 0.05 vs. all, n= 7).
The real-time PCR showed an increase of NF-κB-dependent MMP9 expression at 2 h after resuscitation in liver samples obtained from saline-treated animals (H/R+Ctrl), compared with all other groups (P < 0.05, Figure 9C). The expression of MMP9 was not increased at any time in the H/R+EtOH (Figure 9C).
These results indicate that H/R induced NF-κB activation and expression of NF-κB-dependent MMP9. These effects were strongly influenced by ethanol that inhibited NF-κB activation and NF-κB-mediated expression of MMP9 after H/R.
Discussion
In the present study, we evaluated the effect of a binge-like ethanol treatment, 12 h before the onset of H/R. Previous studies using doses of 5 g·kg−1 of ethanol clearly demonstrated ethanol-dependent changes in the liver, that is, decreased graft survival and disturbance of hepatic microcirculation as well as increased triglyceride content (Ylikahri et al., 1972; Zhong et al., 1996; Enomoto et al., 2000) This is consistent with our study where a fatty liver was induced by this dose of ethanol (Figure 1). Taken together, these data provide a valid basis for using ethanol at a dose of 5 g·kg−1 to study the effects of an acute fatty liver in inflammatory conditions, that is, with the H/R procedure. Interestingly, the BAC showed an early peak 1 and 2 h after alcohol gavage with a rapid decline to almost normal levels by 10–12 h after the gavage, results that correspond to BAC measurements in the present study (Wendell and Thurman, 1979; Rivera et al., 1998). Our results showed that alcohol intoxication reduced mortality after H/R from 78% in the saline-treated group to 22% (Figure 2). Hepatocellular damage at 2 h after H/R, was aggravated by ethanol, with subsequent hepatocellular recovery (Figures 3 and 4). In addition, the ethanol-intoxicated group showed enhanced systemic IL-1β levels, while IL-6 levels were reduced 2 h after H/R (Figure 5). Ethanol also reduced hepatic gene expression of local proinflammatory markers IL-6 and TNF-α as well as the hepatic expression of ICAM-1 and hepatic PMNL infiltration as compared with the saline-treated group early after H/R (Figures 6–8). These beneficial effects are associated with an ethanol-dependent inhibition of the activation of NF-κB (Figure 9).
Trauma victims are frequently intoxicated with alcohol (Madan et al., 1999; Hadfield et al., 2001). Several studies report an association of alcohol intoxication with an increased risk of secondary complications due to an impaired host immune response (Faunce et al., 1997; Messingham et al., 2002; Zhang et al., 2002; Boe et al., 2003; Zambell et al., 2004; Greiffenstein et al., 2007). However, consequences of acute alcohol intoxication on the inflammatory changes after H/R have not been systematically assessed. Both acute and chronic alcohol abuse and H/R have been shown to induce hepatocellular dysfunction. With regard to the liver, an additive effect of acute alcohol intoxication and H/R on liver function, leading to increased risk for hepatic failure after injury, has been suggested (Phelan et al., 2002). In parallel to these findings, we also showed enhanced elevation of the liver enzyme ALT, as well as increased necrotic areas, an effect that was confined to the early post-resuscitation period, at 2 h after completion of resuscitation, in ethanol-gavaged animals (Figure 4), suggesting that acute ethanol intoxication contributes to early liver injury. Of note, the early resolution of markers of liver injury was associated with the lower inflammatory response after binge-like ethanol treatment before H/R. This contrasted with the late rise in hepatocellular damage at 12 and 24 h after H/R in saline-gavaged animals, effects associated with a stronger inflammatory response. However, although H/R increased serum LDH as a marker of general cellular damage, this was not affected by ethanol consumption combined with H/R. Taken together, these data indicate that acute ethanol ingestion was a significant contributor to early hepatic damage after H/R.
