

Abstract
BACKGROUND AND PURPOSE
Increased firing of the glutamatergic pathway between the subthalamic nucleus and substantia nigra pars reticulata (SNpr) contributes to the abnormal firing of motor circuits and subsequent motor deficits seen in Parkinson's disease. Broad spectrum agonist-induced activation of presynaptic group III metabotropic glutamate (mGlu) receptors within the SNpr reduced glutamate release and reversed akinesia in the reserpine-treated rat model of Parkinson's disease. Here, we have sought to identify which subtypes of group III mGlu receptor in the SNpr were responsible for these beneficial effects.
EXPERIMENTAL APPROACH
The ability of the mGlu4 positive allosteric modulator, N-phenyl-7-(hydroxyminocyclopropa[b]chromen-1a-carboxamide) (PHCCC), the mGlu7 allosteric agonist, N,N′-dibenzhydrylethane-1,2-diamine dihydrochloride (AMN082) and the mGlu8-selective agonist (S)-3,4-dicarboxyphenylglycine [(S)-3,4-DCPG] to inhibit KCl-evoked [3H]-D-aspartate release was examined in vitro in rat nigral prisms. Reversal of akinesia in reserpine-treated rats was also assessed following intranigral injection of these agents.
KEY RESULTS
PHCCC and AMN082 inhibited [3H]-D-aspartate release by 42% and 53%, respectively when given alongside a sub-threshold concentration of the broad spectrum group III agonist, L-2-amino-4-phosphonobutyrate (L-AP4; 1 µM). In contrast (S)-3,4-DCPG failed to inhibit [3H]-D-aspartate release. All three agents also reversed reserpine-induced akinesia although only the effects of PHCCC and AMN082 were inhibited by pre-treatment with the group III antagonist (RS)-α-cyclopropyl-4-phosphonophenylglycine (CPPG).
CONCLUSIONS AND IMPLICATIONS
These findings reveal that targeting SNpr mGlu4 or mGlu7 receptors, but not mGlu8 receptors, provided relief from akinesia in the reserpine-treated rat model of Parkinson's disease, most likely reflecting inhibition of excess glutamate release in this region.
Keywords: akinesia, group III mGlu receptor, metabotropic glutamate receptors, mGlu4, mGlu7, mGlu8, Parkinson's disease, reserpine, substantia nigra, symptomatic relief
Introduction
Current dopaminergic treatments for Parkinson's disease are very effective in the short term but produce debilitating side-effects with long-term use including on-off fluctuations in efficacy and the appearance of uncontrollable dyskinesia. The motor deficits seen in Parkinson's disease such as postural instability, akinesia and tremor result primarily from degeneration of dopaminergic neurones in the substantia nigra pars compacta (SNpc). This degeneration produces a marked reduction in striatal dopamine innervation, which produces downstream changes in basal ganglia firing culminating in increased activity of the GABAergic output regions, the substantia nigra pars reticulata (SNpr) and internal globus pallidus, which leads to inhibition of thalamocortical feedback and ensuing motor deficits. The increased activity of the output regions may be initiated and maintained through increased innervation from the glutamatergic subthalamic nucleus (STN), which is known to exhibit increased firing in idiopathic Parkinson's disease (Hutchison et al., 1998; Benazzouz et al., 2002). It follows that reducing this glutamatergic drive may help combat the motor symptoms in Parkinson's disease. In support of this suggestion, surgical subthalamotomy or deep brain stimulation to dampen down firing of the STN provide symptomatic relief in patients (see Peppe et al., 2004; Alvarez et al., 2005). Given the cost, limited accessibility and inherent risks of neurosurgery, pharmacological inhibition of glutamate release from STN terminals in the SNpr is an attractive alternative strategy worth pursuing in order to achieve similar symptomatic benefits. Group III metabotropic glutamate (mGlu) receptors are considered promising targets to achieve such an effect (see Hopkins et al., 2009; Duty, 2010).
Group III mGlu receptors comprise four subtypes: mGlu4, mGlu6, mGlu7 and mGlu8 (receptor nomenclature follows Alexander et al., 2011) with all but mGlu6 playing important neuromodulatory roles in the brain (Conn and Pin, 1997). These receptors couple through Gi/Go and are found predominantly, though not exclusively, on presynaptic elements of both GABAergic and glutamatergic synapses, where their activation has already been shown to reduce transmitter release in regions such as the thalamus, superior colliculus and globus pallidus (Turner and Salt, 1999; Pothecary et al. 2002; MacInnes and Duty, 2008). More recently we demonstrated that activation of group III mGlu receptors in the SNpr, using broad spectrum agonists such as L-2-amino-4-phosphonobutyrate (L-AP4) and O-phospho-L-serine (L-SOP) reduces glutamate release in the SNpr both in vitro and in vivo (Austin et al., 2010). These findings were consistent with previous electrophysiological reports of L-AP4-mediated inhibition of STN-evoked excitatory post synaptic currents (EPSCs) in the SNpr (Wittmann et al., 2001).
In addition, we have demonstrated that intranigral injection of L-AP4 or L-SOP reverses akinesia in the reserpine-treated rat model of Parkinson's disease (MacInnes et al., 2004; Austin et al., 2010). The efficacy of broad spectrum group III mGlu agonists has also been demonstrated against haloperidol-induced catalepsy following injection into the SNpr (Konieczny et al., 2007). Taken together, these transmitter release and behavioural studies suggest that group III mGlu receptors are promising targets for alleviation of akinetic symptoms in Parkinson's disease. In situ hybridization studies have revealed mRNA encoding mGlu4, mGlu7 and mGlu8 in the STN (Messenger et al., 2002) and immunoreactivity for at least mGlu4, and mGlu7 is confirmed within the SNpr, mGlu4 having been further localized to presynaptic elements of excitatory asymmetric synapses (Kosinski et al., 1999; Corti et al., 2002). Thus at least two subtypes of mGlu receptor may underlie these responses and therefore offer useful pharmacological targets to reduce glutamate release in the SNpr in Parkinson's disease.
The aims of the present study were therefore to establish which group III mGlu receptors were able to produce the desired inhibition of transmitter release in the SNpr using an in vitro nigral slice preparation and to examine whether activation of these receptors in vivo could provide relief of akinesia in the reserpine-treated rat model. The findings of the study support a role for mGlu4 and mGlu7 receptors in bringing about these actions, but rule out any contribution from mGlu8 receptors.
Methods
Animals
All animal care and experimental procedures conformed to the UK Animals (Scientific Procedures) Act, 1986 and every effort was made to minimize animal numbers and suffering. Male Sprague Dawley rats (B & K, UK) weighing 270–300 g were used in these studies. Food and water were provided ad libitum. Animals were housed in a temperature- and humidity-controlled environment with a 12 h light/dark cycle.
