

Abstract
BACKGROUND AND PURPOSE
Orexin receptors potently signal to lipid messenger systems, and our previous studies have suggested that PLD would be one of these. We thus wanted to verify this by direct measurements and clarify the molecular mechanism of the coupling.
EXPERIMENTAL APPROACH
Orexin receptor-mediated PLD activation was investigated in CHO cells stably expressing human OX1 orexin receptors using [14C]-oleic acid-prelabelling and the transphosphatidylation assay.
KEY RESULTS
Orexin stimulation strongly increased PLD activity – even more so than the phorbol ester TPA (12-O-tetradecanoyl-phorbol-13-acetate), a highly potent activator of PLD. Both orexin and TPA responses were mediated by PLD1. Orexin-A and -B showed approximately 10-fold difference in potency, and the concentration–response curves were biphasic. Using pharmacological inhibitors and activators, both orexin and TPA were shown to signal to PLD1 via the novel PKC isoform, PKCδ. In contrast, pharmacological or molecular biological inhibitors of Rho family proteins RhoA/B/C, cdc42 and Rac did not inhibit the orexin (or the TPA) response, nor did the molecular biological inhibitors of PKD. In addition, neither cAMP elevation, Gαi/o nor Gβγ seemed to play an important role in the orexin response.
CONCLUSIONS AND IMPLICATIONS
Stimulation of OX1 receptors potently activates PLD (probably PLD1) in CHO cells and this is mediated by PKCδ but not other PKC isoforms, PKDs or Rho family G-proteins. At present, the physiological significance of orexin-induced PLD activation is unknown, but this is not the first time we have identified PKCδ in orexin signalling, and thus some specific signalling cascade may exist between orexin receptors and PKCδ.
Keywords: orexin, OX1 receptor, PLD, PKC, PKD, Rho
Introduction
Orexin receptors OX1 and OX2 (nomenclature follows the BJP Guide to Receptors and Channels, Alexander et al., 2011) are typical class A GPCRs. Their native ligands orexin-A and -B are neuropeptides that were classically proposed to control appetite and sleep/wakefulness, but have since also been found to influence many other aspects of neurophysiology such as hormone release, stress and drug-seeking behaviour (Boutrel et al., 2005; Harris et al., 2005; reviewed in Kukkonen et al., 2002). Orexins were first discovered in the hypothalamus but were subsequently observed outside the CNS, including the gastrointestinal tract, kidney, lung and heart (reviewed in Kukkonen et al., 2002; Heinonen et al., 2008). However, their physiological action outside the CNS is not well understood yet.
Orexin receptors appear promiscuous in their signalling. Orexins often depolarize CNS neurons via inhibition of K+ channels or activation of some non-selective cation channels (reviewed in Kukkonen and Åkerman, 2005). Activation of similar receptor-operated channels has been investigated extensively in cultured cells but neither the channels nor the activating signals have thus far been unequivocally identified (Lund et al., 2000; Larsson et al., 2003; Näsman et al., 2006; Peltonen et al., 2009; Turunen et al., 2010; reviewed in Kukkonen and Åkerman, 2005). Ca2+ influx, both in primary cells and in cell lines, is potently activated by orexin receptors, probably through these channels (Lund et al., 2000; Näsman et al., 2006; Ekholm et al., 2007; Peltonen et al., 2009; reviewed in Kukkonen and Åkerman, 2005). Ca2+ elevation is also induced by PLC-triggered Ca2+ release (Lund et al., 2000; Holmqvist et al., 2002; Karteris et al., 2005; Johansson et al., 2008). In addition, orexins very potently activate arachidonic acid release via PLA2 or other (phospho)lipases (Turunen et al., 2010). Indirect evidence has also implicated PLD as a target for orexin signalling (Johansson et al., 2008). Other, probably less potent responses include positive or negative regulation of AC and ERK and p38 MAPK pathways and cell death (Malendowicz et al., 1999; Hilairet et al., 2003; Rouet-Benzineb et al., 2004; Holmqvist et al., 2005; Karteris et al., 2005; Spinazzi et al., 2005; Ammoun et al., 2006a,b; Voisin et al., 2008; Ramanjaneya et al., 2009).
PLD hydrolyzes phosphatidylcholine (PC) to generate choline and the intracellular messenger phosphatidic acid (PA). PA activates type I phosphatidylinositol-4-phosphate 5-kinases (PIP5K), which are primarily needed for PIP2 (phosphatidylinositol-4,5-bisphosphate) production (reviewed in van den Bout and Divecha, 2009). PIP2 is required for membrane binding of PLD as well as for PLD activity, and thus PA-stimulated PIP2 production may constitute a feed-forward loop for PLD activation. Many other proteins possess PA-binding domains, including the kinases mTOR and Raf-1 (reviewed in Jenkins and Frohman, 2005; Wang et al., 2006). PA can be converted to another second messenger, DAG, in a single step by the PA phosphohydrolase activity displayed by lipin isoforms and lipid phosphate phosphatases (Huang et al., 2011; reviewed in Brindley and Pilquil, 2009; Khalil et al., 2010). DAG activates, among other proteins, classical and novel PKC isoforms (c- and nPKC, respectively), PKD isoforms, RasGRPs, munc13, chimaerins and TRPC channels (reviewed in Carrasco and Merida, 2007; Venkatachalam and Montell, 2007). An alternative pathway from PA leads to an extracellular messenger (GPCR ligand), lysophosphatidic acid, catalysed by PA-PLA2 or -PLA1 activity.
There are two classical mammalian isoforms of PLD, PLD1 and PLD2, which exhibit differences in subcellular localization and regulation (see, e.g. Hammond et al., 1995; Colley et al., 1997). PLD1 is activated by PKC (both by phosphorylation and by direct interaction) (Hammond et al., 1997; Sung et al., 1999; Zhang et al., 1999; reviewed in Exton, 1998). In addition, the members of the monomeric G-protein families of Rho (Rho, Rac, cdc42) and Arf (Arf1-6) are thought to be the major stimulants of PLD1 (Hammond et al., 1997; reviewed in Jenkins and Frohman, 2005); some studies also implicate the Ras family protein RalA (see, e.g. Jiang et al., 1995). Regulation of PLD2 is less clear, but may involve some of the same stimuli as PLD1 (reviewed in Jenkins and Frohman, 2005). The known regulation of PLD includes many possible ways of signalling from GPCRs, most obviously via PKC and guanine nucleotide exchange factors for Rho family G-proteins (reviewed in Oude Weernink et al., 2007; Aittaleb et al., 2010).
We have previously reported indirect potential evidence for the involvement of PLD in orexin signalling by measuring orexin receptor-stimulated production of DAG (Johansson et al., 2008). DAG may be one of the major signal substances produced in orexin receptor signalling, with possible action on, for example, the receptor-operated channels (reviewed in Kukkonen and Åkerman, 2005). We therefore sought, in this study, to directly examine PLD activation and determine the underlying mechanisms. We report that orexin receptors potently activate PLD with an absolute dependence on nPKC, probably PKCδ. In contrast, no evidence was obtained for the involvement of the Rho family G-proteins or PKD.
Methods
Test system used
CHO-hOX1 cells, expressing human OX1 receptors (Lund et al., 2000), were cultured in Ham's F12 medium (Gibco, Paisley, UK) supplemented with 100 U·mL−1 penicillin G (Sigma Chemical Co., St Louis, MO, USA), 80 U·mL−1 streptomycin (Sigma), 400 µg·mL−1 geneticin (G418; Gibco) and 10% (v/v) fetal calf serum (Gibco) at 37°C in 5% CO2 in an air-ventilated humidified incubator on plastic culture dishes (56 cm2 bottom area; Greiner Bio-One GmbH, Frickenhausen, Germany). For the PLD experiments, the cells were cultured on six-well plates (9.6 cm2 well bottom area; Greiner) and for the PLC and cAMP assays, on 24-well plates (Greiner). CHO cells were chosen for the studies due to previously performed systematic characterization of OX1 receptor signal transduction in these cells (see Introduction) and the easy manipulation of these cells.
Plasmids and transfection
CHO-hOX1 cells were transiently transfected to introduce inhibitors of the Rho family members Rho, Rac and cdc42 and PKD. The Rho family inhibitor plasmids used were: pCMV-Myc J3 RhoA(T19N) (dominant-negative RhoA; inhibits RhoA, -B and -C), pEGFP-C1 Rhotekin-RBD (Rho-binding domain of Rhotekin; inhibits RhoA, -B and -C), pEF-C3 (Clostridium botulinum C3-exoenzyme; inactivates RhoA, -B and -C), pCMV-Myc J3 Rac1(T17N) (dominant-negative Rac1; inhibits Rac1, -2 and -3), pEGFP-C1 POSH RBD (Rac-binding domain of POSH; inhibits Rac1, -2 and -3), pCMV-Myc J3 Cdc42(T17N) (dominant-negative cdc42; inhibits cdc42) and pEGFP-C1 Pak1 PBD (p21-binding domain of Pak1; inhibits Rac1, -2 and -3 and cdc42). All dominant-negative Rho, Rac and cdc42 plasmids, pEGFP-C1 POSH RBD and pEGFP-C1 Rhotekin-RBD were from Dr Krister Wennerberg [Institute for Molecular Medicine Finland (FIMM), Helsinki, Finland], pEF-C3 from Alan Hall (Memorial Sloan-Kettering Cancer Center, New York, NY, USA) and pEGFP-C1 Pak1 PBD from Jonathan Chernoff (Fox Chase Cancer Center, Philadelphia, PA, USA). PKD was inhibited in the same manner by dominant-negative (ATP-site mutant) plasmids pEGFP-PKD1-KD (K618N), pEGFP-PKD2-KD (K580A) and pEGFP-PKD3-K605A, of which the two first were from Dr Vivek Malhotra (Center for Genomic Regulation, Barcelona, Spain) and the last one from Dr Osvaldo Rey (University of California at Los Angeles, USA) (Liljedahl et al., 2001; Yeaman et al., 2004; Rey et al., 2006). The free G-protein Gβγ-subunits were sequestered using expression of pcDNA Gtr-α (α-subunit of rod transducin; referred here as Gαt) or pcDNAIII T8-βARK [a fusion of the extracellular and transmembrane part of CD8 and the C-terminus of the human βARK1 (β-adrenoceptor kinase 1)] (Crespo et al., 1995), which were from Guthrie cDNA Resource Center (currently Missouri S&T cDNA Resource Center; http://www.cdna.org) and Dr J. Silvio Gutkind (National Institutes of Health, Bethesda, MD USA), respectively. pcDNA Gβ1 and pcDNA Gγ2 were from Guthrie cDNA Resource Center.
CHO cells on six-well plates were grown to 40–50% confluence and transfected in Ham's F12 with 100 µL of OPTI-MEM (Gibco), FuGeneHD (Roche, Mannheim, Germany) and DNA mixture [with 0.208 µg·cm2 DNA and 0.52 µL·cm2 FuGeneHD (optimized conditions)]. pEGFP-C1 (Clontech, Palo Alto, CA, USA; 10% of the total DNA) was used as a marker to follow transfection efficacy if the construct itself did not contain GFP. Transfection efficacy was calculated to be approximately 60–80%. Control cells were mock-transfected with empty plasmids. Transfection time was 24 or 48 h; 24 h transfection was used to avoid the apparent detrimental effect of some constructs on cell viability.