H/R-induced hepatic injury was accompanied by elevated circulating and hepatic cytokine expression. During haemorrhage, NF-κB is activated to promote tissue injury and augment the inflammatory cascade, modulating IL-6 and TNF-α production (Rushing and Britt, 2008). With regard to the liver, NF-κB is activated after haemorrhagic shock followed by resuscitation, and this activation is associated with increased IL-6 gene expression (Hierholzer et al., 1998; Gaddipati et al., 2003). Consistent with these observations, we detected an activation of the NF-κB inhibitor IκBα at 2 h after resuscitation in the liver. The increase in hepatic MMP9 expression confirmed the NF-κB activation with subsequent production of NF-κB-dependent proteins (Figure 9). Furthermore, early after H/R, both circulatory IL-6 and hepatic IL-6, as well as TNF-α mRNA levels were increased, suggesting an augmented inflammatory response after H/R in our model (Figures 5 and 6). IL-6 and TNF-α are early response cytokines not only after haemorrhagic shock (Bahrami et al., 1997; Lehnert et al., 2008; Lee et al., 2009; Relja et al., 2010). Down-regulation of IL-6 by anti-IL-6 antibodies and in IL-6 knockout mice resulted in diminished liver damage and improved bile production after trauma/haemorrhage in rats undergoing H/R (Meng et al., 2001; Toth et al., 2004). Treatment with anti-TNF-α antibodies reduced organ injury and improved survival in rats after haemorrhagic shock followed by resuscitation (Bahrami et al., 1997). Ethanol inhibited cytokine responses under acute inflammatory conditions such as induction of LPS shock or after H/R (Nelson et al., 1989; Phelan et al., 2002). Consistent with these findings, we found that ethanol markedly reduced serum IL-6 levels as well as hepatic IL-6 and TNF-α mRNA expression. Furthermore, the ethanol-dependent anti-inflammatory effects after H/R were associated with decreased hepatic IκBα activation and MMP-9 expression, both indicating an inhibition of NF-κB activation in our model (Figures 7–9). Interestingly, early serum IL-1β levels were enhanced in ethanol-intoxicated rats after H/R (Figure 5). Given the transient increase in IL-1β, this may be due to an altered post-translational processing of this cytokine into its active form by the caspase-1 inflammasome. However, we studied the activity of circulating immune cells by evaluating LPS-stimulated IL-1β production in whole blood obtained 12 h after oral gavage with EtOH or saline (stimulation with 10 ng/ml LPS for 24 h). Whole blood from saline-treated animals released significantly more IL-1β compared with whole blood from ethanol-gavaged rats. Moreover, ethanol markedly reduced IL-1β production in whole blood stimulated with LPS (data not shown). Nevertheless, hepatic IL-1β mRNA expression increased 72 h after H/R and was not diminished by ethanol exposure, probably because this time was beyond the duration of action of ethanol in this model (Figure 6). These data demonstrate that ethanol affects various cytokines differentially, resulting in an early anti-inflammatory effect of acute ethanol intoxication.
Several studies attribute the anti-inflammatory effects of alcohol to a decreased chemokine production and neutrophil recruitment (Szabo and Mandrekar, 2009). ICAM-1 expression is necessary for transendothelial migration and adherence of neutrophils to parenchymal cells. Neutrophil infiltration in the liver plays an important role in hepatic injury after H/R, hepatic ischaemia/reperfusion and LPS shock (Xu et al., 1994). Furthermore, ICAM-1 production depends on NF-κB activation (Zingarelli, 2005). In our study, release of proinflammatory cytokines and the upregulation of ICAM-1 after H/R correlated with increased hepatic adhesion and infiltration with neutrophils and enhanced hepatic damage (Figures 3–8). Acute ethanol ingestion was associated with reduced activation of NF-κB and down-regulation of ICAM-1 expression with subsequently decreased neutrophil infiltration in the liver (Figures 7–9).
Besides acute alcohol intoxication itself, an acute withdrawal syndrome occurring 8–16 h after alcohol intoxication (‘hangover’) is associated with a reduced proinflammatory response in animals and humans (Szabo et al., 1994; Gottesfeld et al., 2002; Kim et al., 2003; Greiffenstein and Molina, 2008; Zhang et al., 2009). In accordance with these results, we also found 12 h after ethanol gavage, a decreased IL-1β production in LPS-stimulated whole blood (data not shown). Therefore, a ‘hangover’ may influence the pathophysiology after H/R via anti-inflammatory effects and counteract the proinflammatory changes induced by H/R. Further studies should evaluate potential differences in the activation status of the immune system prior to an experimental insult in a ‘hangover’ state.
Taken together, we have demonstrated a reduction in mortality and early systemic and local inflammation in acutely ethanol-intoxicated rats, undergoing resuscitated blood loss, accompanied by a faster resolution of liver damage. Furthermore, the anti-inflammatory effects of ethanol were also manifested in reduced ICAM-1 expression and neutrophil infiltration and were associated with the stabilization of the NF-κB inhibitor IκBα.