Experimental protocol for [3H]-D-aspartate release in the substantia nigra
Release experiments were carried out using the non-metabolized tritiated analogue of glutamate, [3H]-D-aspartate, as previously described (Austin et al., 2010). Animals were killed by stunning and cervical dislocation, the brains rapidly removed and the substantia nigra (SN) (containing both SNpc and SNpr segments) dissected out and chopped into 350 µm prisms. Tissues pooled from three animals per experimental run were equilibrated for 45 min in Krebs solution (composition in mM: NaCl 134, CaCl 1.3, MgSO4 1, NaHCO3 25, KCl 5, KH2PO4 1.24, glucose 10) at pH 7.4, aerated (95% O2/5% CO2) and maintained at 37°C. Tissues were then loaded for 45 min in Krebs solution containing 33 µM [3H]-D-aspartate (0.222 MBq), washed three times in Krebs solution and finally suspended in 1.2 mL Krebs solution before being transferred in 100 µL aliquots into a 12-chamber Brandel perfusion system (Semat, St Albans, UK) with a 1 mL·min−1 perfusion rate. Following a 1 h equilibration, 2 min perfusate fractions were collected into vials containing 3 mL of scintillation fluid (Optiphase Hi-Safe 2). Basal [3H]-D-aspartate release was determined over the first three fraction (6 min) and followed by 2 min stimulation (S1) with Krebs solution containing 25 mM KCl (with equimolar reduction of NaCl to 114 mM) to evoke [3H]-D-aspartate release. An 18 min washout period was then followed by a second, 25 mM KCl stimulation for 2 min (S2). The mGlu4 positive allosteric modulator (PAM), PHCCC (Maj et al., 2003; 1–100 µM), the mGlu7-selective agonist, AMN082 (Mitsukawa et al., 2005; 1–100 µM), the mGlu8-selective agonist (S)-3,4-DCPG (Thomas et al., 2001; 0.03–10 µM) or vehicle were added to the perfusate 8 min prior to and during S2. Where noted, drugs were applied in combination with a sub-threshold concentration of L-AP4 (1 µM; taken from Austin et al., 2010). L-AP4 and (S)-3,4-DCPG were dissolved in Krebs solution while PHCCC and AMN082 were made up in a final solution of 1% DMSO in Krebs solution. Release fractions and remaining tissues were analysed for [3H]-D-aspartate content by liquid scintillation spectroscopy.
Experimental protocol for monitoring reversal of akinesia in reserpine-treated rats following intranigral drug administration
A total of 128 rats were used for these studies, which were performed as described in Austin et al. (2010). Under general anaesthesia (isoflurane, 5% induction and 3% maintenance in 95% O2/5% CO2) 23-gauge stainless-steel guide cannulae were stereotaxically implanted 2 mm above the SNpr [AP, −5.5 mm; ML, ±2.2 mm; VM, −6.3 mm from Bregma (Paxinos and Watson, 1998)]. Bilateral cannulation was performed to double the chances of obtaining a patent cannula in each animal, thereby reducing overall numbers required. Cannulae were secured in place with dental cement and microscrews anchored into the skull before placing 30-gauge stainless-steel stylets inside to prevent blockage. A minimum of 4 days post cannulation, animals were rendered akinetic by administration of reserpine [4 mg·kg−1, s.c.; prepared as 5 mL of a 4 mg·mL−1 solution in 18 MΩ water containing 0.85% (v/v) glacial acetic acid] under light anaesthesia (3% isoflurane in 95% O2/5% CO2).
Eighteen hours later, when a stable level of akinesia had developed, manifested as complete loss of locomotor activity, pre-acclimatized animals were replaced into 40 cm diameter, flat-bottomed hemispherical bowls for video-recording of baseline locomotor activity over 30 min. Animals (n= 5–8 per group) then received a single unilateral injection (adjusted to pH 7.4) of PHCCC (3–75 nmol in 2.5 µL PBS with 40% DMSO), AMN082 (3–100 nmol in 2.5 µL PBS with 40% DMSO), (S)-3,4-DCPG (30–300 nmol in 0.5 µL PBS) or corresponding vehicle. Injections (1 µL·min−1) were made, in conscious animals, through a 30-gauge stainless-steel needle that extended 2 mm below the tip of the guide cannulae into the SNpr and was attached via Portex tubing to a 10 µL Hamilton syringe. When examined, the group III mGlu antagonist, CPPG (75 nmol in 2.5 µL PBS; Toms et al., 1996) or vehicle was injected into the SNpr 30 min before the mGlu agonists or allosteric modulators. Following each injection, the needle was left in place for a further 2 min to allow diffusion of the drug from the site of injection. As firing of the STN is increased ∼50% following reserpine treatment (Robledo and Feger, 1991) and this is accompanied by an increase in extracellular glutamate levels in STN target areas (Biggs et al., 1997), endogenous glutamate levels were expected to be sufficient to provide a low level of occupation of the orthosteric site on the group III mGlu receptors. Therefore, in contrast to the above in vitro experiments, both PHCCC and AMN082 were administered to the reserpine-treated rat without L-AP4. Locomotor activity in the form of 360° contraversive rotations representative of unilateral reversal of akinesia was video-recorded for 60 min for subsequent analysis, without knowledge of the treatments. Trypan blue dye was injected through the cannula at the end of the experiment to allow the anatomical location of the probe, and hence site of injection, to be determined. Animals then received a lethal dose of pentobarbital before their brains were removed and rapidly frozen on cooled (−40°C) isopentane. Frozen brains were cryostat sectioned at 30 µm and then analysed by light microscopy. Any animals with incorrect cannula placement (<8% total) were eliminated from the data analysis and, importantly, did not produce any rotational behaviour like that seen in animals with accurately placed cannulae.
Data handling and statistical analysis
All data are expressed as mean ± SEM, where n represents the number of animals in each experimental group. Statistical analyses were performed using GraphPad Prism (version 5.0).
In vitro release
The amount of [3H]-D-aspartate present in each release fraction was calculated as a percentage of the total present in the tissue at the start of the fraction. Graphs of % release over time were thus constructed and the total [3H]-D-aspartate content of S1 and S2 was calculated by integrating the area under the respective peaks using Origin 7 Peak-fitting software (Aston Scientific, Buckinghamshire, UK). The mean S2/S1 ratio was calculated to give an index of drug efficacy by pooling data from at least two independent experimental runs for that condition (minimum of six tissues from six rats). Differences in the S2/S1 ratio between groups were analysed using a one-way anova with a Student Newman-Keuls (SNK) post hoc test.
Reversal of reserpine-induced akinesia
The number of 360° contraversive rotations evoked following intranigral drug administration was counted, in 5 min intervals from videotaped recordings and plotted as a time-course. Differences at each time point between drug and vehicle were analysed using a two-way anova and Bonferroni's post hoc test. The total number of rotations over 60 min was compared between different treatment groups using one-way anova and SNK post hoc test. The ability of CPPG to block responses to PHCCC or AMN082 was analysed using an unpaired two tailed Student's t-test.
Materials
L-AP4, (S)-3,4-DCPG, AMN082, PHCCC and CPPG were all obtained from Tocris Cookson, Bristol, UK. [3H]-D-aspartate was obtained from Amersham, Little Chalfont, Bucks, UK. All other materials were obtained from Sigma-Aldrich, Somerset, UK.
Results
[3H]-D-aspartate release in the SN
Basal release of [3H]-D-aspartate from nigral prisms (0.8–1%) was consistent with previous reports (Austin et al., 2010). The mGlu4 PAM, PHCCC, was applied in conjunction with a sub-threshold concentration of L-AP4 (1 µM), added to occupy the orthosteric, glutamate-binding site on mGlu4. As expected, neither L-AP4 (1 µM) nor PHCCC (1–100 µM) had any effect on basal or 25 mM KCl-evoked [3H]-D-aspartate release when administered alone. In contrast, when given with L-AP4, PHCCC inhibited evoked release. The profile of this inhibition is shown in Figure 1A for the optimum concentration (30 µM PHCCC). PHCCC produced a concentration-dependent inhibition of 25 mM KCl-evoked release (reduced S2/S1 ratio), reaching significance at 3, 30 and 100 µM (one-way anova with SNK post hoc analysis; Figure 1B–E,). Maximal inhibition of 42 ± 11.2% (mean ± SEM, n= 6) was achieved with 100 µM PHCCC.
Figure 1.