PLD assay
Phosphatidic acid can be produced not only by PLD but also by phosphorylation of DAG by DAG kinase or through acylation of lysophosphatidic acid (reviewed in Wang et al., 2006). To address this issue, we utilized the transphosphatidylation assay, which measures only PLD activity (reviewed in Morris et al., 1997). PLDs normally utilize water in the hydrolysis reaction; however, primary alcohols, when present, are preferred substrates, by 1000-fold. In this reaction, a phosphatidylalcohol [here phosphatidylbutanol (PtdBut)], instead of PA, is produced. The phosphatidylalcohol generated is a dead-end product and can therefore be measured in a cumulative manner (reviewed in Morris et al., 1997). For this assay, the cells were labelled with [14C]-oleic acid ([1-14C]-oleic acid) in cell culture medium (Ham's F12) 16 h before the experiments. [14C]-oleic acid loading medium was removed from the cells and normal Ham's F12 added 1 h before the experiment. When inhibitors of signalling were used, the cells were pre-incubated in Ham's F12 containing the inhibitor for an additional 30 min before being stimulated. In the case of GGTI-2133 (a geranylgeranyl transferase I inhibitor), the cells were incubated with the inhibitor (together with the [14C]-oleic acid) for 16 h before the experiment (to reduce the mature activatable pool of the geranylgeranylated G-proteins). The cells were activated by removing the medium from the cells and replacing it with activation solution (Ham's F12 medium containing 0.3% 1-butanol), and the inhibitors (in experiments where they were used) and stimulators [orexin, TPA (12-O-tetradecanoyl-phorbol-13-acetate), KAC1-1 (cPKC peptide activator), KAD1-1 (PKCδ peptide activator), KAE1-1 (PKCε peptide activator), thapsigargin or ionomycin or vehicle only]. In the experiments shown in Figure 10A, HBM (HEPES-buffered medium; 137 mM NaCl, 5 mM KCl, 1 mM CaCl2, 1.2 mM MgCl2, 0.44 mM KH2PO4, 4.2 mM NaHCO3, 10 mM glucose and 20 mM HEPES adjusted to pH 7.4 with NaOH) was used instead, as this allows easier control of the Ca2+ concentrations. 3 µM Ca2+ indicates nominally Ca2+-free HBM (no CaCl2 added, free Ca2+ concentration 2.5–3.3 µM) whereas ≍140 nM Ca2+ was achieved by adding 1.5 mM EGTA to the regular HBM (Johansson et al., 2007; Turunen et al., 2010). After a 30 min incubation, the activation solution was removed and rapidly replaced with 300 µL of ice-cold methanol, and the plates placed on ice. The cells were scraped off and collected in Eppendorf tubes, and each dish washed with another 300 µL of methanol, which was pooled together with the first sample.
Figure 10.
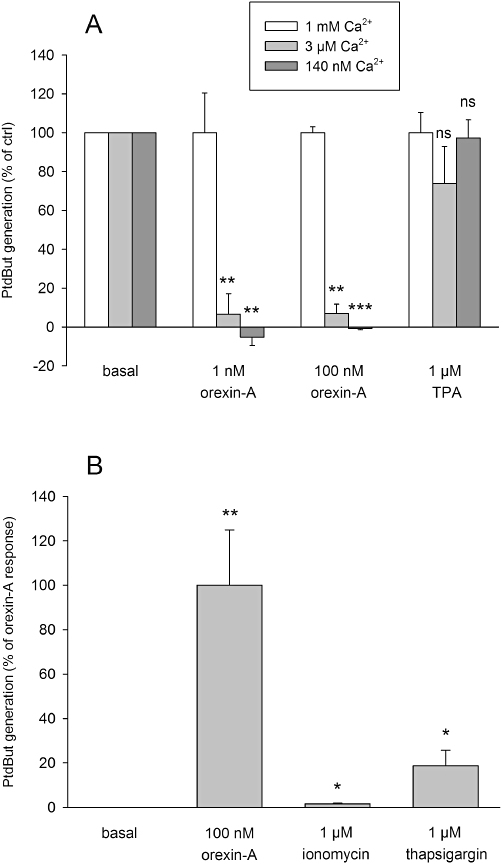
The effect of Ca2+ on PLD activation. (A) The effect of reduction of the extracellular Ca2+ concentration to 3 µM or 140 nM from the regular 1 mM. The comparisons are to the corresponding control (same stimulus in the presence of 1 mM Ca2+). The experiments were performed in HBM instead of cell culture medium. (B) The ability of the Ca2+-elevating compounds, the Ca2+ ionophore, ionomycin and the SERCA (sarco-/endoplasmic reticulum Ca2+ ATPase) inhibitor, thapsigargin, to activate PLD. The comparisons are to basal. ns, not significant; *P < 0.05; **P < 0.01; ***P < 0.001.
Lipid extraction and TLC
Chloroform (500 µL) was added to the Eppendorf tubes. The tubes were vortex-mixed and incubated at room temperature for 15 min. Then 400 µL of water was added, tubes were vortex-mixed again and centrifuged (5 min at 13 500× g). The upper phase was removed and the lower phase containing lipids dried under a stream of nitrogen. Lipids were then dissolved in chloroform : methanol (19:1) and 5 µg of ‘cold’ standard PtdBut (in chloroform) was added to each sample. The samples were applied to TLC plates (Silicagel 60, Merck, Darmstadt, Germany), which had been pretreated with 1% K+-oxalate in [2:3 (v/v) methanol : water] and dried at 110°C for 1 h. Plates were developed in the organic phase of ethylacetate : isooctane : acetic acid : water (11:5:2:10) in an unlined chromatography tank.
Lipid quantification
TLC plates were quantified by two methods, ‘direct reading’ of the radioactivity from the imaging plate exposed with the TLC plate, and conventional scraping of the TLC plate and scintillation counting. Both methods gave similar results. Briefly, after the development, the plates were vacuum-dried and placed overnight with an imaging plate (BAS-MS, Fujifilm, Tokyo, Japan). The imaging plate was scanned with FLA 5100 scanner (Fujifilm) and the band areas and intensities were measured with Nikon NIS-Elements AR. The background was subtracted from the intensity; PtdBut level = (intensityband− intensitybackground) × areaband. The plates were then placed in an iodine vapour tank to visualize the lipid bands. The bands were humidified with water, the PtdBut bands scraped into scintillation vials, and the radioactivity extracted with scintillation cocktail (HiSafe 3, Wallac-PerkinElmer, Turku, Finland) and determined with a Wallac 1414 liquid scintillation counter.
PLC assays
Total inositol phosphate release was measured essentially as described by Johansson et al. (2008). Briefly, membrane phosphoinositides were prelabelled with 3 µCi·mL−1[3H]-inositol ([3H]-myo-inositol) for 20 h, after which the cells were washed and incubated at 37°C containing 10 mM LiCl (to inhibit inositol monophosphatase) for 10 min. The cells were then stimulated with orexin-A for 20–30 min. The reactions were stopped by rapid removal of the medium, addition of 200 µL of 0.4 M ice-cold perchloric acid and freezing. Thawed samples were neutralized with 100 µL of 0.36 M KOH + 0.3 M KHCO3, the insoluble fragments spun down (10 min, 1100× g, +4°C), and the total inositol phosphate fraction isolated by anion-exchange chromatography. The radioactivity of the inositol phosphate fraction was determined by scintillation counting as above.
AC assay
Cellular ATP was prelabelled with 5 µCi·mL−1[3H]-adenine for 2 h in culture medium, after which the cells were washed with PBS and incubated at 37°C in culture medium containing 500 µM IBMX (a cyclic nucleotide PDE inhibitor) for 10 min. The cells were then stimulated for 30 min at 37°C – that is, the same time as in the PLD assay – after which the reactions were stopped by rapid removal of the medium, addition of 300 µL of 0.33 M perchloric acid and freezing. The insoluble fragments of the thawed samples were spun down (10 min, 1100× g, room temperature) and the [3H]-ATP +[3H]-ADP and [3H]-cAMP fractions of the cell extracts isolated by sequential Dowex/alumina chromatography (see, e.g. Holmqvist et al., 2005). Radioactivity was determined using scintillation counting; the conversion of [3H]-ATP to [3H]-cAMP was calculated as a percentage of the total eluted [3H]-ATP +[3H]-ADP.
Data analysis and statistical procedures
All the data are presented as mean ± SE; n refers to the number of batches of cells. Each experiment was performed in duplicate (PtdBut generation), triplicate (cAMP generation) or quadruplicate (inositol phosphate release) at least three times. All the data presented are summarized from at least three batches of cells unless specifically indicated to be representative data from a single experiment. Student's two-tailed t-test with Bonferroni correction for multiple corrections was used in all pairwise comparisons. Significances are as follows: ns (not significant), P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001. Microsoft Excel was used for the nonlinear curve-fitting. The equations used were
| (eq. 1) |
and
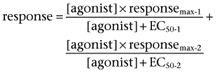 |
(eq. 2) |
Fits obtained with these models were compared using the F-test individually for each batch of cells. The effects of inhibitors on the OX1 receptor or TPA-induced PLD activity were calculated from the formula: PtdBut generation [as % of the ctrl (non-inhibited)]= (stimulantinhibitor− basalinhibitor)/(stimulantctrl− basalctrl) × 100% (Turunen et al., 2010). However, the basal values were often essentially 0. In this manner, the untreated stimulant controls (basal, 1 nM orexin-A, 100 nM orexin-A, 1 µM TPA, 200 nM KAE-1) were set to 100% and complete inhibition to 0%.
Drugs and chemical reagents
Human orexin-A and -B were from NeoMPS (Strasbourg, France), MAFP (methyl arachidonyl fluorophosphonate), PLD1i (PLD1 inhibitor; CAY10593; compound 69 of (Scott et al., 2009); N-[2-[4-(5-chloro-2,3-dihydro-2-oxo-1H-benzimidazol-1-yl)-1-piperidinyl]-1-methylethyl]-2-naphthalenecarboxamide) and PLD2i (PLD2 inhibitor; CAY 10594; compound 72 of (Scott et al., 2009); N-[2-(4-oxo-1-phenyl-1,3,8-triazaspiro[4,5]dec-8-yl)ethyl]-2-naphthalenecarboxamide) were from Cayman Europe (Tallinn, Estonia) and rottlerin (3′-[(8-cinnamoyl-5,7-dihydroxy-2,2-dimethyl-2H-1-benzopyran-6-yl)methyl]-2′,4′,6′-trihydroxy-5′-methylacetophenone), SB-334867 (1-[2-methylbenzoxazol-6-yl]-3-[1,5]naphthyridin-4-yl-urea HCl), thapsigargin and U-73122 (1-(6-[([17b]-3-methoxyestra-1,3,5[10]-trien-17-yl)amino]hexyl)-1H-pyrrole-2,5-dione) from Tocris Cookson Ltd (Avonmouth, UK). Ionomycin, GF109203X (=bisindolylmaleimide I = Gö6850 = 2-[1-(3-dimethylaminopropyl)-1H-indol-3-yl]-3-(1H-indol-3-yl)-maleimide) and Gö6976 (5,6,7,13-tetrahydro-13-methyl-5-oxo-12H-indolo[2,3-a]pyrrolo[3,4-c]carbazole-12-propanenitrile) were from Calbiochem (La Jolla, CA, USA) and GGTI-2133 (N-[[4-(imidazol-4-yl)methylamino]-2-(1-naphthyl)benzoyl]leucine trifluoroacetate salt), forskolin, IBMX, Pertussis toxin (PTx), TPA and EGTA from Sigma-Aldrich (St. Louis, MO, USA). Cell-permeant PKC peptide inhibitors and activators KAC1-1 (cPKC activator), KIC1-1 (cPKC inhibitor), KAD1-1 (PKCδ activator), KAE1-1 (PKCε activator), KIE1-1 (PKCε inhibitor) were from KAI Pharmaceuticals (South San Francisco, CA, USA). PtdBut was from BIOMOL (Enzo Life Sciences, Plymouth Meeting, PA, USA), [14C]-oleic acid and [3H]-myo-inositol (PT6–271) from PerkinElmer (Boston, MA, USA) and [3H]-adenine from Amersham Biosciences (Buckinghamshire, UK).