Acknowledgments
We thank Kerstin Wilhelm and Minhong Wang for outstanding technical assistance. Grant support: The study was supported by DFG MA 1119/3-3.
Glossary
- ALT
alanine aminotransferase
- BAC
blood alcohol concentrations
- H/R:
haemorrhagic shock and resuscitation
- ICAM-1
intercellular adhesion molecule 1
- MODS
multiple organ dysfunction syndrome
- MOF
multiple organ failure
- PMNL
polymorphonuclear leukocytes
Conflicts of interest
The authors declare no conflicts of interest.
References
- Akgur FM, Brown MF, Zibari GB, McDonald JC, Epstein CJ, Ross CR, et al. Role of superoxide in hemorrhagic shock-induced P-selectin expression. Am J Physiol Heart Circ Physiol. 2000;279:H791–H797. doi: 10.1152/ajpheart.2000.279.2.H791. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels, 5th edition. Br J Pharmacol. 2011;164:S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagby GJ, Zhang P, Stoltz DA, Nelson S. Suppression of the granulocyte colony-stimulating factor response to Escherichia coli challenge by alcohol intoxication. Alcohol Clin Exp Res. 1998;22:1740–1745. [PubMed] [Google Scholar]
- Bahrami S, Yao YM, Leichtfried G, Redl H, Marzi I, Schlag G. Significance of TNF in hemorrhage-related hemodynamic alterations, organ injury, and mortality in rats. Am J Physiol. 1997;272:H2219–H2226. doi: 10.1152/ajpheart.1997.272.5.H2219. [DOI] [PubMed] [Google Scholar]
- Baue AE, Durham R, Faist E. Systemic inflammatory response syndrome (SIRS), multiple organ dysfunction syndrome (MODS), multiple organ failure (MOF): are we winning the battle? Shock. 1998;10:79–89. doi: 10.1097/00024382-199808000-00001. [DOI] [PubMed] [Google Scholar]
- Boe DM, Nelson S, Zhang P, Bagby GJ. Acute ethanol intoxication suppresses lung chemokine production following infection with Streptococcus pneumoniae. J Infect Dis. 2001;184:1134–1142. doi: 10.1086/323661. [DOI] [PubMed] [Google Scholar]
- Boe DM, Nelson S, Zhang P, Quinton L, Bagby GJ. Alcohol-induced suppression of lung chemokine production and the host defense response to Streptococcus pneumoniae. Alcohol Clin Exp Res. 2003;27:1838–1845. doi: 10.1097/01.ALC.0000095634.82310.53. [DOI] [PubMed] [Google Scholar]
- Botha AJ, Moore FA, Moore EE, Kim FJ, Banerjee A, Peterson VM. Postinjury neutrophil priming and activation: an early vulnerable window. Surgery. 1995;118:358–364. doi: 10.1016/s0039-6060(05)80345-9. [DOI] [PubMed] [Google Scholar]
- Cai B, Brunner M, Wang H, Wang P, Deitch EA, Ulloa L. Ethyl pyruvate improves survival in awake hemorrhage. J Mol Med. 2009;87:423–433. doi: 10.1007/s00109-009-0441-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockerill GW, McDonald MC, Mota-Filipe H, Cuzzocrea S, Miller NE, Thiemermann C. High density lipoproteins reduce organ injury and organ dysfunction in a rat model of hemorrhagic shock. FASEB J. 2001;15:1941–1952. doi: 10.1096/fj.01-0075com. [DOI] [PubMed] [Google Scholar]
- Emanuele NV, Lapaglia N, Emanuele MA. Impact of acute and chronic ethanol exposure on prolactin in both male and female rats. Endocrine. 2001;16:29–37. doi: 10.1385/ENDO:16:1:29. [DOI] [PubMed] [Google Scholar]
- Enomoto N, Ikejima K, Yamashina S, Enomoto A, Nishiura T, Nishimura T, et al. Kupffer cell-derived prostaglandin E(2) is involved in alcohol-induced fat accumulation in rat liver. Am J Physiol Gastrointest Liver Physiol. 2000;279:G100–G106. doi: 10.1152/ajpgi.2000.279.1.G100. [DOI] [PubMed] [Google Scholar]