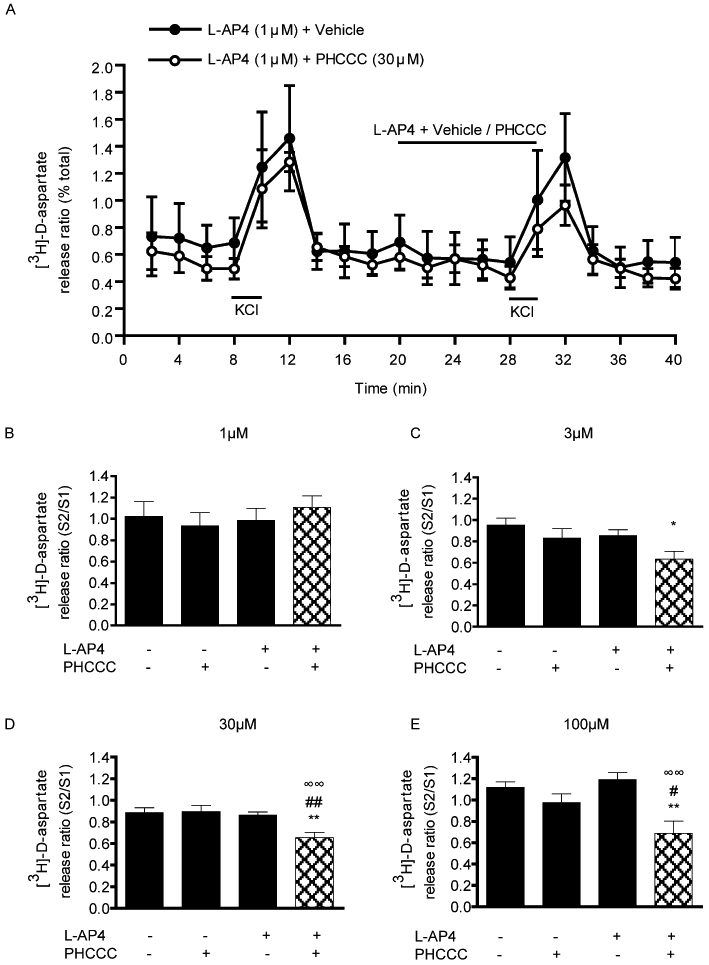
Effect of the mGlu4 positive allosteric modulator, PHCCC, on 25 mM KCl-evoked [3H]-D-aspartate release in rat nigral tissue prisms. (A) Release profile showing the effect of 30 µM PHCCC, given with a sub-threshold concentration of L-AP4 (1 µM), on release evoked by the second KCl stimulus (S2). Horizontal bars indicate the periods of contact with KCl or drug/vehicle. Data are mean ± SEM (n= 3 from a single experimental run). (B–E) Graphs of S2/S1 ratio showing concentration-dependent effects of PHCCC, combined with L-AP4 (1 µM), on [3H]-D-aspartate release. Data are mean ± SEM (n= 6). *P < 0.05, **P < 0.01, significantly different from vehicle; #P < 0.05, ##P < 0.01, significantly different from PHCCC alone; ∞∞P < 0.01, significantly different from L-AP4 alone. The presence (+) or absence (−) of drugs is indicated.
The effect of selective activation of mGlu7 receptors was tested using AMN082. Although originally described as an allosteric agonist that binds to a site within the transmembrane domain, distal from the glutamate-binding site (Mitsukawa et al., 2005), when administered alone in the present dual stimulation release paradigm, AMN082 had no effect on evoked [3H]-D-aspartate release across a wide concentration range (0.01–100 µM; data not shown), which extended beyond its estimated EC50 of 0.064–0.290 µM. Given that allosteric agonists may have potentiating effects when given with orthosteric agonists (Conn and Niswender, 2006), we next examined the effects of AMN082 in combination with the sub-threshold concentration of L-AP4 (1 µM), previously used to probe the potentiating effects of PHCCC. As expected, neither L-AP4 (1 µM) nor AMN082 (1–100 µM) had any effect on basal or 25 mM KCl-evoked [3H]-D-aspartate release when administered alone. However, when combined with the sub-threshold concentration of L-AP4, AMN082 inhibited 25 mM KCl-evoked release. The profile of this inhibition is shown in Figure 2A for the optimum concentration (30 µM). Under these conditions, AMN082 produced a concentration-dependent inhibition of release, reaching significance at 30 and 100 µM (one-way anova with SNK post hoc analysis; Figure 2B–E) with maximal inhibition of 53 ± 9.8% (mean ± SEM, n= 6) produced by 30 µM AMN082.
Figure 2.
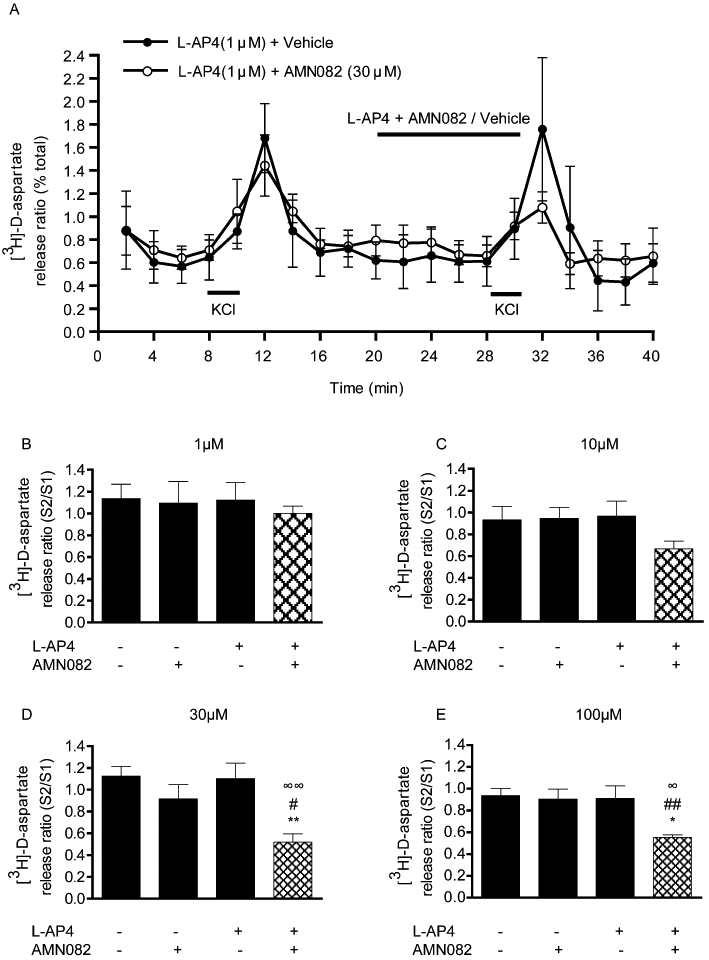
Effect of the mGlu7 allosteric modulator, AMN082, on 25 mM KCl-evoked [3H]-D-aspartate release in rat nigral tissue prisms. (A) Release profile showing the effect of 100 µM AMN082, given with a sub-threshold concentration of L-AP4 (1 µM), on release evoked by the second KCl stimulus (S2). Horizontal bars indicate the periods of contact with KCl or drug/vehicle. Data are mean ± SEM (n= 3 from a single experimental run). (B–E) Graphs of S2/S1 ratio showing concentration-dependent effects of AMN082 on [3H]-D-aspartate. Data are mean ± SEM (n= 6). *P < 0.05, **P < 0.01, significantly different from vehicle; #P < 0.05, ##P < 0.01, significantly different from AMN082 alone; ∞P < 0.05, ∞∞P < 0.01, significantly different from L-AP4 alone. The presence (+) or absence (−) of drugs is indicated.
The effect of selective activation of mGlu8 receptors was probed using (S)-3,4-DCPG. Over a wide concentration range (0.03–10 µM) spanning the estimated EC50 of 31 nM (Thomas et al., 2001) (S)-3,4-DCPG had no significant effects on either basal or 25 mM KCl-evoked [3H]-D-aspartate release (Figure 3A,B). Any action as a positive modulator was also ruled out since (S)-3,4-DCPG (0.3–30 µM) also failed to inhibit release when given in conjunction with the sub-threshold concentration of 1 µM L-AP4 (data not shown).