Results
OX1 stimulation potently and efficaciously activates PLD1
In resting cells, very little PtdBut was detected when viewing the imaging plates; thus, the basal PLD activity was very low (Figure 1A). However, when the cells were stimulated with orexin-A (Figure 1A,B) or TPA (as shown in Figure 4C) a robust PtdBut production was observed. A weak production of PtdBut was sometimes detected even with 0.01 nM orexin-A. Orexin-A was approximately 10-fold more potent than orexin-B, as would be expected for the OX1 receptor (Figure 1B; see Discussion). The concentration–response curves in each experiment with both orexin-A and -B were very shallow, and could therefore be fitted with a single-site equations only with a slope factor (Hill coefficient) <1 [Methods, eq. (1)] or with a two-site equation without slope factor [Methods, eq. (2)]. For orexin-A, in all four experiments performed, the two-site fit was significantly better than the single-site fit with slope factor (P < 0.05 or P < 0.01; F-test). For orexin-B, in three of six experiments, the two-site fit was significantly better than the single-site fit with slope factor (P < 0.01 or P < 0.001; F-test) and in three resting ones it was not. With this result, we considered the two-site fit to be generally favoured. The averaged analysis data are presented in Table 1. Figure 1B presents data from a single representative experiment in order to demonstrate that the biphasic shape of the curve is not an artefact resulting from averaging of data from different experiments. In contrast, the concentration–response curves for PLC activation with orexin-A or orexin-B were not biphasic, although sometimes slightly shallow (Figure 1C).
Figure 1.
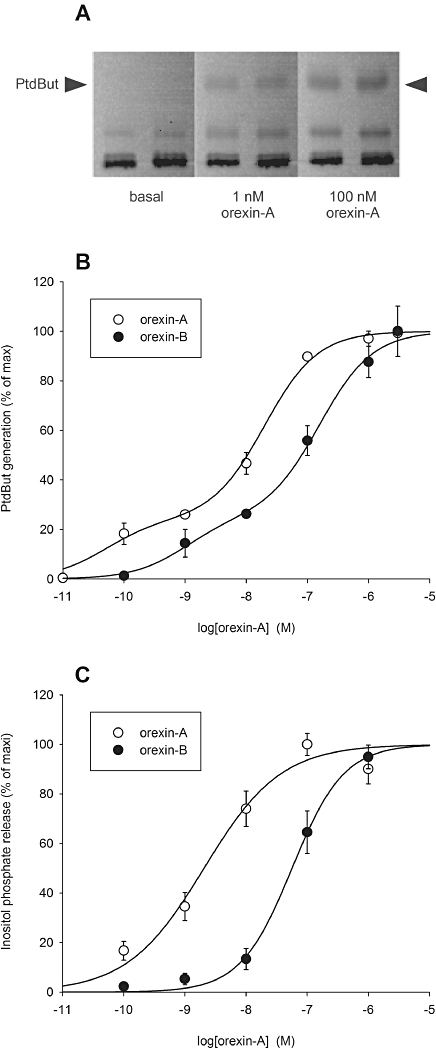
Orexins strongly stimulate PLD and PLC activation. (A) PtdBut generation as viewed on the radioactivity image of a TLC plate read from the imaging plate. The results are from a representative experiment. (B) Concentration–response curves of PLD activation for orexin-A and -B. The parameters for the two-site fit (eq. 2): orexin-A: pEC50-1= 10.3 (24%), pEC50-2= 7.7 ± 0.2 (76%); orexin-B: pEC50-1= 9.0 (25%), pEC50-2= 6.8 (75%). Data from a single representative experiment are presented in order to demonstrate that the biphasic shape of the curve is not an artefact resulting from averaging of data from different experiments. (C) Concentration–response curves of PLC activation for orexin-A and -B (n= 4). The parameters for the single-site fit with slope factor (eq. 1): orexin-A: pEC50= 8.7, nH= 0.69; orexin-B: pEC50= 7.2, nH= 1.0.
Figure 4.
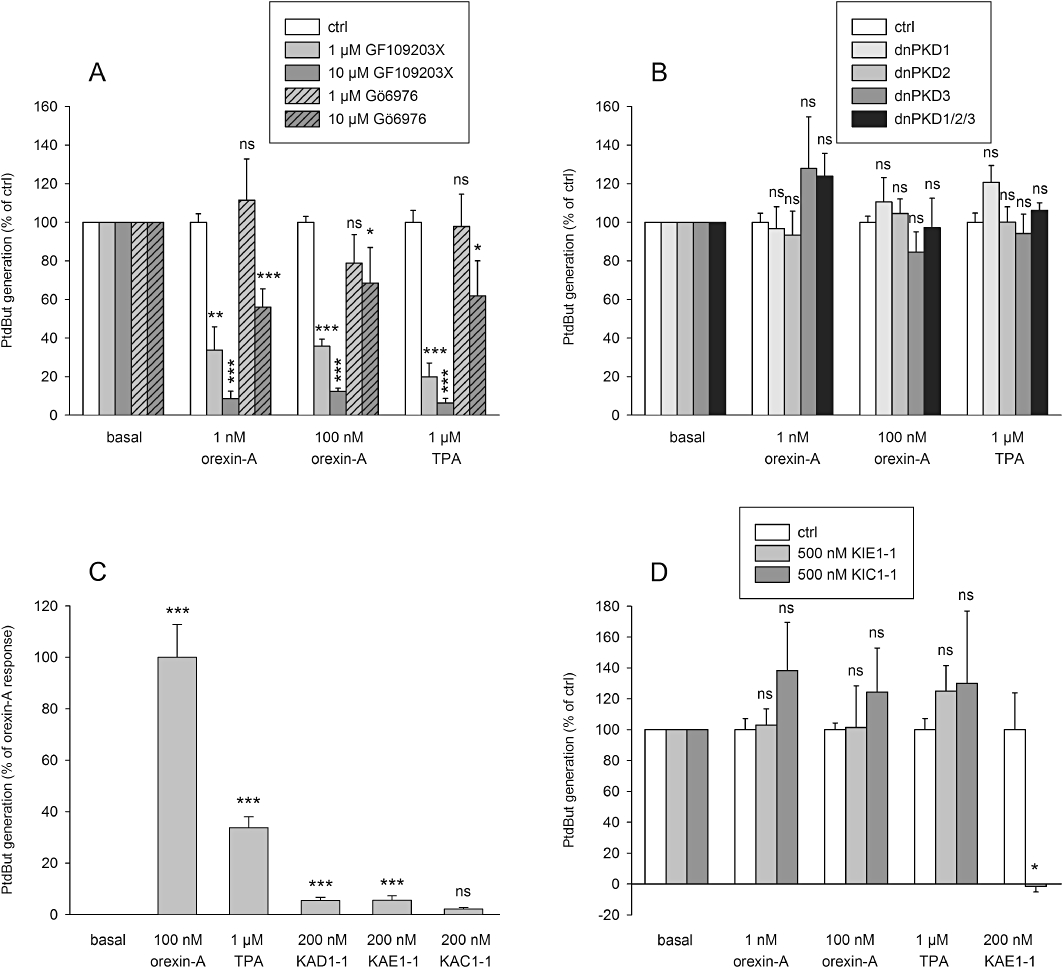
The role of PKC and PKD in PLD stimulation. (A) The effect of the nPKC inhibitor, GF109203X, and the cPKC and PKD inhibitor, Gö6076, on orexin- and TPA-induced PLD activities. (B) The effect of dominant-negative PKD constructs on orexin- and TPA-induced PLD activities. (C) PLD activity in response to the peptide PKC activators of PKCδ (KAD1-1), PKCε (KAE1-1) and cPKC (KAC1-1) as compared with orexin-A and TPA. (D) The effect of the peptide PKC inhibitors of PKCε (KIE1-1) and cPKC (KIC1-1) on orexin-, TPA- and KAE1-1-induced PLD activity. Please note that the effect of KIC1-1 on KAE1-1 was not tested, and thus that column is altogether missing. The comparisons are to the corresponding controls (same stimuli in the absence of the inhibitor) (A, B, D) or to the basal (C). ns, not significant; *P < 0.05; **P < 0.01; ***P < 0.001.
Table 1.
Results from the analysis of the concentration–response data for orexin-A and orexin-B
| 1-site | 2-site | |||||
|---|---|---|---|---|---|---|
| pEC50 | nHill | pEC50-1 | Fraction (%) | pEC50-2 | Fraction (%) | |
| Orexin-A | 7.65 ± 0.25 | 0.62 ± 0.15 | 10.5 ± 0.1 | 21 ± 6 | 7.2 ± 0.2 | 79 ± 6 |
| Orexin-B | 6.65 ± 0.21 | 0.81 ± 0.17 | 9.2 ± 0.2 | 18 ± 6 | 6.3 ± 0.2 | 82 ± 6 |
n is 4 for orexin-A and 6 for orexin-B.
Due to the apparent presence of two components in the orexin-A response, we decided to further study the molecular mechanism of the orexin-A response utilizing 1 and 100 nM orexin-A. The OX1-selective antagonist SB-334867 concentration-dependently inhibited the response (not shown) and full inhibition of the response to both 1 and 100 nM orexin-A was obtained with 10 µM SB-334867 (Figure 2). This confirmed that the response was mediated by the OX1 receptors.
Figure 2.
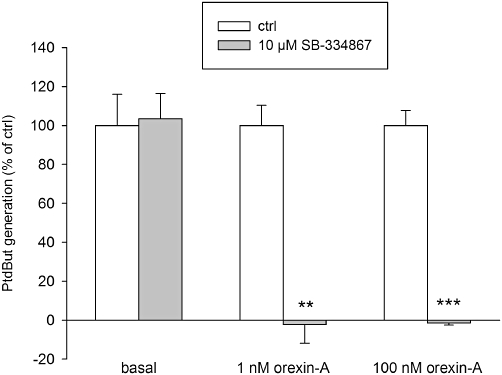
The OX1 orexin receptor antagonist SB-334867 effectively blocks the orexin-A-induced PLD activity. The comparisons are to the corresponding controls (same stimuli in the absence of the inhibitor). **P < 0.01; ***P < 0.001.
We next tested whether the response was mediated by PLD1 or PLD2 using pharmacological inhibitors of each isoform (here referred as PLD1i and PLD2i, respectively). Neither inhibitor shows absolute selectivity for either isoform, but PLD1i has been suggested to exhibit higher selectivity (20- to 160-fold for PLD1 over PLD2) as compared with PLD2i (10- to 40-fold for PLD2 over PLD1) (Scott et al., 2009). PLD2i is less potent than PLD1i (Scott et al., 2009), and was therefore applied at 3.3-fold higher concentrations to compensate for this. Still, PLD1i was much more potent as an inhibitor for both orexin and TPA responses (Figure 3), suggesting that PLD1 is the sole isoform mediating orexin- and TPA-induced PtdBut generation. PLD2i modestly inhibited the orexin response at 1 µM, but this was probably a result of its partial inhibition of PLD1 at this concentration (Scott et al., 2009).
