

Abstract
BACKGROUND AND PURPOSE
It is thought that the anti-inflammatory effects of glucocorticoids (GCs) are largely due to GC receptor (GR)-mediated transrepression of NF-κB and other transcription factors, whereas side effects are caused by activation of gene expression (transactivation). Selective GR modulators (SGRMs) that preferentially promote transrepression should retain anti-inflammatory properties whilst causing fewer side effects. Contradicting this model, we found that anti-inflammatory effects of the classical GC dexamethasone were partly dependent on transactivation of the dual specificity phosphatase 1 (DUSP1) gene. We wished to determine whether anti-inflammatory effects of SGRMs are also mediated by DUSP1.
EXPERIMENTAL APPROACH
Dissociated properties of two SGRMs were confirmed using GR- and NF-κB-dependent reporters, and capacity to activate GC-responsive elements of the DUSP1 gene was tested. Effects of SGRMs on the expression of DUSP1 and pro-inflammatory gene products were assessed in various cell lines and in primary murine Dusp1+/+ and Dusp1−/−macrophages.
KEY RESULTS
The SGRMs were able to up-regulate DUSP1 in several cell types, and this response correlated with the ability of the compounds to suppress COX-2 expression. Several anti-inflammatory effects of SGRMs were ablated or significantly impaired in Dusp1−/− macrophages.
CONCLUSIONS AND IMPLICATIONS
Like dexamethasone, SGRMs appear to exert anti-inflammatory effects partly via the up-regulation of DUSP1. This finding has implications for how potentially therapeutic novel GR ligands are identified and assessed.
Keywords: immunopharmacology, nuclear receptors, steroids/neurosteroids, anti-inflammatory drugs, gene transcription, inflammation
Introduction
For more than half a century, synthetic glucocorticoids (GCs) have been extensively used to treat chronic inflammatory diseases such as rheumatoid arthritis, asthma and inflammatory bowel diseases (Barnes, 2006; Hillier, 2007). The basis of their therapeutic action is the impairment, in most cell types, of the expression of pro-inflammatory genes. Various unpredictable and occasionally life-threatening side effects of GCs have been documented since their earliest clinical use (Schacke et al., 2002). These include osteoporosis and diabetes mellitus, atrophy of skin and muscle, hypertension and increased susceptibility to infection. Nevertheless, GCs are still the cornerstone of treatment for many diseases. At the same time, major research initiatives attempt to separate the desired anti-inflammatory effects of GCs from their side effects (Schacke et al., 2007; Hudson et al., 2008; De Bosscher et al., 2010).
GCs modulate gene expression via the GC receptor (GR), a transcription factor belonging to the nuclear hormone receptor superfamily (Newton, 2000; Tuckermann et al., 2005). Lipophilic ligands such as the endogenous GC cortisol or the synthetic GC dexamethasone (dex) diffuse across the cell membrane and bind to GR in the cytoplasm. This promotes the release of GR from a large complex of chaperone proteins and its migration to the nucleus. In most but not all cases, transcriptional activation by GR (transactivation) is dependent on homodimerization, which is mediated by a short motif adjacent to the first of two zinc finger DNA-binding motifs. GR homodimers recognize sequences related to the idealized, palindromic consensus AGAACAnnnTGTTCT (GC response element or GRE).
A second physiologically important function of GR is to inhibit transcription via a mechanism known as transrepression (Kassel and Herrlich, 2007; De Bosscher et al., 2010; Glass and Saijo, 2010). In this case, GR does not bind directly to DNA but instead is recruited to DNA via direct or indirect interactions with other transcription factors, notably members of the activating protein 1 (AP-1) and NF-κB families, both of which play important roles in the expression of pro-inflammatory genes. The presence of GR at AP-1 or NF-κB binding sites is thought to inhibit transcriptional activation by impairing recruitment of transcriptional co-activators, or by promoting recruitment of co-repressors.