Figure 3.
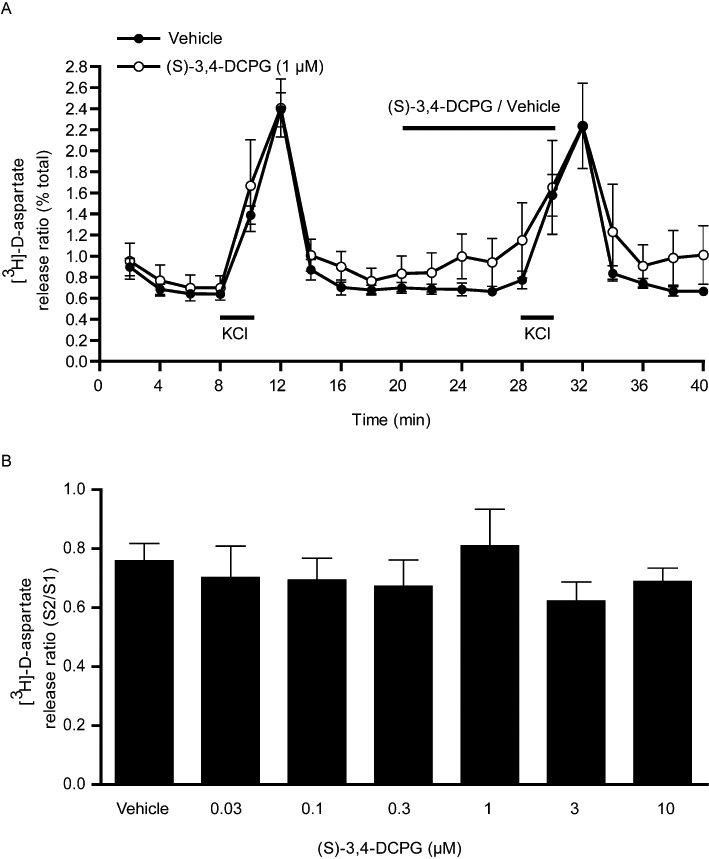
Effect of the mGlu8 agonist (S)-3,4-DCPG, on 25 mM KCl-evoked [3H]-D-aspartate release in rat nigral tissue prisms. (A) Release profile showing the effect of (S)-3,4-DCPG (1 µM) on release evoked by the second KCl stimulus (S2). Horizontal bars indicate the periods of contact with KCl or drug/vehicle. Data are mean ± SEM (n= 3 from a single experimental run). (B) Graph of S2/S1 ratio showing lack of effect of increasing concentrations of (S)-3,4-DCPG on [3H]-D-aspartate release. Data are mean ± SEM (n= 6).
Reversal of akinesia following intranigral group III mGlu receptor stimulation
The antiparkinsonian effects of the mGlu4 PAM, PHCCC and the mGlu7 allosteric agonist, AMN082 were explored in vivo without the need for co-administration of a sub-threshold dose of L-AP4. Unilateral intranigral injection of both agents produced a marked reversal of reserpine-induced akinesia, the profiles of which are shown for the maximally effective doses in Figure 4. PHCCC (75 nmol) produced net contraversive rotations, which were significantly increased compared with vehicle within 5 min of injection and remained so for up to 40 min (two-way anova with Bonferroni's post hoc test) (Figure 4A). Intranigral injection of AMN082 (100 nmol) also produced net contraversive rotations, which were significantly increased compared with vehicle between 15 and 30 min (two-way anova with Bonferroni's post hoc test) (Figure 4B). In both cases, animals treated with vehicle alone remained fully akinetic throughout the recording period. Quantification of the rotations produced in 60 min revealed that responses to both PHCCC (3–75 nmol) and AMN082 (3–100 nmol) were dose-dependent (Figure 4C,D), reaching a maximum of 141 ± 28 rotations with 75 nmol PHCCC (mean ± SEM, n= 8; P < 0.01 vs. vehicle) and 226 ± 75 rotations with 100 nmol AMN082 (mean ± SEM, n= 6; P < 0.05 vs. vehicle) both using one-way anova with SNK post hoc test. The involvement of group III receptor activation in these responses was assessed using the group III mGlu-selective antagonist, CPPG that binds to the orthosteric site and is therefore expected to compete with endogenous glutamate for binding (Toms et al., 1996). Administration of CPPG (75 nmol) alone produced no change in net contraversive rotations (data not shown). However, pre-treatment with CPPG significantly inhibited the subsequent responses to both 75 nmol PHCCC and 100 nmol AMN082 by approximately 70% (Figure 4E,F, P < 0.01 vs. vehicle pre-treatment; unpaired two-tailed t-test).
Figure 4.
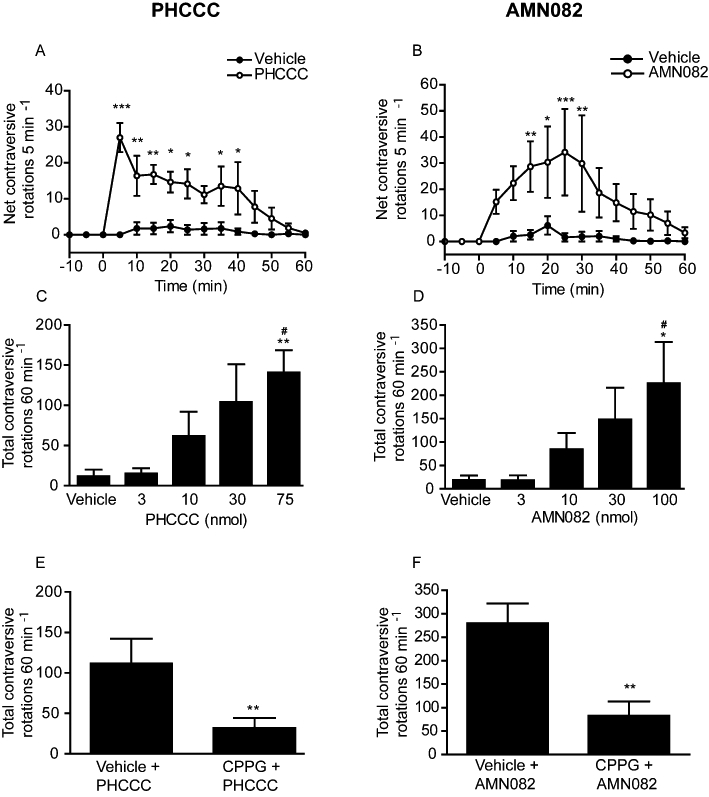
Effect of unilateral injection of the mGlu4 positive allosteric modulator, PHCCC (A,C,E) or the mGlu7 allosteric modulator, AMN082 (B,D,F) in the SNpr on reversal of akinesia in the reserpine-treated rat. (A,B) Time-course of rotational activity induced by the maximally effective dose of 75 nmol PHCCC or 100 nmol AMN082. Data are mean ± SEM (n= 8, PHCCC or n= 6, AMN082). *P < 0.05, **P < 0.01, ***P < 0.001, significantly different from vehicle. (C,D) Effect of increasing doses of PHCCC or AMN082 on total rotational activity over 60 min. Data are mean ± SEM (n= 8, PHCCC or n= 6 AMN082). *P < 0.05, **P < 0.01, significantly different from vehicle; #P < 0.05, significantly different from 3 nmol doses. (E,F) Effect of pre-treatment with CPPG (75 nmol) on the rotational response to PHCCC (75 nmol) or AMN082 (100 nmol). Data are mean ± SEM (n= 8, PHCCC or n= 6 AMN082). **P < 0.01, significantly different from vehicle pre-treatment.