Figure 3.
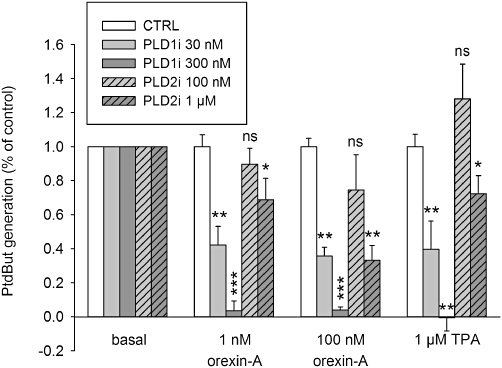
The effect of the PLD isoform-selective inhibitors, PLD1i and PLD2i, on orexin- and TPA-induced PLD activities. The comparisons are to the corresponding controls (same stimuli in the absence of the inhibitor). ns, not significant; *P < 0.05; **P < 0.01; ***P < 0.001.
PLD1 activation by OX1 and TPA is mediated by PKCδ
GF109203X potently inhibits both the conventional PKCs (cPKC; PKCα, -βI, -βII, -γ) and novel PKCs (nPKC; PKCδ, -ε, -η, -θ) whereas Gö6976 should only inhibit the cPKC subfamily (Martiny-Baron et al., 1993; Zang et al., 1994). GF109203X and Gö6976 were used to determine which subtypes might be involved in the activating signal from OX1 to PLD (Figure 4A). GF109203X inhibited both orexin- and TPA-induced PLD activity very potently: 1 µM resulted in more than half-maximal inhibition and 10 µM produced essentially complete inhibition (Figure 4A). In contrast, Gö6976 was weaker, with some inhibition seen only at 10 µM (Figure 4A).
Gö6976, unlike GF109203X, is also able to inhibit PKD enzymes (at least PKD1 and PKD3) (Gschwendt et al., 1996; Chen et al., 2005), and it has been suggested that orexin receptors stimulate PKD1 and -3 (Peltonen et al., 2010). We transfected CHO cells with dominant-negative constructs of PKD1, -2 and -3 to examine if the inhibitory effect of Gö6976 on PLD activation (Figure 4A) results from PKD inhibition. The PKD constructs, alone or together, did not inhibit orexin-A-mediated PLD activation (Figure 4B).
These results suggest that nPKCs play a major part in the response but cPKCs may also be involved. We have previously shown that CHO-hOX1 cells express only the δ- and ε-isoforms of nPKCs; PKCδ, specifically, is suggested to be activated very potently in OX1 receptor signalling to AC (Holmqvist et al., 2005). To more selectively evaluate the role of specific PKC isoforms in orexin receptor signalling to PLD, we turned to RACKs (receptors for activated C-kinase)-based PKC activator and inhibitor peptides (reviewed in Churchill et al., 2009). Activators of PKCδ and PKCε (KAD1-1 and KAE1-1 respectively) produced small but significant PLD activation (Figure 4C). KAC1-1, activator of cPKCs, produced still a smaller stimulation of PLD activity. This suggests that both of the nPKC isoforms expressed, PKCδ and -ε, and also cPKCs, are able to induce PLD activity in CHO cells. However, inhibitors of PKCε and cPKCs, KIE1-1 and KIC1-1, respectively, did not inhibit orexin- or TPA-induced PLD activation (Figure 4D). As a control to this, KIE1-1 inhibited KAE1-1-induced PLD activation (Figure 4D). Thus, orexin receptor stimulation (or TPA)-mediated signalling to PLD does not seem to be mediated via PKCε or any cPKC.
As neither PKCε, cPKCs nor PKDs appear to play any part in the orexin-induced PLD response, it seems likely that the other nPKC isoform expressed in CHO-hOX1 cells, PKCδ, underlies the orexin (and TPA) response. Unfortunately, the selective peptide inhibitor of PKCδ, KID1-1 (KAI-9803; also from KAI Pharmaceuticals), is not available. We therefore turned to another small-molecular PKCδ inhibitor, rottlerin. Rottlerin produced a concentration-dependent inhibition of the orexin and TPA responses (Figure 5), with a potency similar to its reported IC50 for PKCδ (Gschwendt et al., 1994).
Figure 5.
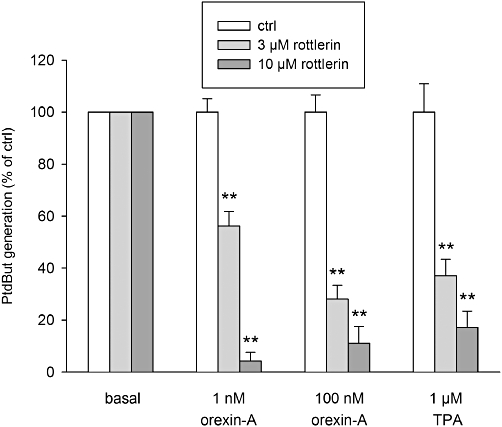
The PKCδ inhibitor, rottlerin, produces a concentration-dependent inhibition of the PLD responses to orexin-A and TPA. The comparisons are to the corresponding control (same stimulus in the absence of the inhibitor). **P < 0.01.
PKCδ activation by OX1 does not occur via PLC or arachidonic acid
We next investigated the mechanism through which PKCδ activation stimulates PLD via OX1 receptors. The most likely translocating and activating stimulus for nPKC is DAG as we previously showed this to occur in CHO cells with high potency upon OX1 receptor stimulation (Ammoun et al., 2006a; Johansson et al., 2008). Both PLC (directly) and PLD (via PA) activities are able to generate DAG (see Introduction) and both appear to drive membrane translocation of PKC in CHO cells (Johansson et al., 2008). PLC is activated very much in the same orexin concentration range as PLD (Johansson et al., 2008), which was also verified in this study (Figure 1C). To evaluate the role of PLC in PLD activation, we exposed the cells to the PLC inhibitor U-73122. No inhibition of PLD was seen (Figure 6) although PLC was fully inhibited (not shown). Arachidonic acid has also been suggested to facilitate PKC activation (Shirai et al., 1998; Cho and Stahelin, 2006), so, next we treated CHO cells with the PLA2 inhibitor MAFP, which fully blocks orexin-A-induced arachidonic acid release (Turunen et al., 2010). Similar to U-73122, MAFP did not inhibit PLD activation in response to orexin-A (Figure 6).
Figure 6.
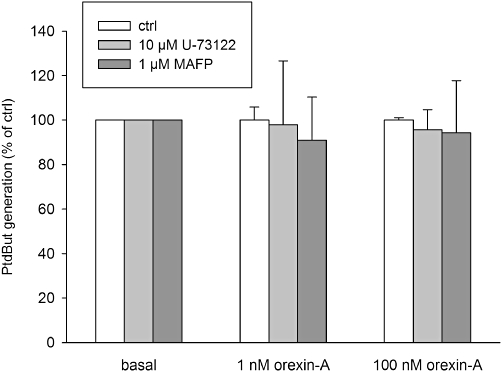
Investigations on the activation mechanisms for the PKC in PLD activation utilizing the PLC inhibitor U-73122 and the PLA2 inhibitor MAFP. Neither treatment produced significant inhibition as compared with the corresponding controls.
Rho family GTP-binding proteins are not involved in PLD1 activation by OX1
There is extensive evidence implicating involvement of Rho family proteins in PLD activation. Geranylgeranyl transferase I inhibitors inhibit membrane localization of all geranylgeranylated proteins and therefore activation of all geranylgeranylated (but not farnesylated) monomeric G-proteins. Rho family proteins are exclusively geranylgeranylated, and we tested their involvement in the orexin signalling to PLD using GGTI-2133 (Vasudevan et al., 1999). Isoprenylation is a part of the protein maturation and we therefore incubated the cells for 24 h with GGTI-2133 to inhibit this in the early phase. Cell shape changes observed during this time indicated that the inhibitor was effective (not shown); a longer incubation time was not performed due to concerns regarding toxicity. GGTI-2133 (100 nM) did not decrease the ability of orexin-A or TPA to activate PLD (Figure 7A). However, a non-significant effect was observed at 1 µM (Figure 7A). We therefore further focused on the Rho family using more selective inhibitors. Inhibition of the classical Rho family members, Rac1/2/3 or cdc42, with dominant-negative constructs did not have significant effect (Figure 7B). However, inhibition of RhoA/B/C with dominant-negative RhoA had a small effect (Figure 7B). Nonetheless, inhibitors considered to be even more selective, such as C. botulinum C3-exoenzyme (RhoA/B/C), Rhotekin-RBD (RhoA/B/C), POSH-PBD (Rac1/2/3) and Pak1-PBD (Rac1/2/3 and cdc42), did not inhibit orexin-A- or TPA-mediated PLD activation (Figure 7C).
Figure 7.
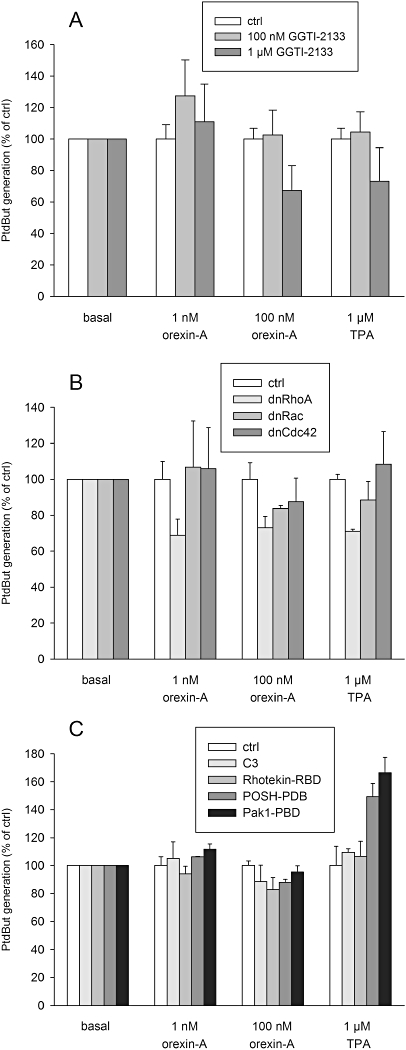
Investigations on the involvement of the Rho family monomeric G-proteins in the PLD response to orexin and TPA. (A) The effect of the geranylgeranyl transferase I inhibitor GGTI-2133. The cells were incubated with GGTI-2133 for 24 h before the assay. (B) The effect of the dominant-negative (dn) Rho, Rac and cdc42 constructs. The cells were transfected with the constructs 24 h before the assay. (C) The effect of C3-exoenzyme (C3; inhibitor of Rho), Rhotekin-RBD (inhibitor of Rho), POSH-PBD (inhibitor of Rac) and Pak1-PBD (inhibitor of Rac and Cdc42). The cells were transfected with the constructs 24 h before the assay. No treatment/transfection produced significant inhibition as compared with the corresponding controls.