It is often stated that the anti-inflammatory effects of GCs are largely mediated by transrepression, whereas side effects are largely mediated by transactivation. If this is correct, it may be possible to improve upon classical GCs by identifying novel ligands of GR that selectively promote its transrepressive function rather than its transactivating function (Schacke et al., 2007; Berlin, 2010; De Bosscher et al., 2010; Newton et al., 2010). Such compounds are known as dissociated GR ligands, selective GR agonists (SEGRAs) or modulators (SGRMs). They are predicted to retain anti-inflammatory effects of classical GCs like dex but cause fewer or less severe side effects. Typically, SGRMs have been identified from drug libraries first on the basis of affinity for GR and second on the basis of effects on reporter constructs. Transactivation is tested against well-known GC target genes such as tyrosine aminotransferase (TAT), or against constructs that contain well-characterized GC responsive promoters or multimerized GR binding sites. Transrepression is tested using promoters that contain AP-1 and/or NF-κB binding sites and are activated by pro-inflammatory stimuli. Alternatively, reporters containing multimerized AP-1 or NF-κB binding sites may be used. As reviewed elsewhere (Schacke et al., 2007; Berlin, 2010; De Bosscher et al., 2010), a number of interesting compounds have been identified using this basic approach. Recently described examples include ZK216348 and LGD-552, which are non-steroidal GR ligands (Schacke et al., 2004; Humphrey et al., 2006; Miner et al., 2007; Lopez et al., 2008).
As well as directly inhibiting expression of pro-inflammatory genes by means of transrepression, GCs can exert indirect therapeutic effects, via the up-regulation of several anti-inflammatory genes (Clark, 2007; Newton and Holden, 2007). For example, many cell types respond to GCs by expressing dual specificity phosphatase (DUSP1), an enzyme that dephosphorylates and inactivates both p38 MAPK and JNK (Abraham and Clark, 2006; Owens and Keyse, 2007). The up-regulation of DUSP1 has been suggested to contribute to destabilization of pro-inflammatory mRNAs (Lasa et al., 2001; 2002; Quante et al., 2008) and to the inhibition of AP-1 and NF-κB function (Diefenbacher et al., 2008; Bladh et al., 2009; Cho and Kim, 2009; King et al., 2009). Correspondingly, many of the anti-inflammatory effects of GCs are impaired in macrophages derived from Dusp1−/− mice, or cells in which DUSP1 has been down-regulated using RNA interference (Abraham et al., 2006; Furst et al., 2007; Issa et al., 2007; Kang et al., 2008; Quante et al., 2008; King et al., 2009). In vivo anti-inflammatory effects of dex were dependent on DUSP1 in experimental models of acute inflammation (Abraham et al. 2006; Wang et al., 2008), asthma (Li et al., 2010) and rheumatoid arthritis (our unpublished results). There are therefore problems with a paradigm that equates anti-inflammatory effects of GCs with transrepression and not transactivation. In fact, such a model is not strongly supported by experimental evidence. For example, there is no genetically modified mouse strain that clearly demonstrates a separation between side effects and transactivation on one hand, and transrepression and anti-inflammatory effects on the other. A knock-in mouse strain expressing a dimerization defective mutant of GR (known as GRdim) was initially thought to provide evidence of just such a mechanistic separation between therapeutic and harmful effects (Tuckermann et al., 1999), but emerging complexities in the phenotype of the GRdim mouse now undermine rather than support the paradigm (Kleiman and Tuckermann, 2007; Frijters et al., 2010; Rauch et al., 2010). Recent results also show that, although SGRMs were identified on the basis of impaired transcriptional activation, they may be quite capable of inducing expression of DUSP1 and other genes with anti-inflammatory roles (Chivers et al., 2006; Janka-Junttila et al., 2006; Lopez et al., 2008; Newton et al., 2010). We therefore asked whether anti-inflammatory effects of SGRMs may actually be dependent on the induction of DUSP1. The answer to this question will have an important impact on how novel GR ligands with improved therapeutic indices are discovered, and how their properties are to be understood.