Despite showing a lack of efficacy in vitro, the mGlu8 agonist (S)-3,4-DCPG (0.3–300 nmol) produced a significant dose-dependent reversal of akinesia in the reserpine-treated rat. This response was, however, much shorter in duration than that seen with PHCCC or AMN082, with a significant increase in contraversive rotations compared with vehicle seen for the maximally effective dose (300 nmol) only between 5 and 10 min post injection (P < 0.01; two-way anova with Bonferroni's post hoc test) (Figure 5A). The effects of (S)-3,4-DCPG were dose-dependent, but reached significance only at the highest dose tested (300 nmol; one-way anova with SNK post hoc test) (Figure 5B). However, 30 min pre-treatment with the specific group III receptor antagonist CPPG failed to inhibit the rotational responses to either 100 nmol (S)-3,4-DCPG (data not shown) or 300 nmol (S)-3,4-DCPG (Figure 5C).
Figure 5.
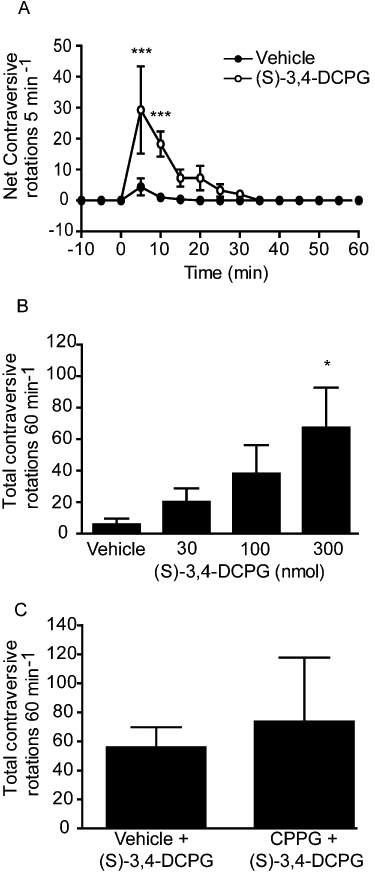
Effect of unilateral injection of the mGlu8 agonist (S)-3,4-DCPG, in the SNpr on reversal of akinesia in the reserpine-treated rat. (A) Time-course of locomotor activity induced by the maximally effective dose of 300 nmol (S)-3,4-DCPG. Data are mean ± SEM (n= 5). ***P < 0.001, significantly different from vehicle. (B) Effect of increasing doses of (S)-3,4-DCPG on total rotational activity over 60 min. Data are mean ± SEM (n= 4). *P < 0.05, significantly different from vehicle. (C) Effect of pre-treatment with CPPG (75 nmol) on the rotational response to (S)-3,4-DCPG (300 nmol). Data are mean ± SEM (n= 5).
Discussion
Restricting the excess glutamate release in the SNpr may offer symptomatic relief in Parkinson's disease. We recently reported that activation of presynaptic group III mGlu receptors using the broad spectrum agonists L-SOP or L-AP4 inhibited depolarization-evoked glutamate release in the SNpr both in vitro and in vivo (Austin et al., 2010). Moreover, in the reserpine-treated rat model of Parkinson's disease, injection of L-SOP or L-AP4 into the SNpr reversed akinesia (MacInnes et al., 2004; Austin et al., 2010). The present study extends these findings to show that selective targeting of mGlu4 and mGlu7 receptors, but not mGlu8 receptors is sufficient to reduce glutamate release from nigral tissue in vitro and to bring about reversal of reserpine-induced akinesia.
Effects of group III mGlu receptor activation on glutamate release in the SN
The presence of mGlu4 receptors on the desired excitatory terminals in the SNpr has been confirmed using electron microscopy (Corti et al., 2002). The localization of mRNA encoding mGlu4 receptors in the STN (Messenger et al., 2002), coupled with the fact that glutamatergic efferents from the STN comprise the major excitatory input into the SNpr (Parent and Hazrati, 1995), points to these mGlu4 receptors being located on STN terminals in this region. The present study reveals that these receptors are functional as their activation using the mGlu4 PAM, PHCCC in combination with a sub-threshold concentration of L-AP4 to activate the orthosteric site on the receptor inhibits release of the glutamate analogue, [3H]-D-aspartate, from SNpr tissue prisms in vitro. These data are consistent with previous reports that, in combination with a low concentration of L-AP4, PHCCC inhibits STN-evoked EPSCs in the SNpr (Valenti et al., 2005). The present effects were triggered between 1 and 3 µM, in agreement with an estimated EC50 of PHCCC at mGlu4 receptors of around 3.7 µM (Marino et al., 2003). While at these concentrations, PHCCC may also reportedly inhibit mGlu1 receptors (IC50 3 µM; Annoura et al., 1996), such an action is ruled out here since PHCCC had no effect in the absence of L-AP4, supporting an allosteric potentiation mechanism. These findings indicate that mGlu4 receptors in the SNpr present an effective target for restricting glutamate release from STN terminals in Parkinson's disease.
The presence of mGlu7 receptors on STN terminals in the SNpr has not been directly demonstrated. However, given that mGlu7 mRNA is found within the STN, it is very likely that at least some of the mGlu7 immunoreactivity present in the SNpr reflects receptors on presynaptic STN terminals (Kosinski et al., 1999; Messenger et al., 2002; Figure S1). When added alone, the mGlu7-selective allosteric agonist, AMN082 failed to modify [3H]-D-aspartate release. However, when given alongside a sub-threshold concentration of L-AP4, AMN082 produced a concentration-dependent inhibition of release. AMN082 has also been shown to potentiate L-AP4-mediated inhibition of EPSCs in the rat basolateral amygdala (Ugolini et al., 2008), further supporting a potentiating rather than a direct agonist action of AMN082. These electrophysiological effects, like those on [3H]-D-aspartate release, were produced at concentrations of AMN082 (10 µM and above) higher than the estimated EC50 (0.26 µM). Indeed, 10 µM AMN082 does induce moderate increases in GTPγ35S binding in cells expressing mGlu4 (18%) and mGlu8 (20%) receptors, albeit with much more marked activation (140%) seen in cells expressing mGlu7 receptors (Mitsukawa et al., 2005). Therefore, while the present effects of AMN082 are most likely mGlu7-mediated, a small contribution from other group III mGlu receptors cannot be ruled out.
Contrary to the finding with PHCCC and AMN082, the mGlu8-selective agonist (S)-3,4-DCPG failed to inhibit release over a wide concentration range when applied either alone or in combination with L-AP4. Although peak KCl-evoked release was higher in these studies (2.4%, compared with 1.5% and 1.7% in the studies with PHCCC or AMN082, respectively), this higher peak would not mask any inhibitory effect of (S)-3,4-DCPG, since inhibition of a similarly high peak release was achieved withL-SOP and L-AP4 (Austin et al., 2010). These data therefore indicate a clear lack of involvement of mGlu8 receptors in modulating glutamate release in the SNpr. Given that STN efferents co-excite both segments of the SN (SNpr and SNpc), these findings are consistent with the lack of effect of mGlu8 receptors in mediating inhibition of STN-evoked EPSCs in the SNpc (Valenti et al., 2005) and may reflect the very low levels of mGlu8 immunoreactivity present in the SN(Figure S1). Collectively, these data imply that mGlu4 and mGlu7, but not mGlu8 receptors may be ideally placed to restrict, pharmacologically, excess glutamate release in the SNpr and thereby provide symptomatic relief in Parkinson's disease.