AC activation is not involved in PLD1 stimulation
AC is activated by orexin receptor stimulation in CHO cells (Holmqvist et al., 2005). However, as previously shown (Holmqvist et al., 2005), the response occurs with very low potency in non-Gs-stimulated cells and essentially no response was seen with 10 nM orexin-A (Figure 8A; see Discussion), which potently activates PLD (compare with Figure 1B). The strong AC stimulator, forskolin, did not activate PLD (Figure 8B). We therefore conclude that cAMP elevation is not involved in the activation of PLD by orexin receptor stimulation, which is consistent with the finding that PKCδ lies upstream of AC activation in orexin receptor signalling.
Figure 8.
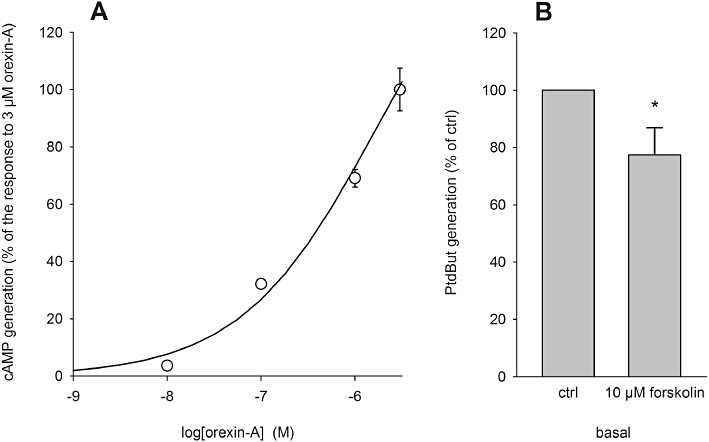
Investigations on the involvement of cAMP in the PLD response to orexin. (A) Orexin receptor stimulation of AC. The results are from a representative experiment. In the lack of apparent saturation in the concentration range used, the data are normalized to the response with 3 µM orexin-A. (B) Basal PLD activity in response to the AC activator forskolin. *P < 0.05.
Gi/o-protein signalling is not involved in PLD1 stimulation
It has been suggested that orexin receptors also engage Gi/o-proteins in their signalling (Holmqvist et al., 2005; Karteris et al., 2005). The Gi/o-inactivator, PTx (100 ng·mL–1, 24 h), only very weakly inhibited the PLD response to 100 nM orexin-A, strongly suggesting that Gi/o-proteins are not significantly involved in this signalling pathway (Figure 9A). Gi/o-proteins usually underlie Gβγ-signalling, but Gβγ may also be derived from other G-proteins. We thus evaluated this possibility by overexpressing both Gβγ-subunits and Gβγ-scavengers. Overexpressed Gβ1γ2 alone did not stimulate PLD (Figure 9B). The well-known Gβγ-scavengers Gαt and T8-βARK only weakly affected orexin (and TPA) activation of PLD (Figure 9B). We thus consider it unlikely that Gβγ-signalling significantly contributes to PLD activation via orexin receptors.
Figure 9.
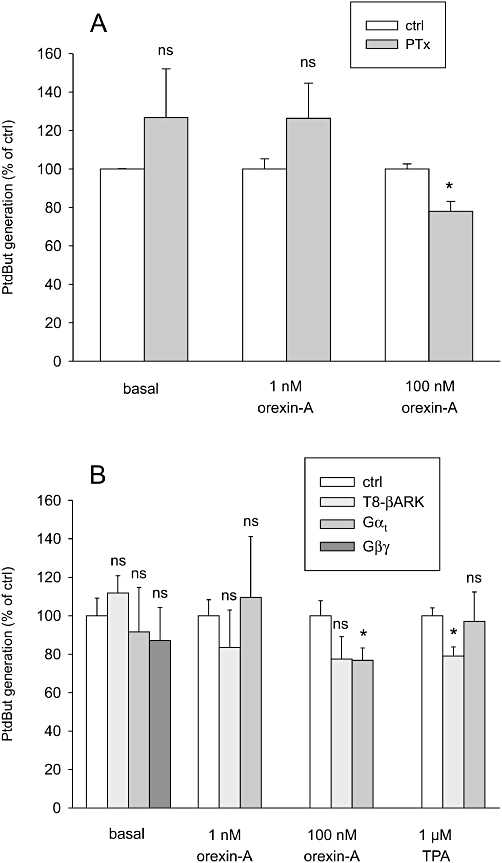
Investigations on the involvement of Gαi//o- and Gβγ-proteins in the PLD stimulation. (A) The effect of PTx pretreatment (100 ng·mL–1, 24 h) on the orexin response. (B) The effect of exogenous Gβ1γ2- and Gβγ-scavengers Gαt and T8-βARK on PLD activity. The cells were transfected with the constructs 24 h before the assay. The comparisons are to the corresponding controls (same stimuli in the ctrl cells). ns, not significant; *P < 0.05.
The role of Ca2+ signalling in the PLD1 activation
OX1 receptor stimulation induces a marked rise in intracellular Ca2+ concentration via both Ca2+ influx and Ca2+ release; especially significant seems to be the receptor-operated Ca2+ influx (see Introduction). Reduction of the extracellular Ca2+ concentration to 2–3 µM essentially abolished orexin-A-induced PLD stimulation, but had no effect on the TPA response (Figure 10A). Ca2+ influx, especially via the receptor-operated pathway, has previously been shown to be an obligatory requirement for coupling of orexin receptors to many responses including PLC (Lund et al., 2000; Johansson et al., 2007), ERK (Ammoun et al., 2006a) and arachidonic acid release (Turunen et al., 2010). In contrast, Ca2+ elevation as such, as induced by ionomycin or thapsigargin, was a much poorer stimulant of PLD than orexin receptor stimulation (or TPA) (Figure 10B).
Discussion and conclusions
In the present study, we showed that the OX1 receptor is a potent and strong activator of PLD, more potent than the regular strong stimulator phorbol ester (TPA). Thus PA joins the lipid messengers previously shown to be produced by orexin receptor stimulation, DAG and arachidonic acid (Johansson et al., 2008; Turunen et al., 2010). PA also reveals a new way for DAG to be produced in the setting of orexin stimulation (Johansson et al., 2008). All of the lipid messengers were produced at very low orexin receptor activation levels, suggesting that they may have major significance for orexin responses in the physiological context.
The pharmacological inhibitors used to determine the identity of the PLD isoform involved in orexin and TPA signalling are not absolutely selective (Scott et al., 2009), therefore the concentration used was of utmost importance. The most selective inhibitor, PLD1i, in the concentration range shown to be essentially PLD1-specific (Scott et al., 2009), produced a concentration-dependent – and at a higher concentration full – inhibition of the orexin and TPA responses. In contrast, the less selective inhibitor, PLD2i, produced weak but significant inhibition of the stimulated PLD responses only at a high concentration, 3 µM, where significant inhibition of PLD1 is likely to have occurred (Scott et al., 2009). The results with the pharmacological inhibitors thus suggest that PLD1 is the enzyme activated by stimulation of orexin receptors, and also by TPA. However, because of the relatively weak selectivity of the inhibitors, it is impossible to completely rule out a minor contribution by PLD2. Interestingly, orexin-A at 100 nM was clearly more inhibited by PLD2i than orexin at 1 nM or TPA. Thus, it is possible that PLD2 is activated at higher orexin concentrations and contributes to the response to some extent, but this is difficult to reconcile with the finding that PLD1i produced more than half-maximal inhibition at 100 nM and almost full inhibition at 1 µM. One possibility is that PLD1 and PLD2 work in concert and that PLD1 activation is required for PLD2 activation. This could occur via PLD1-enhanced PIP2 production (PA → PIP5K), which might counteract the PLC-induced PIP2 breakdown.
The concentration–response curves for PLD activation by orexin-A and -B were shallow, and determined to be, in most cases, significantly biphasic. Similar biphasic concentration–response curves have been seen with respect to arachidonic acid release (Turunen et al., 2010), whereas other orexin receptor responses, like Ca2+ elevation and AC and PLC activation in CHO cells do not show this (Lund et al., 2000; Ammoun et al., 2003; Holmqvist et al., 2005; Johansson et al., 2008). Biphasic concentration–response curves could originate from heterogeneity of the receptor population or from the signalling level. However, the receptor population in CHO cells should be homogeneous as the human OX1 receptors are recombinantly expressed in these cells and this was confirmed by the inhibitory potency of the OX1 receptor antagonist, SB-334867, and the finding that orexin-A and -B showed similar biphasic responses. Therefore, it is more likely that the biphasic response occurs at the level of intracellular signalling. Hence, we measured the signal pathway in each phase of the PLD activation response using 1 nM orexin-A (high potency component) and 100 nM (high + low potency component). However, in contrast to orexin-stimulated arachidonic acid release where different signal pathways appear to be active in the different phases (Turunen et al., 2010), we did not observe major differences in the molecular mechanism behind the PLD response at different orexin concentrations (see below). The only notable difference was the stronger inhibition of the response to 100 nM orexin-A than 1 nM orexin-A by PLD2i as discussed above. Regulation of PLD2 is less clear than that of PLD1, but PLD2, unlike PLD1, has been shown to be activated by mono- and polyunsaturated fatty acids (Kim et al., 1999; Sarri et al., 2003). Stimulation of OX1 receptors in CHO cells efficiently activates arachidonic acid release (Turunen et al., 2010), and higher concentrations of arachidonic acid (at higher orexin concentrations) could enhance PLD2 activity. However, we found that complete inhibition of arachidonic acid release by MAFP did not inhibit the orexin receptor-induced PLD activity. Although PLD2 probably is involved in the response to high orexin concentrations, the response at the low end of the orexin response curve is more physiologically relevant, and there is little doubt that this would not be solely mediated by PLD1.
PKC is the signal transduction component required for orexin-induced PLD activity. Potent inhibition with GF109203X and only a weak inhibition with high concentration of Gö6976 indicates that nPKC is responsible for this orexin response. It is likely that the small inhibition seen with 10 µM Gö6976 is due to reduced specificity for cPKC at this high concentration, as the pharmacological analysis with peptide PKC activators showed that in CHO cells, nPKC isoforms are able to activate PLD, while the cPKCs do not seem to do this. Also the general c- and nPKC activator, TPA, only induced nPKC activity and not cPKC activity towards PLD. Similarly, in these cells Holmqvist et al. (2005) found that in the presence of active Gαs, TPA stimulated AC solely in an nPKC (specifically PKCδ)-dependent manner and only if TPA stimulation was combined with an elevated Ca2+ (thapsigargin), did cPKC become involved. Interestingly, orexin-A, although inducing a strong Ca2+ influx, only relies on nPKC with respect to both AC (Holmqvist et al., 2005) and PLD stimulation (present study). We have previously shown that, of nPKCs, only PKCδ and -ε are expressed in CHO cells (Holmqvist et al., 2005). KIE1-1, the peptide inhibitor for PKCε, is active against PKCε in CHO cells; it completely inhibits orexin-induced PKCε translocation to the membrane at 1 µM (Holmqvist et al., 2005) and it also fully blocked the KAE1-1-induced, but not orexin- or TPA-induced, PLD activation. The PKCδ inhibitor, rottlerin, blocked the cAMP response to orexin-A [in the presence of activated Gαs; (Holmqvist et al., 2005)] as well as the PLD response to orexin-A and TPA. Therefore, it is likely that PKCδ mediates the orexin and TPA responses in CHO cells. Rottlerin has previously been suggested to be an unreliable inhibitor of PKCδ and also to inhibit other targets (reviewed in Soltoff, 2007). However, our data obtained with different peptide PKC activators and inhibitors, as presented above, and the fact that we have previously shown that rottlerin does not display non-specific actions on the AC signalling in CHO cells (Holmqvist et al., 2005), makes us confident about the identification of PKCδ as the isoform responsible for PLD activation in the current study.