Methods
Mice and reagents
All animal procedures were performed under United Kingdom Home Office regulations and with local Ethical Review Committee approval. Dusp1−/− mice were generated as described previously (Dorfman et al., 1996). They were backcrossed against C57BL/6 for 10 generations to generate a colony with almost pure (99.9%) C57BL/6 genetic background, genotype at the Dusp1 locus being determined by a PCR assay. The same breeding programme was used to generate a Dusp1+/+ colony with equivalent genetic background. Age- and sex-matched Dusp1+/+ and Dusp1−/− animals were used to generate bone marrow-derived macrophages (BMMs) by differentiation from BM haematopoietic stem cells for 5 days in RPMI 1640 medium supplemented with 10% FCS, 100 U·mL−1 penicillin, 100 µg·mL−1 streptomycin (PAA) and 10 ng·mL−1 M-CSF (Peprotech, London, UK). This method routinely generates macrophages of at least 85% purity (Lari et al., 2007). Dulbecco's modified Eagle's medium (DMEM, PAA) supplemented with 10% FCS was used for the culture of the HeLa, RAW264.7 and A549 cell lines. Stably transfected A549 cells were maintained with additional 0.2 mg·mL−1 of G-418 (Peprotech). All cells were maintained in a humidified atmosphere of 5% CO2 at 37°C and seeded to confluence (unless otherwise stated) in FCS supplemented medium the day before the experiment. Cells were treated with different doses of dex (Sigma, Dorset, UK), Cpd1 and Cpd2 (Roche, Palo Alto, CA, USA) or vehicle [dimethyl sulfoxide (DMSO) Sigma] and stimulated with 1 ng·mL−1 IL-1β (made in house) or 10 ng·mL−1 LPS (Alexis Biochemicals, Exeter, UK).
Plasmids, transfection and luciferase assays
The GRE A549 reporter line (pGL3.neo.TATA.2GRE) was a gift from R Newton (University of Calgary). The NF-κB-dependent reporter containing three κB binding sites linked to a TATA box and a firefly luciferase coding sequence (3κBtkluc) was also generously provided by R Newton. pGL3b-Hs.-4.8, pGL3p-Mm-29GRR, -Hs-1.3GRR and -Hs-4.6GRR were as previously described (Tchen et al., 2010). Cells were seeded to approximately 50% confluence the day before the experiment then transiently transfected using Superfect (Qiagen, Crawley, UK) with 200 ng of firefly luciferase reporters as indicated plus 100 ng of Renilla luciferase expression vector and pBluescript (Agilent Technologies, Edinburgh, UK) as carrier to make the total quantity of DNA up to 1 µg. Following transfection, cells were treated with vehicle (0.1% DMSO), Dex, Cpd1 or Cpd2 as described. Cells were harvested, and luciferase activities were measured using the dual luciferase reporter assay kit (Promega, Southampton, UK) and Microbeta luminometer (PerkinElmer Life Sciences, Seer Green, UK). Firefly luciferase activities were normalized against Renilla luciferase activities.
Western blotting
Whole cell lysates were harvested in ice-cold lysis buffer [50 mM Tris–HCl (pH 7.5), 250 mM NaCl, 3 mM EGTA, 3 mM EDTA, 1% Triton X-100, 10% glycerol, 0.5% NP40, 10 mM NaF, 1 mM sodium orthovanadate, 2 mM dithiothreitol (DTT), 1 mM phenylmethylsulphonyl fluoride, 3 µg·mL−1 aprotinin, 23 µM E64]. Lysates were clarified by centrifugation at 13 000×g for 1 min at 4°C, and protein concentrations were measured using Bradford assay. Equal amounts of total protein were loaded on SDS-PAGE gels. After electrophoresis samples were transferred to PVDF membranes (PerkinElmer Life Science), probed with anti-DUSP1, anti-COX-2 and anti-tubulin primary antibodies (Santa Cruz, Heidelberg, Germany; Cayman Chemical, Tallinn, Estonia and Sigma, respectively) then with appropriate horseradish peroxidase-coupled secondary antibodies (Dako, Cambridge, UK). Proteins were detected using the enhanced chemiluminescence system (GE Healthcare, Buckinghamshire, UK). COX-2 protein expression was estimated by scanning densitometry of Western blots using a calibrated imaging densitomer (GS-710; Bio-Rad Laboratories, Hertfordshire, UK) and the Phoretix ID software.
Measurement of cytokine expression
Supernatants were collected and stored at −20°C until used for measurement of cytokine proteins. IL-12p40 protein was detected by elisa, using an eBioscience kit, according to the manufacturer's instructions. IL-6, TNF-α and CXCL-1 proteins were detected simultaneously using the xMap Technology from Luminex® and 96-well filter plates (Millipore, Dundee, UK). Briefly, colour-coded Bio-plex beads (Bio-Rad Laboratories), or microspheres, were coupled to antibodies against IL-6, TNF-α and CXCL-1 (R&D Systems, Abingdon, UK) using an amine coupling kit from Biacore Life Sciences (Buckinghamshire, UK). Beads were coated with 50 µg·mL−1 of the primary antibodies; standards and samples were then added and left overnight at 4°C. After addition of 0.5 µg·mL−1 of the corresponding biotinylated secondary antibodies and streptavidin-PE (Peprotech), cytokines were detected by laser excitation of each internal dye identifying the different microspheres, using the Luminex® 100 Total System.