- Faunce DE, Gregory MS, Kovacs EJ. Effects of acute ethanol exposure on cellular immune responses in a murine model of thermal injury. J Leukoc Biol. 1997;62:733–740. doi: 10.1002/jlb.62.6.733. [DOI] [PubMed] [Google Scholar]
- Gaddipati JP, Sundar SV, Calemine J, Seth P, Sidhu GS, Maheshwari RK. Differential regulation of cytokines and transcription factors in liver by curcumin following hemorrhage/resuscitation. Shock. 2003;19:150–156. doi: 10.1097/00024382-200302000-00011. [DOI] [PubMed] [Google Scholar]
- Gluckman SJ, MacGregor RR. Effect of acute alcohol intoxication on granulocyte mobilization and kinetics. Blood. 1978;52:551–559. [PubMed] [Google Scholar]
- Gottesfeld Z, Moore AN, Dash PK. Acute ethanol intake attenuates inflammatory cytokines after brain injury in rats: a possible role for corticosterone. J Neurotrauma. 2002;19:317–326. doi: 10.1089/089771502753594882. [DOI] [PubMed] [Google Scholar]
- Greiffenstein P, Molina PE. Alcohol-induced alterations on host defense after traumatic injury. J Trauma. 2008;64:230–240. doi: 10.1097/TA.0b013e318158a4ad. [DOI] [PubMed] [Google Scholar]
- Greiffenstein P, Mathis KW, Stouwe CV, Molina PE. Alcohol binge before trauma/hemorrhage impairs integrity of host defense mechanisms during recovery. Alcohol Clin Exp Res. 2007;31:704–715. doi: 10.1111/j.1530-0277.2007.00355.x. [DOI] [PubMed] [Google Scholar]
- Hadfield RJ, Mercer M, Parr MJ. Alcohol and drug abuse in trauma. Resuscitation. 2001;48:25–36. doi: 10.1016/s0300-9572(00)00315-4. [DOI] [PubMed] [Google Scholar]
- von Heymann C, Langenkamp J, Dubisz N, Dossow V, Schaffartzik W, Kern H, et al. Posttraumatic immune modulation in chronic alcoholics is associated with multiple organ dysfunction syndrome. J Trauma. 2002;52:95–103. doi: 10.1097/00005373-200201000-00017. [DOI] [PubMed] [Google Scholar]
- Hierholzer C, Harbrecht B, Menezes JM, Kane J, MacMicking J, Nathan CF, et al. Essential role of induced nitric oxide in the initiation of the inflammatory response after hemorrhagic shock. J Exp Med. 1998;187:917–928. doi: 10.1084/jem.187.6.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hietbrink F, Koenderman L, Rijkers G, Leenen L. Trauma: the role of the innate immune system. World J Emerg Surg. 2006;1:15. doi: 10.1186/1749-7922-1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzer K, Schubel F, Konietzny P, Wilhelm K, Bechstein WO, Henrich D. Interleukin 8 mRNA gene expression in peripheral and intra-abdominal neutrophils during human secondary peritonitis. Shock. 2005;23:501–506. [PubMed] [Google Scholar]
- Kim DJ, Kim W, Yoon SJ, Choi BM, Kim JS, Go HJ, et al. Effects of alcohol hangover on cytokine production in healthy subjects. Alcohol. 2003;31:167–170. doi: 10.1016/j.alcohol.2003.09.003. [DOI] [PubMed] [Google Scholar]
- Kozan R, Ayyildiz M, Yildirim M, Agar E. The effect of alpha-tocopherol in the acute ethanol intake and its withdrawal on penicillin-induced epilepsy. Acta Neurobiol Exp (Wars) 2009;69:177–188. doi: 10.55782/ane-2009-1743. [DOI] [PubMed] [Google Scholar]
- Lau A, Dossow V, Sander M, MacGuill M, Lanzke N, Spies C. Alcohol use disorder and perioperative immune dysfunction. Anesth Analg. 2009;108:916–920. doi: 10.1213/ane.0b013e318193fd89. [DOI] [PubMed] [Google Scholar]
- Lee CC, Lee RP, Subeq YM, Lee CJ, Chen TM, Hsu BG. Fluvastatin attenuates severe hemorrhagic shock-induced organ damage in rats. Resuscitation. 2009;80:372–378. doi: 10.1016/j.resuscitation.2008.12.003. [DOI] [PubMed] [Google Scholar]