It has been suggested that PLD is activated by PKC either by phosphorylation or a direct, phosphorylation-independent, interaction (Hammond et al., 1997; Sung et al., 1999; Zhang et al., 1999; reviewed in Exton, 1998). GF109203X is an ATP-site inhibitor and, therefore, probably only inhibits kinase activity-dependent functions. Therefore, we assume that the action of TPA and orexin receptor activation on PLD requires the protein kinase activity of nPKC. TPA is a DAG analogue and is therefore likely to induce direct DAG-like activation of PKC, whereas the orexins could act by inducing either the production of DAG, phosphorylation or arachidonic acid release, the latter of which could also contribute to the TPA response. However, inhibitors of PLC activity, U-73122, and arachidonic acid release, MAFP (Turunen et al., 2010), did not inhibit the response to orexin (or TPA). Although U-73122 has been shown to be an unreliable inhibitor of PLC and also toxic to, among other cells, CHO (reviewed in Taylor and Broad, 1998, also own unpublished findings), we demonstrated that it was able to inhibit orexin-induced PLC activity in CHO, but not TPA- or orexin-induced PLD activity. It therefore seems likely that PLC is not involved in PKCδ activation. However, PLD itself also produces DAG, and thus PLD could constitute a positive feedback loop for its own activation. The other external signal regulating nPKC activation (and PKCδ in particular) is tyrosine phosphorylation by, for example, Src family kinases or c-Abl (reviewed in Yoshida, 2007). OX1 receptors have been suggested to activate Src (Ammoun et al., 2006a; Voisin et al., 2008). Further studies are required to elucidate the activation mechanism of PKCδ. If both PA-derived DAG-mediated positive feedback cycle and tyrosine phosphorylation were driving the PKCδ activity but in different orexin concentration ranges, this could explain the two phases of the concentration–response curves. Interestingly, Thr505 phosphorylation of PKCδ has recently been found to increase upon OX1 receptor stimulation in HEK-293 cells (Peltonen et al., 2010), probably corresponding to autophosphorylation occurring secondary to some primary activation cue. It is also possible that PKC-PLD signalling occurs by a phosphorylation-independent mechanism, as TPA was shown to induce PKC signalling to PLD without the requirement of kinase activity, and binding of GF109203X might also affect the conformation of PKC occluding kinase activity-independent signalling.
The Rho family of G-proteins have been shown to be important activators of PLD (at least PLD1). This may occur via direct binding to PLD1 (Yamazaki et al., 1999) or indirectly via some Rho-dependent kinase-mediated phosphorylation (Schmidt et al., 1999). GPCRs differ in their signalling mechanisms to PLD. Both fully Rho- or PKC-dominated signalling as well as mixtures of these have been seen (Du et al., 2000; Servitja et al., 2003; reviewed in Powner and Wakelam, 2002). In the present study we unequivocally showed that the Rho family members RhoA/B/C, Rac1/2/3 or cdc42 are not required for PLD activation by orexin-A or TPA in CHO cells. However, the data were obtained by using different selective inhibitors expressed in CHO cells and several isoforms may have to be inhibited simultaneously if the Rho family isoforms have overlapping roles in the process. Also the response to 100 nM orexin-A was slightly, but not significantly, inhibited by 1 µM GGTI-2133. Therefore, it is possible that the Rho family proteins, although not primarily activated by orexin receptors, become engaged in the replenishment of PIP2 levels, reduced by PLC activity in orexin signalling; Rho proteins are known to activate PIP5K (van den Bout and Divecha, 2009). It has even been suggested that the major effect of Rho proteins on PLD occurs through PIP5K (Oude Weernink et al., 2007).
The results of the present study showing that OX1 receptors activate PLD was anticipated from previous indirect evidence indicating that PLD inhibition blocks the high potency component of DAG generation (Johansson et al., 2008). However, whereas Johansson et al. (2008) found that inhibiting PLD blocked only a minor component of the DAG generation, the present results indicate that PLD activity is operative in a very wide orexin concentration range. PLC is also activated in the same orexin concentration range as PLD (see Johansson et al., 2008, see also Figure 1C), and, therefore, PLC may work as a major DAG producer in this system. It is also possible that the protein domain probes used as fluorescent DAG sensors by Johansson et al. (2008) saturate in their maximum response and therefore the absolute magnitudes of the DAG responses cannot be measured. Nevertheless, orexin receptors produced significant amounts of DAG even at low levels of receptor activity; therefore, DAG constitutes a promising candidate for transduction of physiological orexin responses to, for instance, non-selective cation channels (Kukkonen and Åkerman, 2005). It should be noted that the composition of the DAG produced from PLC and PLD activities is different, and may have different signalling profiles. However, DAG is not the only signal transducer derived from PLD activity. PA should also be considered, and both PLD1 and -2 have been shown to enhance choline availability and incorporation into ACh in a neuronal cell line (Zhao et al., 2001). Orexinergic neurons innervate cholinergic neurons of the basal forebrain and increase cortical ACh release (Eggermann et al., 2001; Fadel et al., 2005). The same is seen in other CNS loci (Wu et al., 2004; Kim et al., 2009; Cid-Pellitero and Garzon, 2011; reviewed in Arrigoni et al., 2010; Okumura and Nozu, 2011). Therefore, orexins, in addition to their more immediate excitatory effects on cholinergic neurons, may also stimulate ACh synthesis.
Orexin signalling to PLD1 distinguishes itself from the TPA signalling only by its requirement for extracellular Ca2+; this is probably due to the previously discovered requisite receptor-operated Ca2+ influx for coupling of orexin receptors to a number of intracellular signal pathways (Holmqvist et al., 2005; Ammoun et al., 2006a; Johansson et al., 2007; Turunen et al., 2010). As these pathways, for example, PLC, PKCδ, ERK and AC, are not directly stimulated by Ca2+ influx, or only to a very weak extent, we have suggested that the action of Ca2+ influx takes place at the proximal level of orexin receptor signalling. However, the extracellular Ca2+ could affect both the intracellular signalling pathways and orexin binding and we are in process of developing an intact-cell real-time binding assay for orexin receptors to elucidate this.
In conclusion, we have shown that OX1 receptors potently activate PLD (probably PLD1) in CHO cells and this is mediated by PKCδ but not other PKC isoforms, PKDs or Rho family G-proteins. Direct stimulation of PKC isoforms with the phorbol ester TPA apparently signals to PLD1 via the same effector, PKCδ. As the PLD activation occurs very potently, it is likely to occur in native systems and PLD may thus be a significant producer of the signalling molecules DAG and PA that serve as effectors for intracellular orexin responses. One of the most interesting tissue responses to orexin receptor activation to be examined with respect to DAG and PA is the activation of the non-selective cation channels, central for the orexin-induced neuronal excitation. Orexinergic neurons also innervate many types of cholinergic neurons, and the choline liberated by PLD activity may enhance the function of these. However, the current study was performed in a recombinant system where the receptor expression level is high and the cell type significantly different from CNS neurons. Thus, the physiological significance of orexin-induced PLD activation is as yet unresolved, but the recently developed potent and selective PLD inhibitors will find much use in future studies. Unfortunately, no similar potent, selective and non-toxic inhibitor for PLC is available, hampering comparison of these signalling systems.
Acknowledgments
We gratefully acknowledge Dr Osvaldo Rey (University of California at Los Angeles, USA), Dr Vivek Malhotra (Center for Genomic Regulation, Barcelona, Spain), Dr Krister Wennerberg [Institute for Molecular Medicine Finland (FIMM), Helsinki, Finland], Dr Alan Hall (Memorial Sloan-Kettering Cancer Center, New York, NY, USA), Dr Jonathan Chernoff (Fox Chase Cancer Center, Philadelphia, PA, USA) and Dr J. Silvio Gutkind (National Institutes of Health, Bethesda, MD, USA) for kindly supplying us with plasmids. We also thank Dr Krister Wennerberg and Dr Michael J. Courtney (University of Eastern Finland, Kuopio, Finland) for help in obtaining some of the plasmids. We are very grateful to Dr Claude Leray for helpful advices regarding TLC techniques. Pirjo Puroranta is acknowledged for technical assistance. This study was supported by the Academy of Finland, the Magnus Ehrnrooth Foundation and the University of Helsinki Research Funds (JPK), as well as the National Institutes of Health (NIH; grants GM071520 and GM084251; MAF). J.P. was supported by the Drug Discovery Graduate School (DDGS).
Glossary
- βARK1
β-adrenoceptor kinase 1
- c-
and nPKC, conventional and novel PKC, respectively
- GF109203X
(bisindolylmaleimide I, Gö6850), 2-(1-[3-dimethylaminopropyl]-1H-indol-3-yl)-3-(1H-indol-3-yl)-maleimide
- GGTI-2133
N-([4-(imidazol-4-yl)methylamino]-2-[1-naphthyl]benzoyl)leucine trifluoroacetate salt
- Gö6976
5,6,7,13-tetrahydro-13-methyl-5-oxo-12H-indolo(2,3-a)pyrrolo(3,4-c)carbazole-12-propanenitrile
- HBM
HEPES-buffered medium
- KAC1-1
a peptide cPKC activator
- KIC1-1
a peptide cPKC inhibitor
- KAD1-1
a peptide PKCδ activator
- KAE1-1
a peptide PKCε activator, KIE1-1, a peptide PKCε inhibitor
- MAFP
methyl arachidonyl fluorophosphonate
- PA
phosphatidic acid
- pEC50
–logEC50
- PIP2
phosphatidylinositol-4,5-bisphosphate
- PIP5K
type I phosphatidylinositol-4-phosphate 5-kinase
- PLD1i
PLD1 inhibitor (N-[2-[4-(5-chloro-2,3-dihydro-2-oxo-1H-benzimidazol-1-yl)-1-piperidinyl]-1-methylethyl]-2-naphthalenecarboxamide), PLD2i, PLD2 inhibitor (N-[2-(4-oxo-1-phenyl-1,3,8-triazaspiro[4,5]dec-8-yl)ethyl]-2-naphthalenecarboxamide)
- PtdBut
phosphatidylbutanol
- probenecid
p-(dipropylsulphamoyl) benzoic acid
- PTx
Pertussis toxin
- RACK
receptor for activated C-kinase
- rottlerin
3′-([8-cinnamoyl-5,7-dihydroxy-2,2-dimethyl-2H-1-benzopyran-6-yl]methyl)-2′,4′,6′-trihydroxy-5′-methylacetophenone]
- SB-334867
1-(2-methylbenzoxazol-6-yl)-3-(1,5)naphthyridin-4-yl-urea HCl
- TPA
12-O-tetradecanoyl-phorbol-13-acetate
- U-73122
1-(6-[([17b]-3-methoxyestra-1,3,5[10]-trien-17-yl)amino]hexyl)-1H-pyrrole-2,5-dione
Conflicts of interest
None.