Reversal of akinesia in the reserpine-treated rat model of Parkinson's disease following subtype-specific activation of group III mGlu receptors in the SNpr
The reserpine-treated rat was chosen to model parkinsonian akinesia because depletion of dopamine in these animals increases the firing rate of the STN comparable to that seen in Parkinson's disease (Robledo and Feger, 1991; Hutchison et al., 1998; Benazzouz et al., 2002) and elevates glutamate release in the basal ganglia output regions (Biggs et al., 1997). Moreover, direct injection of the broad spectrum agonists, L-AP4 and L-SOP into the SNpr has been shown to reverse akinesia in this model (Macinnes et al., 2004; Austin et al., 2010). Here, reversal of reserpine-induced akinesia was produced following direct injection of the mGlu4 PAM, PHCCC or the mGlu7 allosteric agonist AMN082 into the SNpr. The ability of both PHCCC and AMN082 to act in the absence of a sub-threshold dose of L-AP4 (which was needed for efficacy in vitro), most likely reflects the presence within SNpr of reserpinized rats of sufficient levels of endogenous glutamate to occupy the orthosteric sites on both mGlu4 and mGlu7 receptors (Biggs et al., 1997).
The effects of both PHCCC and AMN082 were inhibited approximately 70% by the selective group III mGlu antagonist, CPPG. Although it remains to be seen whether higher doses of CPPG (>75 nmol) would have inhibited the residual response, this sensitivity to CPPG confirms that the majority of these responses are mediated through group III mGlu receptors. This confirmation is essential for interpretation of the effects of AMN082, given recent preliminary reports that AMN082 is rapidly metabolized in vivo, with its metabolite displaying binding affinity for a wide range of targets including the transporters for dopamine (DAT), noradrenaline (NET) and 5-HT (SERT) (Brodbeck et al., 2010; Sukoff-Rizzo et al., 2010). The direct injection into the SNpr in this study is most likely to circumvent many of these issues with metabolite formation and, coupled with the ability of CPPG to reverse the present effects of AMN082, points to these being produced through mGlu7 receptors.
A role for mGlu4 receptors in bringing about antiparkinsonian responses has been suggested previously on the basis of attenuation of reserpine-induced akinesia by i.c.v. injections of PHCCC or by a second mGlu4 PAM, VU0155041 (Marino et al., 2003; Niswender et al., 2008). Although the site at which these previous actions were mediated was not examined, the authors suggested this was the globus pallidus (analogous to the human globus pallidus externus) where PHCCC potentiates L-AP4-mediated inhibition of striatal-evoked inhibitory post synaptic currents (IPSCs) (Marino et al., 2003). Supporting this suggestion, direct intrapallidal injections of the broad spectrum group III agonists L-SOP or ACPT-I reversed reserpine-induced akinesia or haloperidol-induced catalepsy (MacInnes et al., 2004; Konieczny et al., 2007), while intrapallidal infusion of the mGlu4 agonist, LSP1-2111 reversed akinesia in a bilateral 6-hydroxy dopamine (6-OHDA) lesion model of Parkinson's disease and reverses haloperidol-induced catalepsy (Beurrier et al., 2009). This antiparkinsonian action may reflect inhibition of GABA release within the globus pallidus, as demonstrated in vivo with L-AP4 and L-SOP (MacInnes and Duty, 2008), which is predicted to normalize firing within the indirect basal ganglia circuit. The present study now exposes an additional complementary site of action through which mGlu4 modulators offer antiparkinsonian relief, the SNpr and indicates that a reduction in glutamate release, as shown here in vitro, may underlie this efficacy. Although L-AP4-mediated activation of group III mGlu receptors in the SNpr in vivo similarly leads to inhibition of glutamate release, further studies with mGlu4-selective agents would undoubtedly strengthen this conclusion. It is important to qualify that within the SNpr group III mGlu receptors also exist on presynaptic terminals of the GABAergic striatonigral pathway, where their activation inhibits striatal-evoked IPSCs within the SNpr (Wittmann et al., 2001). Such an action is predicted to worsen, rather than alleviate parkinsonian symptoms by further increasing firing of an already overactive SNpr. However, Wittmann et al. (2002) demonstrated that in brain slices obtained from reserpine-treated rats and bathed continuously in reserpine in vitro, or in control slices incubated with haloperidol, while the ability of L-AP4 to inhibit STN-evoked EPSCs in the SNpr was retained, its ability to inhibit striatal-evoked IPSCs in this region was lost. Thus, under parkinsonian conditions where marked dopamine depletion is evident, actions at group III mGlu receptors on glutamatergic STN terminals in the SNpr appear to override those on GABAergic striatal terminals in this region, as evidenced by the present reversal of akinesia following intra-SNpr injections of both PHCCC and AMN082, and previous observations with L-SOP and L-AP4 (Austin et al., 2010). Indeed, beneficial actions following intranigral injections of broad spectrum group III agonists are also seen in other animal models of Parkinson's disease where dopaminergic transmission is severely compromised such as the haloperidol-induced catalepsy model ((Konieczny et al., 2007) and in animals bearing a full unilateral 6-OHDA lesion of the nigrostriatal tract (Figure S2). In contrast, in animal models of Parkinson's disease where some dopaminergic transmission remains, the action of group III mGlu agonists on GABAergic striatonigral terminals appears to predominate. Thus, Lopez et al. (2007) reported that in 6-OHDA treated rats bearing a partial (∼ 60%) lesion, injection of ACPT-I into the SNpr leads to a worsening of akinesia. Targeting of group III mGlu receptors in the SNpr may therefore have greater impact in the later stages of the disease and further studies with mGlu4- and mGlu8-selective agents in 6-OHDA treated rats bearing lesions of between 60% and 100% will help to clarify this.
The present study also supports a favourable role for mGlu7 receptors in the SNpr to bring about relief of akinesia. A recent study reported that AMN082 (0.1–0.5 nmol) reverses haloperidol-induced catalepsy following injection into the striatum, but not when injected into the globus pallidus (Greco et al., 2010). Therefore, as for mGlu4 receptors, it appears there are multiple complimentary sites of action for mGlu7 agents in the basal ganglia circuitry, though these effects are both likely to reflect inhibition of glutamate release – from overactive corticostriatal or subthalamonigral terminals. As noted for mGlu4, further in vivo microdialysis studies to back up the present in vitro demonstration of mGlu7 receptor-mediated inhibition of glutamate release in the SNpr would strengthen this conclusion. It should be noted that the effective doses in the SNpr (100–300 nmol) are far higher than those required to reverse haloperidol-induced catalepsy in the striatum (0.1–0.5 nmol). While these discrepancies may reflect the different sites of injection and animal models of Parkinson's disease used in the two studies, the high doses of AMN082 required to reverse reserpine-induced akinesia may be suggestive of the involvement of other group III mGlu receptors such as mGlu4, which may be activated at this level (Mitsukawa et al., 2005) and which would be blocked by the non-selective group III mGlu antagonist, CPPG. Although suggestive of a promising role for mGlu7 receptors in the symptomatic treatment of Parkinson's disease, the availability of more selective mGlu7 agonists or potentiators is awaited in order to corroborate these findings.
Consistent with the lack of effect of the mGlu8 agonist (S)-3,4-DCPG in the release studies described above, mGlu8 receptors appear unlikely targets for providing antiparkinsonian relief in the reserpine-treated rat. Contrary to expectations, injection of (S)-3,4-DCPG into the SNpr did produce a dose-dependent reversal of akinesia. However, unlike the responses to PHCCC and AMN082, this effect of (S)-3,4-DCPG was not inhibited by pre-treatment with CPPG, suggesting it was driven through an off-target action of (S)-3,4-DCPG, independent of group III mGlu receptor activation. An action on targets other than mGlu8 also seems likely given the very low level of mGlu8 immunoreactivity witnessed in the SN in our preliminary studies (Figure S1). Although the target of (S)-3,4-DCPG actions has still not been revealed, an additional low affinity non-mGlu, non-iGlu receptor target was proposed for this drug when it was first characterized (Thomas et al., 2001). The present findings are consistent with those of Lopez et al. (2007) who found a similar lack of efficacy of (S)-3,4-DCPG against haloperidol-induced catalepsy following intranigral administration, an effect mirrored by systemic injection of the racemate (R,S)-3,4-DCPG (Ossowska et al., 2004). Furthermore, this lack of target potential of mGlu8 receptors in providing antiparkinsonian relief is not unique to the SNpr; a lack of efficacy of (S)-3,4-DCPG in reversing 6-OHDA-induced akinesia has also been reported following intrapallidal injection (Beurrier et al., 2009). Taken together these findings suggest that, unlike mGlu4 or mGlu7 receptors, mGlu8 receptors appear to hold no potential as targets in the SNpr for bringing about symptomatic relief in Parkinson's disease.