- Lehnert M, Relja B, Sun-Young LV, Schwestka B, Henrich D, Czerny C, et al. A peptide inhibitor of C-jun N-terminal kinase modulates hepatic damage and the inflammatory response after hemorrhagic shock and resuscitation. Shock. 2008;30:159–165. doi: 10.1097/SHK.0b013e31815dd623. [DOI] [PubMed] [Google Scholar]
- Li G, Keyl PM, Smith GS, Baker SP. Alcohol and injury severity: reappraisal of the continuing controversy. J Trauma. 1997;42:562–569. doi: 10.1097/00005373-199703000-00032. [DOI] [PubMed] [Google Scholar]
- Liaw WJ, Chen TH, Lai ZZ, Chen SJ, Chen A, Tzao C, et al. Effects of a membrane-permeable radical scavenger, Tempol, on intraperitoneal sepsis-induced organ injury in rats. Shock. 2005;23:88–96. doi: 10.1097/01.shk.0000145937.70085.89. [DOI] [PubMed] [Google Scholar]
- Loft S, Olesen KL, Dossing M. Increased susceptibility to liver disease in relation to alcohol consumption in women. Scand J Gastroenterol. 1987;22:1251–1256. doi: 10.3109/00365528708996472. [DOI] [PubMed] [Google Scholar]
- Madan AK, Yu K, Beech DJ. Alcohol and drug use in victims of life-threatening trauma. J Trauma. 1999;47:568–571. doi: 10.1097/00005373-199909000-00026. [DOI] [PubMed] [Google Scholar]
- Meng ZH, Dyer K, Billiar TR, Tweardy DJ. Essential role for IL-6 in postresuscitation inflammation in hemorrhagic shock. Am J Physiol Cell Physiol. 2001;280:C343–C351. doi: 10.1152/ajpcell.2001.280.2.C343. [DOI] [PubMed] [Google Scholar]
- Messingham KA, Faunce DE, Kovacs EJ. Alcohol, injury, and cellular immunity. Alcohol. 2002;28:137–149. doi: 10.1016/s0741-8329(02)00278-1. [DOI] [PubMed] [Google Scholar]
- Molina PE, Zambell KL, Norenberg K, Eason J, Phelan H, Zhang P, et al. Consequences of alcohol-induced early dysregulation of responses to trauma/hemorrhage. Alcohol. 2004;33:217–227. doi: 10.1016/j.alcohol.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Moore FA, Sauaia A, Moore EE, Haenel JB, Burch JM, Lezotte DC. Postinjury multiple organ failure: a bimodal phenomenon. J Trauma. 1996;40:501–510. doi: 10.1097/00005373-199604000-00001. [DOI] [PubMed] [Google Scholar]
- Nanji AA, Jokelainen K, Fotouhinia M, Rahemtulla A, Thomas P, Tipoe GL. Increased severity of alcoholic liver injury in female rats: role of oxidative stress, endotoxin, and chemokines. Am J Physiol Gastrointest Liver Physiol. 2001;281:G1348–G1356. doi: 10.1152/ajpgi.2001.281.6.G1348. [DOI] [PubMed] [Google Scholar]
- Nelson S, Bagby GJ, Bainton BG, Summer WR. The effects of acute and chronic alcoholism on tumor necrosis factor and the inflammatory response. J Infect Dis. 1989;160:422–429. doi: 10.1093/infdis/160.3.422. [DOI] [PubMed] [Google Scholar]
- Partrick DA, Moore FA, Moore EE, Barnett CC, Jr, Silliman CC. Neutrophil priming and activation in the pathogenesis of postinjury multiple organ failure. New Horiz. 1996;4:194–210. [PubMed] [Google Scholar]
- Passos JF, Saretzki G, Zglinicki T. DNA damage in telomeres and mitochondria during cellular senescence: is there a connection? Nucleic Acids Res. 2007;35:7505–7513. doi: 10.1093/nar/gkm893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelan H, Stahls P, Hunt J, Bagby GJ, Molina PE. Impact of alcohol intoxication on hemodynamic, metabolic, and cytokine responses to hemorrhagic shock. J Trauma. 2002;52:675–682. doi: 10.1097/00005373-200204000-00010. [DOI] [PubMed] [Google Scholar]
- Redl H, Gasser H, Schlag G, Marzi I. Involvement of oxygen radicals in shock related cell injury. Br Med Bull. 1993;49:556–565. doi: 10.1093/oxfordjournals.bmb.a072630. [DOI] [PubMed] [Google Scholar]