References
- Aittaleb M, Boguth CA, Tesmer JJ. Structure and function of heterotrimeric G protein-regulated Rho guanine nucleotide exchange factors. Mol Pharmacol. 2010;77:111–125. doi: 10.1124/mol.109.061234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th Edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammoun S, Holmqvist T, Shariatmadari R, Oonk HB, Detheux M, Parmentier M, et al. Distinct recognition of OX1 and OX2 receptors by orexin peptides. J Pharmacol Exp Ther. 2003;305:507–514. doi: 10.1124/jpet.102.048025. [DOI] [PubMed] [Google Scholar]
- Ammoun S, Johansson L, Ekholm ME, Holmqvist T, Danis AS, Korhonen L, et al. OX1 orexin receptors activate extracellular signal-regulated kinase (ERK) in CHO cells via multiple mechanisms: the role of Ca2+ influx in OX1 receptor signaling. Mol Endocrinol. 2006a;20:80–99. doi: 10.1210/me.2004-0389. [DOI] [PubMed] [Google Scholar]
- Ammoun S, Lindholm D, Wootz H, Åkerman KE, Kukkonen JP. G-protein-coupled OX1 orexin/hcrtr-1 hypocretin receptors induce caspase-dependent and -independent cell death through p38 mitogen-/stress-activated protein kinase. J Biol Chem. 2006b;281:834–842. doi: 10.1074/jbc.M508603200. [DOI] [PubMed] [Google Scholar]
- Arrigoni E, Mochizuki T, Scammell TE. Activation of the basal forebrain by the orexin/hypocretin neurones. Acta Physiol (Oxf) 2010;198:223–235. doi: 10.1111/j.1748-1716.2009.02036.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Bout I, Divecha N. PIP5K-driven PtdIns(4,5)P2 synthesis: regulation and cellular functions. J Cell Sci. 2009;122:3837–3850. doi: 10.1242/jcs.056127. [DOI] [PubMed] [Google Scholar]
- Boutrel B, Kenny PJ, Specio SE, Martin-Fardon R, Markou A, Koob GF, et al. Role for hypocretin in mediating stress-induced reinstatement of cocaine-seeking behavior. Proc Natl Acad Sci USA. 2005;102:19168–19173. doi: 10.1073/pnas.0507480102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brindley DN, Pilquil C. Lipid phosphate phosphatases and signaling. J Lipid Res. 2009;50:S225–S230. doi: 10.1194/jlr.R800055-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasco S, Merida I. Diacylglycerol, when simplicity becomes complex. Trends Biochem Sci. 2007;32:27–36. doi: 10.1016/j.tibs.2006.11.004. [DOI] [PubMed] [Google Scholar]
- Chen J, Lu G, Wang QJ. Protein kinase C-independent effects of protein kinase D3 in glucose transport in L6 myotubes. Mol Pharmacol. 2005;67:152–162. doi: 10.1124/mol.104.004200. [DOI] [PubMed] [Google Scholar]
- Cho W, Stahelin RV. Membrane binding and subcellular targeting of C2 domains. Biochim Biophys Acta. 2006;1761:838–849. doi: 10.1016/j.bbalip.2006.06.014. [DOI] [PubMed] [Google Scholar]
- Churchill EN, Qvit N, Mochly-Rosen D. Rationally designed peptide regulators of protein kinase C. Trends Endocrinol Metab. 2009;20:25–33. doi: 10.1016/j.tem.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cid-Pellitero ED, Garzon M. Hypocretin1/OrexinA axon targeting of laterodorsal tegmental nucleus neurons projecting to the rat medial prefrontal cortex. Cereb Cortex. 2011 doi: 10.1093/cercor/bhr070. doi: 10.1093/cercor/bhr070. [DOI] [PubMed] [Google Scholar]
- Colley WC, Sung TC, Roll R, Jenco J, Hammond SM, Altshuller Y, et al. Phospholipase D2, a distinct phospholipase D isoform with novel regulatory properties that provokes cytoskeletal reorganization. Curr Biol. 1997;7:191–201. doi: 10.1016/s0960-9822(97)70090-3. [DOI] [PubMed] [Google Scholar]
- Crespo P, Cachero TG, Xu N, Gutkind JS. Dual effect of beta-adrenergic receptors on mitogen-activated protein kinase. Evidence for a beta gamma-dependent activation and a G alpha s-cAMP-mediated inhibition. J Biol Chem. 1995;270:25259–25265. doi: 10.1074/jbc.270.42.25259. [DOI] [PubMed] [Google Scholar]
- Du G, Altshuller YM, Kim Y, Han JM, Ryu SH, Morris AJ, et al. Dual requirement for rho and protein kinase C in direct activation of phospholipase D1 through G protein-coupled receptor signaling. Mol Biol Cell. 2000;11:4359–4368. doi: 10.1091/mbc.11.12.4359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggermann E, Serafin M, Bayer L, Machard D, Saint-Mleux B, Jones BE, et al. Orexins/hypocretins excite basal forebrain cholinergic neurones. Neuroscience. 2001;108:177–181. doi: 10.1016/s0306-4522(01)00512-7. [DOI] [PubMed] [Google Scholar]
- Ekholm ME, Johansson L, Kukkonen JP. IP(3)-independent signalling of OX(1) orexin/hypocretin receptors to Ca(2+) influx and ERK. Biochem Biophys Res Commun. 2007;353:475–480. doi: 10.1016/j.bbrc.2006.12.045. [DOI] [PubMed] [Google Scholar]
- Exton JH. Phospholipase D. Biochim Biophys Acta. 1998;1436:105–115. doi: 10.1016/s0005-2760(98)00124-6. [DOI] [PubMed] [Google Scholar]
- Fadel J, Pasumarthi R, Reznikov LR. Stimulation of cortical acetylcholine release by orexin A. Neuroscience. 2005;130:541–547. doi: 10.1016/j.neuroscience.2004.09.050. [DOI] [PubMed] [Google Scholar]
- Gschwendt M, Muller HJ, Kielbassa K, Zang R, Kittstein W, Rincke G, et al. Rottlerin, a novel protein kinase inhibitor. Biochem Biophys Res Commun. 1994;199:93–98. doi: 10.1006/bbrc.1994.1199. [DOI] [PubMed] [Google Scholar]
- Gschwendt M, Dieterich S, Rennecke J, Kittstein W, Mueller HJ, Johannes FJ. Inhibition of protein kinase C mu by various inhibitors. Differentiation from protein kinase c isoenzymes. FEBS Lett. 1996;392:77–80. doi: 10.1016/0014-5793(96)00785-5. [DOI] [PubMed] [Google Scholar]
- Hammond SM, Altshuller YM, Sung TC, Rudge SA, Rose K, Engebrecht J, et al. Human ADP-ribosylation factor-activated phosphatidylcholine-specific phospholipase D defines a new and highly conserved gene family. J Biol Chem. 1995;270:29640–29643. doi: 10.1074/jbc.270.50.29640. [DOI] [PubMed] [Google Scholar]
- Hammond SM, Jenco JM, Nakashima S, Cadwallader K, Gu Q, Cook S, et al. Characterization of two alternately spliced forms of phospholipase D1. Activation of the purified enzymes by phosphatidylinositol 4,5-bisphosphate, ADP-ribosylation factor, and Rho family monomeric GTP-binding proteins and protein kinase C-alpha. J Biol Chem. 1997;272:3860–3868. doi: 10.1074/jbc.272.6.3860. [DOI] [PubMed] [Google Scholar]
- Harris GC, Wimmer M, Aston-Jones G. A role for lateral hypothalamic orexin neurons in reward seeking. Nature. 2005;437:556–559. doi: 10.1038/nature04071. [DOI] [PubMed] [Google Scholar]
- Heinonen MV, Purhonen AK, Makela KA, Herzig KH. Functions of orexins in peripheral tissues. Acta Physiol (Oxf) 2008;192:471–485. doi: 10.1111/j.1748-1716.2008.01836.x. [DOI] [PubMed] [Google Scholar]
- Hilairet S, Bouaboula M, Carriere D, Le Fur G, Casellas P. Hypersensitization of the Orexin 1 receptor by the CB1 receptor: evidence for cross-talk blocked by the specific CB1 antagonist, SR141716. J Biol Chem. 2003;278:23731–23737. doi: 10.1074/jbc.M212369200. [DOI] [PubMed] [Google Scholar]
- Holmqvist T, Åkerman KEO, Kukkonen JP. Orexin signaling in recombinant neuron-like cells. FEBS Lett. 2002;526:11–14. doi: 10.1016/s0014-5793(02)03101-0. [DOI] [PubMed] [Google Scholar]
- Holmqvist T, Johansson L, Östman M, Ammoun S, Åkerman KE, Kukkonen JP. OX1 orexin receptors couple to adenylyl cyclase regulation via multiple mechanisms. J Biol Chem. 2005;280:6570–6579. doi: 10.1074/jbc.M407397200. [DOI] [PubMed] [Google Scholar]
- Huang H, Gao Q, Peng XX, Choi S-Y, Sarma K, Ren H, et al. piRNA-associated germline nuage formation and spermatogenesis require MitoPLD pro-fusogenic mitochondrial-surface lipid signaling. Dev Cell. 2011;20:376–387. doi: 10.1016/j.devcel.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins GM, Frohman MA. Phospholipase D: a lipid centric review. Cell Mol Life Sci. 2005;62:2305–2316. doi: 10.1007/s00018-005-5195-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Luo JQ, Urano T, Frankel P, Lu Z, Foster DA, et al. Involvement of Ral GTPase in v-Src-induced phospholipase D activation. Nature. 1995;378:409–412. doi: 10.1038/378409a0. [DOI] [PubMed] [Google Scholar]
- Johansson L, Ekholm ME, Kukkonen JP. Regulation of OX(1) orexin/hypocretin receptor-coupling to phospholipase C by Ca(2+) influx. Br J Pharmacol. 2007;150:97–104. doi: 10.1038/sj.bjp.0706959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson L, Ekholm ME, Kukkonen JP. Multiple phospholipase activation by OX(1) orexin/hypocretin receptors. Cell Mol Life Sci. 2008;65:1948–1956. doi: 10.1007/s00018-008-8206-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karteris E, Machado RJ, Chen J, Zervou S, Hillhouse EW, Randeva HS. Food deprivation differentially modulates orexin receptor expression and signalling in the rat hypothalamus and adrenal cortex. Am J Physiol Endocrinol Metab. 2005;288:E1089–E1100. doi: 10.1152/ajpendo.00351.2004. [DOI] [PubMed] [Google Scholar]
- Khalil MB, Blais A, Figeys D, Yao Z. Lipin – the bridge between hepatic glycerolipid biosynthesis and lipoprotein metabolism. Biochim Biophys Acta. 2010;1801:1249–1259. doi: 10.1016/j.bbalip.2010.07.008. [DOI] [PubMed] [Google Scholar]
- Kim JH, Kim Y, Lee SD, Lopez I, Arnold RS, Lambeth JD, et al. Selective activation of phospholipase D2 by unsaturated fatty acid. FEBS Lett. 1999;454:42–46. doi: 10.1016/s0014-5793(99)00745-0. [DOI] [PubMed] [Google Scholar]
- Kim J, Nakajima K, Oomura Y, Wayner MJ, Sasaki K. Electrophysiological effects of orexins/hypocretins on pedunculopontine tegmental neurons in rats: an in vitro study. Peptides. 2009;30:191–209. doi: 10.1016/j.peptides.2008.09.023. [DOI] [PubMed] [Google Scholar]
- Kukkonen JP, Åkerman KEO. Intracellular signal pathways utilized by the hypocretin/orexin receptors. In: de Lecea L, Sutcliffe JG, editors. Hypocretins as Integrators of Physiological Signals. Berlin: Springer Science; Business Media; 2005. pp. 221–231. [Google Scholar]