In conclusion, this study reveals that targeting mGlu4 receptors in the SNpr can bring about relief of akinesia in the reserpine-treated rat model of Parkinson's disease and supports inhibition of glutamate release in this region as a key mechanism underpinning this action. While ruling out any such potential for targeting mGlu8 receptors in this region, a role for mGlu7 receptors remains possible and further clarification awaits the introduction of new pharmacological tools for this subtype.
Acknowledgments
P Austin and M Broadstock contributed equally to this work and were in receipt of studentship funding from the Medical Research Council, and Guy's & St Thomas' Charitable Trustees respectively. MJ Betts is funded by a KCL CASE studentship with Lilly Research Laboratories.
Glossary
- 6-OHDA
6-hydroxy dopamine
- AMN082
N,N′-Dibenzhydrylethane-1,2-diamine dihydrochloride
- CPPG
(RS)-α-cyclopropyl-4-phosphonophenylglycine
- (S)-3
4-DCPG, (S)-3,4-dicarboxyphenylglycine
- EPSC
excitatory post synaptic current
- IPSC
inhibitory post synaptic current
- L-AP4
L-2-amino-4-phosphonobutyrate
- L-SOP
O-phospho-L-serine
- mGlu
metabotropic glutamate
- PAM
positive allosteric modulator
- PHCCC
N-phenyl-7-(hydroxyminocyclopropa[b]chromen-1a-carboxamide
- SNpr/pc
substantia nigra pars reticulata/pars compacta
- STN
subthalamic nucleus
Conflict of interest
The authors report no conflict of interest in the content of this manuscript.
Supporting information
Figure S1 Group III metabotropic glutamate(mGlu) receptor immunoreactivity in the striatum and substantianigra pars reticulata (SNpr) of naive rat brain. Representativephotomicrographs of coronal sections are shown for (A)mGlu4, (B) mGlu7 and (C) mGlu8.Boxes on whole brain sections depict regions shown at 50×magnification on right side images. Scale bars denote 200 µmand arrows define the dorsal border of the SN.
Figure S2 Effect of a unilateral injection ofthe broad spectrum group III mGlu agonist, L-serine-O-phosphate(L-SOP), on rotational behaviour in rats bearing a full 6-OHDAlesion of the nigrostriatal tract. Animals were prepared with afull 6-OHDA lesion as described in Messenger et al. (2002).Two weeks post lesion, animals were cannulated above the SNpr asdescribed in the main text of this paper. Five days later, animalsreceived a single unilateral injection of L-SOP (750 nmol in 2.5µL PBS) or vehicle (2.5 µL PBS) into the SNpr and thenumber of contraversive rotations was quantified over 30 min. L-SOPproduced a significant degree of contraversive rotations comparedwith vehicle, indicating clear antiparkinsonian efficacy in thisfully lesioned 6-OHDA model of Parkinson's disease. Thisresponse was consistent with the antiparkinsonian response producedby 750 nmol L-SOP in the reserpine-treated rat model ofParkinson's disease (Austin et al., 2010). Data aremean ± SEM (n = 6 rats per group). ***Indicates asignificant difference (P < 0.001) compared with vehicle(unpaired two-tailed t-test).
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th edn. Br J Pharmacol. 2011;164:S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez L, Macias R, Lopez G, Alvarez E, Pavon N, Rodriguez-Oroz MC, et al. Bilateral subthalamotomy in Parkinson's disease: initial and long-term response. Brain. 2005;128:570–583. doi: 10.1093/brain/awh397. [DOI] [PubMed] [Google Scholar]
- Annoura H, Fukunaga A, Uesugi M, Tatsuoka T, Horikawa Y. A novel class of antagonists for metabotropic glutamate receptors, 7-(hydroxyimino)cyclopropa[b]chromen-1a-carboxylates. Bioorg Med Chem Lett. 1996;6:763–766. [Google Scholar]
- Austin PJ, Betts MJ, Broadstock M, O'Neill MJ, Mitchell SN, Duty S. Symptomatic and neuroprotective effects following activation of nigral group III metabotropic glutamate receptors in rodent models of Parkinson's disease. Br J Pharmacol. 2010;160:1741–1753. doi: 10.1111/j.1476-5381.2010.00820.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benazzouz A, Breit S, Koudsie A, Pollak P, Krack P, Benabid AL. Intraoperative microrecordings of the subthalamic nucleus in Parkinson's disease. Mov Disord. 2002;17(Suppl. 3):S145–S149. doi: 10.1002/mds.10156. [DOI] [PubMed] [Google Scholar]
- Beurrier C, Lopez S, Révy D, Selvam C, Goudet C, Lhérondel M, et al. Electrophysiological and behavioural evidence that modulation of metabotropic glutamate receptor 4 with a new agonist reverses experimental parkinsonism. FASEB J. 2009;23:3619–3628. doi: 10.1096/fj.09-131789. [DOI] [PubMed] [Google Scholar]
- Biggs CS, Fowler LJ, Whitton PS, Starr MS. Extracellular levels of glutamate and aspartate in the entopeduncular nucleus of the rat determined by microdialysis: regulation by striatal dopamine D2 receptors via the indirect striatal output pathway? Brain Res. 1997;753:163–175. doi: 10.1016/s0006-8993(97)00033-4. [DOI] [PubMed] [Google Scholar]
- Brodbeck RM, Sotty F, Pu X, Uberti M, Isaac LK, Schulenburg T, et al. 2010. Characterisation of AMN082: a rich pharmacology which does not appear to include a measurable stimulation of mGlu7 receptorSFN Abstract, Nov 2010: 159.23.