Measurement of mRNAs
Total cellular RNA was isolated using the RNAeasy® Mini Kit (QIAGEN, Crawley, UK), performing the recommended on-column DNase treatment step. mRNA levels were measured by quantitative real-time PCR using One-Step RT qPCR Mix from Eurogentec (Seraing, Belgium) and TaqMan probes purchased from Applied Biosystems (Paisley, UK) (mouse GAPDH Mm99999915_g1, DUSP1 Mm00457274_g1, COX2 Mm00478374_m1, human GAPDH Hs99999905_g1, DUSP1 Hs00610256_g1, COX-2 Hs00153133_m1). The PCRs were performed in a total volume of 10 µL. The amplification condition consists of an initial reverse transcriptase step of 30 min at 48°C, and 10 min denaturation at 95°C followed by 45 cycles of 3 s at 95°C and 30 s at 60°C. Rotor-Gene 3000 (Corbett Research Ltd, Crawley, UK) was used to quantify the mRNAs. Changes in abundance were assessed by the comparative threshold cycle (ΔCt) method and normalized against GAPDH (measured by the same method).
Statistics and calculation of dissociation indices
Statistical analysis was performed using one-sample t-test or anova with the Bonferroni's post-test for multiple comparisons. All tests were performed using Prism software version 5 (GraphPad, La Jolla, CA, USA). A P-value <0.05 was considered significant.
Dissociation indices were calculated for Cpd1 and Cpd2, based on their potencies, in other words, the EC50 values for activation of a GRE-dependent reporter and inhibition of an NF-κB-dependent reporter, relative to the corresponding values for dex.
Dissociation indices were also calculated on the basis of the efficacies of Cpd1 and Cpd2 for activation of the GRE-dependent reporter and repression of the NF-κB-dependent reporter, relative to the corresponding values for dex.
 |
Results
Compounds
The two compounds used in this study were selected from patents registered by two companies that have led research into SGRMs. Compound 1 (Jaroch et al., 2002) is closely related to ZK 216348 (Schacke et al., 2004) and ZK 245186 (Schacke et al., 2009) (Figure 1). In THP-1 cells stimulated with LPS, Cpd1 inhibited the expression of IL-8 with 77% efficacy and EC50 of 4.3 × 10−9 M. For comparison, prednisolone (pred) inhibited expression of IL-8 with 95% efficacy and EC50 of 2.4 × 10−8 M. In the croton oil ear oedema model, pred and Cpd1 were similarly effective at a dose of 30 mg·kg−1, inhibiting inflammation by 81% and 84%. In the same model, pred induced liver tyrsoine aminotransferase (a surrogate marker of metabolic side effects) by 8-fold, Cpd1 by 3.7-fold (Jaroch et al., 2002). Cpd2 (Coghlan et al., 1999; Kym et al., 2003) is very closely related to LGD-5552 (Lopez et al., 2008) (Figure 1). It is highly selective for GR over progesterone receptor (PR), the respective Ki values being 1.5 × 10−9 M and 1.434 × 10−6 M.
Figure 1.
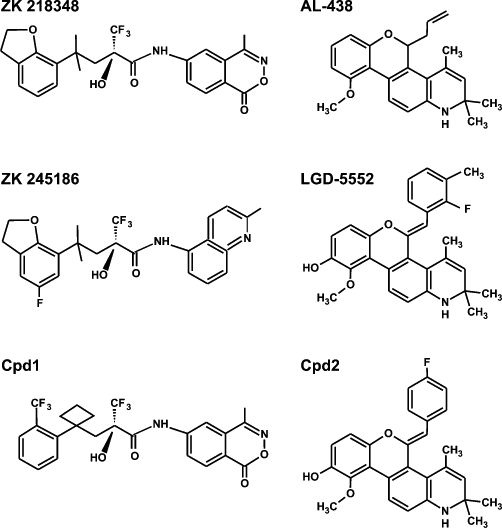
Molecular structures of Cpd1, Cpd2 and near relatives.