- Kukkonen JP, Holmqvist T, Ammoun S, Åkerman KE. Functions of the orexinergic/hypocretinergic system. Am J Physiol Cell Physiol. 2002;283:C1567–C1591. doi: 10.1152/ajpcell.00055.2002. [DOI] [PubMed] [Google Scholar]
- Larsson KP, Åkerman KEO, Magga J, Uotila S, Kukkonen JP, Näsman J, et al. Orexin-A stimulates cholecystokinin release from intestinal neuroendocrine STC-1 cells by a Ca2+-dependent mechanism. Biochem Biophys Res Commun. 2003;309:209–216. doi: 10.1016/s0006-291x(03)01563-8. [DOI] [PubMed] [Google Scholar]
- Liljedahl M, Maeda Y, Colanzi A, Ayala I, Van Lint J, Malhotra V. Protein kinase D regulates the fission of cell surface destined transport carriers from the trans-Golgi network. Cell. 2001;104:409–420. doi: 10.1016/s0092-8674(01)00228-8. [DOI] [PubMed] [Google Scholar]
- Lund PE, Shariatmadari R, Uustare A, Detheux M, Parmentier M, Kukkonen JP, et al. The orexin OX1 receptor activates a novel Ca2+ influx pathway necessary for coupling to phospholipase C. J Biol Chem. 2000;275:30806–30812. doi: 10.1074/jbc.M002603200. [DOI] [PubMed] [Google Scholar]
- Malendowicz LK, Tortorella C, Nussdorfer GG. Orexins stimulate corticosterone secretion of rat adrenocortical cells, through the activation of the adenylate cyclase-dependent signaling cascade. J Steroid Biochem Mol Biol. 1999;70:185–188. doi: 10.1016/s0960-0760(99)00110-7. [DOI] [PubMed] [Google Scholar]
- Martiny-Baron G, Kazanietz MG, Mischak H, Blumberg PM, Kochs G, Hug H, et al. Selective inhibition of protein kinase C isozymes by the indolocarbazole Gö 6976. J Biol Chem. 1993;268:9194–9197. [PubMed] [Google Scholar]
- Morris AJ, Frohman MA, Engebrecht J. Measurement of phospholipase D activity. Anal Biochem. 1997;252:1–9. doi: 10.1006/abio.1997.2299. [DOI] [PubMed] [Google Scholar]
- Näsman J, Bart G, Larsson K, Louhivuori L, Peltonen H, Åkerman KE. The orexin OX1 receptor regulates Ca2+ entry via diacylglycerol-activated channels in differentiated neuroblastoma cells. J Neurosci. 2006;26:10658–10666. doi: 10.1523/JNEUROSCI.2609-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okumura T, Nozu T. Role of brain orexin in the pathophysiology of functional gastrointestinal disorders. J Gastroenterol Hepatol. 2011;26(Suppl. 3):61–66. doi: 10.1111/j.1440-1746.2011.06626.x. [DOI] [PubMed] [Google Scholar]
- Oude Weernink PA, Han L, Jakobs KH, Schmidt M. Dynamic phospholipid signaling by G protein-coupled receptors. Biochim Biophys Acta. 2007;1768:888–900. doi: 10.1016/j.bbamem.2006.09.012. [DOI] [PubMed] [Google Scholar]
- Peltonen HM, Magga JM, Bart G, Turunen PM, Antikainen MS, Kukkonen JP, et al. Involvement of TRPC3 channels in calcium oscillations mediated by OX(1) orexin receptors. Biochem Biophys Res Commun. 2009;385:408–412. doi: 10.1016/j.bbrc.2009.05.077. [DOI] [PubMed] [Google Scholar]
- Peltonen HM, Åkerman KE, Bart G. A role for PKD1 and PKD3 activation in modulation of calcium oscillations induced by orexin receptor 1 stimulation. Biochim Biophys Acta. 2010;1803:1206–1212. doi: 10.1016/j.bbamcr.2010.07.001. [DOI] [PubMed] [Google Scholar]
- Powner DJ, Wakelam MJ. The regulation of phospholipase D by inositol phospholipids and small GTPases. FEBS Lett. 2002;531:62–64. doi: 10.1016/s0014-5793(02)03410-5. [DOI] [PubMed] [Google Scholar]
- Ramanjaneya M, Conner AC, Chen J, Kumar P, Brown JE, Johren O, et al. Orexin-stimulated MAP kinase cascades are activated through multiple G-protein signalling pathways in human H295R adrenocortical cells: diverse roles for orexins A and B. J Endocrinol. 2009;202:249–261. doi: 10.1677/JOE-08-0536. [DOI] [PubMed] [Google Scholar]
- Rey O, Papazyan R, Waldron RT, Young SH, Lippincott-Schwartz J, Jacamo R, et al. The nuclear import of protein kinase D3 requires its catalytic activity. J Biol Chem. 2006;281:5149–5157. doi: 10.1074/jbc.M508014200. [DOI] [PubMed] [Google Scholar]
- Rouet-Benzineb P, Rouyer-Fessard C, Jarry A, Avondo V, Pouzet C, Yanagisawa M, et al. Orexins acting at native OX(1) receptor in colon cancer and neuroblastoma cells or at recombinant OX(1) receptor suppress cell growth by inducing apoptosis. J Biol Chem. 2004;279:45875–45886. doi: 10.1074/jbc.M404136200. [DOI] [PubMed] [Google Scholar]
- Sarri E, Pardo R, Fensome-Green A, Cockcroft S. Endogenous phospholipase D2 localizes to the plasma membrane of RBL-2H3 mast cells and can be distinguished from ADP ribosylation factor-stimulated phospholipase D1 activity by its specific sensitivity to oleic acid. Biochem J. 2003;369:319–329. doi: 10.1042/BJ20021347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M, Voss M, Weernink PA, Wetzel J, Amano M, Kaibuchi K, et al. A role for rho-kinase in rho-controlled phospholipase D stimulation by the m3 muscarinic acetylcholine receptor. J Biol Chem. 1999;274:14648–14654. doi: 10.1074/jbc.274.21.14648. [DOI] [PubMed] [Google Scholar]
- Scott SA, Selvy PE, Buck JR, Cho HP, Criswell TL, Thomas AL, et al. Design of isoform-selective phospholipase D inhibitors that modulate cancer cell invasiveness. Nat Chem Biol. 2009;5:108–117. doi: 10.1038/nchembio.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servitja JM, Masgrau R, Pardo R, Sarri E, von Eichel-Streiber C, Gutkind JS, et al. Metabotropic glutamate receptors activate phospholipase D in astrocytes through a protein kinase C-dependent and Rho-independent pathway. Neuropharmacology. 2003;44:171–180. doi: 10.1016/s0028-3908(02)00361-1. [DOI] [PubMed] [Google Scholar]
- Shirai Y, Kashiwagi K, Yagi K, Sakai N, Saito N. Distinct effects of fatty acids on translocation of gamma- and epsilon-subspecies of protein kinase C. J Cell Biol. 1998;143:511–521. doi: 10.1083/jcb.143.2.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltoff SP. Rottlerin: an inappropriate and ineffective inhibitor of PKCdelta. Trends Pharmacol Sci. 2007;28:453–458. doi: 10.1016/j.tips.2007.07.003. [DOI] [PubMed] [Google Scholar]
- Spinazzi R, Ziolkowska A, Neri G, Nowak M, Rebuffat P, Nussdorfer GG, et al. Orexins modulate the growth of cultured rat adrenocortical cells, acting through type 1 and type 2 receptors coupled to the MAPK p42/p44- and p38-dependent cascades. Int J Mol Med. 2005;15:847–852. [PubMed] [Google Scholar]
- Sung TC, Zhang Y, Morris AJ, Frohman MA. Structural analysis of human phospholipase D1. J Biol Chem. 1999;274:3659–3666. doi: 10.1074/jbc.274.6.3659. [DOI] [PubMed] [Google Scholar]
- Taylor CW, Broad LM. Pharmacological analysis of intracellular Ca2+ signalling: problems and pitfalls. Trends Pharmacol Sci. 1998;19:370–375. doi: 10.1016/s0165-6147(98)01243-7. [DOI] [PubMed] [Google Scholar]
- Turunen PM, Ekholm ME, Somerharju P, Kukkonen JP. Arachidonic acid release mediated by OX(1) orexin receptors. Br J Pharmacol. 2010;159:212–221. doi: 10.1111/j.1476-5381.2009.00535.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasudevan A, Qian Y, Vogt A, Blaskovich MA, Ohkanda J, Sebti SM, et al. Potent, highly selective, and non-thiol inhibitors of protein geranylgeranyltransferase-I. J Med Chem. 1999;42:1333–1340. doi: 10.1021/jm9900873. [DOI] [PubMed] [Google Scholar]
- Venkatachalam K, Montell C. TRP channels. Annu Rev Biochem. 2007;76:387–417. doi: 10.1146/annurev.biochem.75.103004.142819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voisin T, El Firar A, Rouyer-Fessard C, Gratio V, Laburthe M. A hallmark of immunoreceptor, the tyrosine-based inhibitory motif ITIM, is present in the G protein-coupled receptor OX1R for orexins and drives apoptosis: a novel mechanism. FASEB J. 2008;22:1993–2002. doi: 10.1096/fj.07-098723. [DOI] [PubMed] [Google Scholar]
- Wang X, Devaiah SP, Zhang W, Welti R. Signaling functions of phosphatidic acid. Prog Lipid Res. 2006;45:250–278. doi: 10.1016/j.plipres.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Wu M, Zaborszky L, Hajszan T, van den Pol AN, Alreja M. Hypocretin/orexin innervation and excitation of identified septohippocampal cholinergic neurons. J Neurosci. 2004;24:3527–3536. doi: 10.1523/JNEUROSCI.5364-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki M, Zhang Y, Watanabe H, Yokozeki T, Ohno S, Kaibuchi K, et al. Interaction of the small G protein RhoA with the C terminus of human phospholipase D1. J Biol Chem. 1999;274:6035–6038. doi: 10.1074/jbc.274.10.6035. [DOI] [PubMed] [Google Scholar]
- Yeaman C, Ayala MI, Wright JR, Bard F, Bossard C, Ang A, et al. Protein kinase D regulates basolateral membrane protein exit from trans-Golgi network. Nat Cell Biol. 2004;6:106–112. doi: 10.1038/ncb1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K. PKCdelta signaling: mechanisms of DNA damage response and apoptosis. Cell Signal. 2007;19:892–901. doi: 10.1016/j.cellsig.2007.01.027. [DOI] [PubMed] [Google Scholar]
- Zang R, Muller HJ, Kielbassa K, Marks F, Gschwendt M. Partial purification of a type eta protein kinase C from murine brain: separation from other protein kinase C isoenzymes and characterization. Biochem J. 1994;304:641–647. doi: 10.1042/bj3040641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Altshuller YM, Hammond SM, Hayes F, Morris AJ, Frohman MA. Loss of receptor regulation by a phospholipase D1 mutant unresponsive to protein kinase C. EMBO J. 1999;18:6339–6348. doi: 10.1093/emboj/18.22.6339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao D, Frohman MA, Blusztajn JK. Generation of choline for acetylcholine synthesis by phospholipase D isoforms. BMC Neurosci. 2001;2:16. doi: 10.1186/1471-2202-2-16. [DOI] [PMC free article] [PubMed] [Google Scholar]