- Rehm J, Room R, Graham K, Monteiro M, Gmel G, Sempos CT. The relationship of average volume of alcohol consumption and patterns of drinking to burden of disease: an overview. Addiction. 2003;98:1209–1228. doi: 10.1046/j.1360-0443.2003.00467.x. [DOI] [PubMed] [Google Scholar]
- Relja B, Schwestka B, Lee VS, Henrich D, Czerny C, Borsello T, et al. Inhibition of c-Jun N-terminal kinase after hemorrhage but before resuscitation mitigates hepatic damage and inflammatory response in male rats. Shock. 2009;32:509–516. doi: 10.1097/SHK.0b013e3181a2530d. [DOI] [PubMed] [Google Scholar]
- Relja B, Lehnert M, Seyboth K, Bormann F, Höhn C, Czerny C, et al. Simvastatin reduces mortality and hepatic injury after hemorrhage/resuscitation in rats. Shock. 2010;34:46–54. doi: 10.1097/SHK.0b013e3181cd8d05. [DOI] [PubMed] [Google Scholar]
- Reyna TM, Hollis HW, Jr, Hulsebus RC. Alcohol-related trauma. The surgeon's responsibility. Ann Surg. 1985;201:194–197. doi: 10.1097/00000658-198502000-00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivara FP, Jurkovich GJ, Gurney JG, Seguin D, Fligner CL, Ries R, et al. The magnitude of acute and chronic alcohol abuse in trauma patients. Arch Surg. 1993;128:907–912. doi: 10.1001/archsurg.1993.01420200081015. [DOI] [PubMed] [Google Scholar]
- Rivera CA, Bradford BU, Seabra V, Thurman RG. Role of endotoxin in the hypermetabolic state after acute ethanol exposure. Am J Physiol. 1998;275:G1252–G1258. doi: 10.1152/ajpgi.1998.275.6.G1252. [DOI] [PubMed] [Google Scholar]
- Ruiz M, Ewig S, Torres A, Arancibia F, Marco F, Mensa J, et al. Severe community-acquired pneumonia. Risk factors and follow-up epidemiology. Am J Respir Crit Care Med. 1999;160:923–929. doi: 10.1164/ajrccm.160.3.9901107. [DOI] [PubMed] [Google Scholar]
- Rushing GD, Britt LD. Reperfusion injury after hemorrhage: a collective review. Ann Surg. 2008;247:929–937. doi: 10.1097/SLA.0b013e31816757f7. [DOI] [PubMed] [Google Scholar]
- Sauaia A, Moore FA, Moore EE, Moser KS, Brennan R, Read RA, et al. Epidemiology of trauma deaths: a reassessment. J Trauma. 1995;38:185–193. doi: 10.1097/00005373-199502000-00006. [DOI] [PubMed] [Google Scholar]
- Shimizu T, Yu HP, Hsieh YC, Choudhry MA, Suzuki T, Bland KI, et al. Flutamide attenuates pro-inflammatory cytokine production and hepatic injury following trauma-hemorrhage via estrogen receptor-related pathway. Ann Surg. 2007;245:297–304. doi: 10.1097/01.sla.0000232523.88621.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spies CD, Dossow V, Eggers V, Jetschmann G, El-Hilali R, Egert J, et al. Altered cell-mediated immunity and increased postoperative infection rate in long-term alcoholic patients. Anesthesiology. 2004;100:1088–1100. doi: 10.1097/00000542-200405000-00010. [DOI] [PubMed] [Google Scholar]
- Szabo G, Mandrekar P. A recent perspective on alcohol, immunity, and host defense. Alcohol Clin Exp Res. 2009;33:220–232. doi: 10.1111/j.1530-0277.2008.00842.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo G, Mandrekar P, Verma B, Isaac A, Catalano D. Acute ethanol consumption synergizes with trauma to increase monocyte tumor necrosis factor alpha production late postinjury. J Clin Immunol. 1994;14:340–352. doi: 10.1007/BF01546318. [DOI] [PubMed] [Google Scholar]
- Szabo G, Chavan S, Mandrekar P, Catalano D. Acute alcohol consumption attenuates interleukin-8 (IL-8) and monocyte chemoattractant peptide-1 (MCP-1) induction in response to ex vivo stimulation. J Clin Immunol. 1999;19:67–76. doi: 10.1023/a:1020518703050. [DOI] [PubMed] [Google Scholar]