- Conn PJ, Niswender CM. mGluR7's lucky number. Proc Natl Acad Sci USA. 2006;103:251–252. doi: 10.1073/pnas.0510051103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn PJ, Pin J-P. Pharmacology and functions of metabotropic glutamate receptors. Ann Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- Corti C, Aldegheri L, Somogyi P, Ferraguti F. Distribution and synaptic localisation of the metabotropic glutamate receptor 4 (mGluR4) in the rodent CNS. Neuroscience. 2002;110:403–420. doi: 10.1016/s0306-4522(01)00591-7. [DOI] [PubMed] [Google Scholar]
- Duty S. Therapeutic potential of targeting group III metabotropic glutamate receptors in the treatment of Parkinson's disease. Br J Pharmacol. 2010;161:271–287. doi: 10.1111/j.1476-5381.2010.00882.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greco B, Lopez S, van der Putten H, Flor PJ, Amalric M. Metabotropic glutamate 7 receptor subtype modulates motor symptoms in rodent models of Parkinson's disease. J Pharmacol Exp Ther. 2010;332:1064–1071. doi: 10.1124/jpet.109.162115. [DOI] [PubMed] [Google Scholar]
- Hopkins CR, Lindsley CW, Niswender CM. mGluR4-positive allosteric modulation as potential treatment for Parkinson's disease. Future Med Chem. 2009;1:501–513. doi: 10.4155/fmc.09.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchison WD, Allan RJ, Opitz H, Levy R, Dostrovsky JO, Lang AE, et al. Neurophysiological identification of the subthalamic nucleus in surgery for Parkinson's disease. Ann Neurol. 1998;44:622–628. doi: 10.1002/ana.410440407. [DOI] [PubMed] [Google Scholar]
- Konieczny J, Wardas J, Kuter K, Pilc A, Ossowska K. The influence of group III metabotropic glutamate receptor stimulation by (1S,3R,4S)-1-aminocyclopentane-1,3,4-tricarboxylic acid on the parkinsonian like akinesia and striatal proenkephalin and prodynorphin expression in rats. Neuropharmacology. 2007;145:611–620. doi: 10.1016/j.neuroscience.2006.12.006. [DOI] [PubMed] [Google Scholar]
- Kosinski CM, Bradley SR, Conn PJ, Levey AI, Landwehrmeyer GB, Penney JJB, et al. Localisation of metabotropic glutamate receptor 7 mRNA and mGluR7a protein in the rat basal ganglia. J Comp Neurol. 1999;415:266–284. [PubMed] [Google Scholar]
- Lopez S, Turle-Lorenzo N, Acher F, De LE, Mele A, Amalric M. Targeting group III metabotropic glutamate receptors produces complex behavioral effects in rodent models of Parkinson's disease. J Neurosci. 2007;27:6701–6711. doi: 10.1523/JNEUROSCI.0299-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacInnes N, Duty S. Group III metabotropic glutamate receptors act as hetero-receptors modulating GABA release in the globus pallidus in vivo. Eur J Pharmacol. 2008;580:95–99. doi: 10.1016/j.ejphar.2007.10.030. [DOI] [PubMed] [Google Scholar]
- MacInnes N, Messenger MJ, Duty S. Activation of group III metabotropic glutamate receptors in selected regions of the basal ganglia alleviates akinesia in the reserpine-treated rat. Br J Pharmacol. 2004;141:15–22. doi: 10.1038/sj.bjp.0705566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maj M, Bruno V, Dragic Z, Yamamoto R, Battaglia G, Inderbitzin W, et al. (-)-PHCCC, a positive allosteric modulator of mGluR4: characterization, mechanism of action, and neuroprotection. Neuropharmacology. 2003;45:895–906. doi: 10.1016/s0028-3908(03)00271-5. [DOI] [PubMed] [Google Scholar]
- Marino MJ, Williams DL, Jr, O'Brien JA, Valenti O, McDonald TP, Clements MK, et al. Allosteric modulation of group III metabotropic glutamate receptor 4: a potential approach to Parkinson's disease treatment. Proc Natl Acad Sci USA. 2003;100:13668–13673. doi: 10.1073/pnas.1835724100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messenger MJ, Dawson LG, Duty S. Changes in metabotropic glutamate receptor 1-8 gene expression in the rodent basal ganglia motor loop following lesion of the nigrostriatal tract. Neuropharmacology. 2002;43:261–271. doi: 10.1016/s0028-3908(02)00090-4. [DOI] [PubMed] [Google Scholar]
- Mitsukawa K, Yamamoto R, Ofner S, Nozulak J, Pescott O, Lukic S. A selective metabotropic glutamate receptor 7 agonist: activation of receptor signaling via an allosteric site modulates stress parameters in vivo. Proc Natl Acad Sci USA. 2005;102:18712–18717. doi: 10.1073/pnas.0508063102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niswender CM, Johnson KA, Weaver CD, Jones CK, Xiang Z, Luo Q, et al. Discovery, characterisation and antiparkinsonian effect of novel positive allosteric modulators of metabotropic glutamate receptor 4. Mol Pharmacol. 2008;74:1345–1358. doi: 10.1124/mol.108.049551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossowska K, Pietraszek M, Wardas J, Wolfarth S. Potential antipsychotic and extrapyramidal effects of (R,S)-3,4-dicarboxyphenylglycine [(R,S)-3,4-DCPG], a mixed AMPA antagonist/mGluR8 agonist. Pol J Pharmacol. 2004;56:295–304. [PubMed] [Google Scholar]
- Parent A, Hazrati LN. Functional anatomy of the basal ganglia. II. The place of subthalamic nucleus and external pallidum in basal ganglia circuitry. Brain Res Brain Res Rev. 1995;20:128–154. doi: 10.1016/0165-0173(94)00008-d. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. New York: Academic Press; 1998. [Google Scholar]
- Peppe A, Pierantozzi M, Bassi A, Altibrandi MG, Brusa L, Stefani A, et al. Stimulation of the subthalamic nucleus compared with the globus pallidus internus in patients with Parkinson disease. J Neurosurg. 2004;101:195–200. doi: 10.3171/jns.2004.101.2.0195. [DOI] [PubMed] [Google Scholar]
- Pothecary CA, Jane DE, Salt TE. Reduction of excitatory transmission in the retino-collicular pathway via selective activation of mGlu8 receptors by DCPG. Neuropharmacology. 2002;43:231–234. doi: 10.1016/s0028-3908(02)00077-1. [DOI] [PubMed] [Google Scholar]
- Robledo P, Feger J. Acute monoaminergic depletion in the rat potentiates the excitatory effect of the subthalamic nucleus in the substantia nigra pars reticulata but not in the pallidal complex. J Neural Transm Gen Sect. 1991;86:115–126. doi: 10.1007/BF01250572. [DOI] [PubMed] [Google Scholar]
- Sukoff-Rizzo S, Leonard SK, Gilbert A, Dollings P, Smith DL, Zhang M-Y, et al. 2010. The mGluR7 allosteric agonist AMN082 is a monoaminergic agent in disguise!SFN Abstract Nov 2010: 643.28.
- Thomas NK, Wright RA, Howson PA, Kingston AE, Schoepp DD, Jane DE. (S)-3,4-DCPG, a potent and selective mGlu8a receptor agonist, activates metabotropic glutamate receptors on primary afferent terminals in the neonatal rat spinal cord. Neuropharmacology. 2001;40:311–318. doi: 10.1016/s0028-3908(00)00169-6. [DOI] [PubMed] [Google Scholar]
- Toms NJ, Jane DE, Kemp MC, Bedingfield JS, Robinson CS. The effects of (RS)-alpha-cyclopropyl-4-phosphonophenylglycine ((RS)-CPPG), a potent and selective metabotropic glutamate receptor antagonist. Br J Pharmacol. 1996;118:851–854. doi: 10.1111/j.1476-5381.1996.tb15750.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner JP, Salt TE. Group III metabotropic glutamate receptors control corticothalamic synaptic transmission in the rat thalamus in vitro. J Physiol. 1999;519:481–491. doi: 10.1111/j.1469-7793.1999.0481m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugolini A, Large CH, Corsi M. AMN082, an allosteric mGluR7 agonist that inhibits afferent glutamatergic transmission in rat basolateral amygdala. Neuropharmacology. 2008;55:532–536. doi: 10.1016/j.neuropharm.2008.04.020. [DOI] [PubMed] [Google Scholar]
- Valenti O, Mannaioni G, Seabrook GR, Conn PJ, Marino MJ. Group III metabotropic glutamate receptor-mediated modulation of excitatory transmission in the rodent substantia nigra pars compacta dopamine neurons. J Pharmacol Exp Ther. 2005;313:1296–1304. doi: 10.1124/jpet.104.080481. [DOI] [PubMed] [Google Scholar]
- Wittmann M, Marino M, Bradley SR, Conn PJ. Activation of group III mGluRs inhibits GABA-ergic and glutamatergic transmission in the substantia nigra parts reticulata. J Neurophysiol. 2001;85:1960–1968. doi: 10.1152/jn.2001.85.5.1960. [DOI] [PubMed] [Google Scholar]
- Wittmann M, Marino M, Conn PJ. Dopamine modulates the function of group II and group III metabotropic glutamate receptors in the substantia nigra parts reticulata. J Pharmacol Exp Ther. 2002;302:433–441. doi: 10.1124/jpet.102.033266. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.