- Tak PP, Firestein GS. NF-kappaB: a key role in inflammatory diseases. J Clin Invest. 2001;107:7–11. doi: 10.1172/JCI11830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth B, Yokoyama Y, Schwacha MG, George RL, Rue LW, III, Bland KI, et al. Insights into the role of interleukin-6 in the induction of hepatic injury after trauma-hemorrhagic shock. J Appl Physiol. 2004;97:2184–2189. doi: 10.1152/japplphysiol.00499.2004. [DOI] [PubMed] [Google Scholar]
- Treggiari MM, Hudson LD, Martin DP, Weiss NS, Caldwell E, Rubenfeld G. Effect of acute lung injury and acute respiratory distress syndrome on outcome in critically ill trauma patients. Crit Care Med. 2004;32:327–331. doi: 10.1097/01.CCM.0000108870.09693.42. [DOI] [PubMed] [Google Scholar]
- Verma BK, Fogarasi M, Szabo G. Down-regulation of tumor necrosis factor alpha activity by acute ethanol treatment in human peripheral blood monocytes. J Clin Immunol. 1993;13:8–22. doi: 10.1007/BF00920631. [DOI] [PubMed] [Google Scholar]
- Wendell GD, Thurman RG. Effect of ethanol concentration on rates of ethanol elimination in normal and alcohol-treated rats in vivo. Biochem Pharmacol. 1979;28:273–279. doi: 10.1016/0006-2952(79)90515-x. [DOI] [PubMed] [Google Scholar]
- Xu H, Gonzalo JA, St Pierre Y, Williams IR, Kupper TS, Cotran RS, et al. Leukocytosis and resistance to septic shock in intercellular adhesion molecule 1-deficient mice. J Exp Med. 1994;180:95–109. doi: 10.1084/jem.180.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamakawa Y, Takano M, Patel M, Tien N, Takada T, Bulkley GB. Interaction of platelet activating factor, reactive oxygen species generated by xanthine oxidase, and leukocytes in the generation of hepatic injury after shock/resuscitation. Ann Surg. 2000;231:387–398. doi: 10.1097/00000658-200003000-00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ylikahri RH, Kähönen MT, Hassinen I. Modification of metabolic effects of ethanol by fructose. Acta Med Scand Suppl. 1972;542:141–150. doi: 10.1111/j.0954-6820.1972.tb05328.x. [DOI] [PubMed] [Google Scholar]
- Zambell KL, Phelan H, Vande SC, Zhang P, Shellito JE, Molina PE. Acute alcohol intoxication during hemorrhagic shock: impact on host defense from infection. Alcohol Clin Exp Res. 2004;28:635–642. doi: 10.1097/01.alc.0000122104.85971.55. [DOI] [PubMed] [Google Scholar]
- Zhang P, Bagby GJ, Boe DM, Zhong Q, Schwarzenberger P, Kolls JK, et al. Acute alcohol intoxication suppresses the CXC chemokine response during endotoxemia. Alcohol Clin Exp Res. 2002;26:65–73. [PubMed] [Google Scholar]
- Zhang P, Welsh DA, Siggins RW, 2nd, Bagby GJ, Raasch CE, Happel KI, et al. Acute alcohol intoxication inhibits the lineage- c-kit+ Sca-1+ cell response to Escherichia coli bacteremia. J Immunol. 2009;182:1568–1576. doi: 10.4049/jimmunol.182.3.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Z, Connor H, Mason RP, Qu W, Stachlewitz RF, Gao W, et al. Destruction of Kupffer cells increases survival and reduces graft injury after transplantation of fatty livers from ethanol-treated rats. Liver Transpl Surg. 1996;2:383–387. doi: 10.1002/lt.500020509. [DOI] [PubMed] [Google Scholar]
- Zhong Z, Arteel GE, Connor HD, Schemmer P, Chou SC, Raleigh JA, et al. Binge drinking disturbs hepatic microcirculation after transplantation: prevention with free radical scavengers. J Pharmacol Exp Ther. 1999;290:611–620. [PubMed] [Google Scholar]
- Zingarelli B. Nuclear factor-kappaB. Crit Care Med. 2005;33:S414–S416. doi: 10.1097/01.ccm.0000186079.88909.94. [DOI] [PubMed] [Google Scholar]