Dissociated properties of Cpd1 and Cpd2
Dissociated properties of Cpd1 and Cpd2 were determined using reporters with multimerized binding sites for GR and NF-κB. Similar methods have previously been used to identify SGRMs (Vayssiere et al., 1997) and continue to be used as screening tools in the pharmaceutical industry. The transactivation functions of Cpd1 and Cpd2 were assessed using an A549 pulmonary epithelial cell line stably transfected with a luciferase reporter containing two tandem GREs derived from the rat TAT gene upstream of a minimal TATA box from the rabbit β-globin gene (Figure 2A) (Chivers et al., 2006). Dex activated this construct with Amax of 16.4 and EC50 of 4.1 × 10−9 M. The novel GR ligands up-regulated luciferase expression with Amax and EC50 values of 8.4 and 2.9 × 10−8 M (Cpd1); 5.9 and 1.30 × 10−7 M (Cpd2), respectively. Both Cpd1 and Cpd2 therefore have relatively low efficacy and potency in this assay of transcriptional activation. Activation of the GRE reporter by dex, Cpd1 or Cpd2 was effectively blocked by an equivalent concentration of the GR antagonist RU486 (mifepristone), confirming that the SGRMs regulate transcription via GR (Figure 2C). Since Cpd1 and Cpd2 appeared to function as partial agonists of GR-mediated transcription, we considered whether they might impair transcriptional activation by a full agonist such as dex. To address this question, the GRE reporter was activated by 10−8 M dex in the presence of increasing concentrations of Cpd1 or Cpd2, from 10−9 to 10−7 M (Figure 2D). Statistically significant impairment of dex-induced transcription was observed only in the presence of a 10-fold molar excess of Cpd2 over dex.
Figure 2.
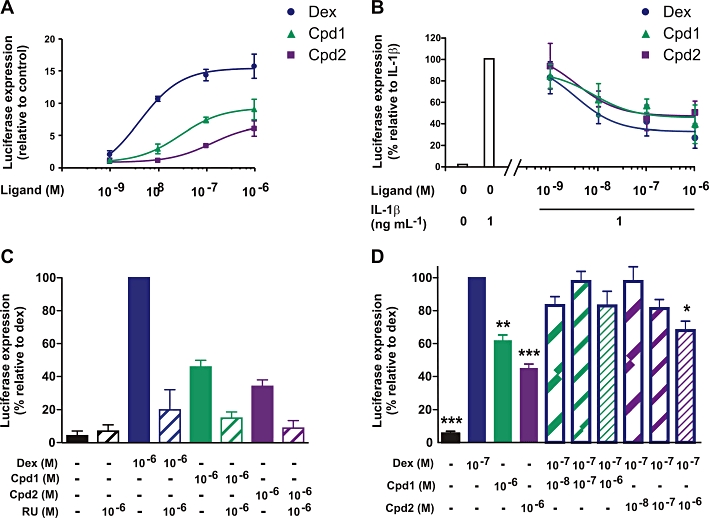
Dissociation parameters and GR dependence of Cpd1 and Cpd2. (A) A549 cells stably transfected with a GRE-dependent reporter were incubated for 6 h with vehicle (0.1% DMSO) or increasing concentrations (10−9 to 10−6 M) of dex, Cpd1 and Cpd2. Cell lysates were prepared, and luciferase activities were measured. Graph indicates mean ± SEM luciferase activities relative to vehicle treated control cells from four independent experiments. (B) A549 cells were transiently transfected with a reporter plasmid expressing firefly luciferase under the control of three tandem NF-κB sites, and with a control plasmid expressing Renilla luciferase. The following day, cells were pretreated for 2 h with increasing concentrations (10−9 to 10−6 M) of dex, Cpd1 and Cpd2, then stimulated with 1 ng·mL−1 of IL-1β for 6 h. Cell lysates were harvested, and firefly and Renilla luciferase activities were measured. Firefly luciferase activities were normalized against Renilla luciferase activities to correct for variations in transfection efficiency. The graph represents % of the activity of the IL-1β-stimulated cells ± SEM from four independent experiments. (C) The A549 GRE reporter cell line was incubated for 6 h with vehicle (0.1% DMSO) or 10−6 M dex, Cpd1 or Cpd2 in the absence or presence of 10−6 M RU486 (RU). Luciferase activities are expressed relative to that of cells treated with dex alone. The graph shows averages from two independent experiments ± SD. (D) The A549 GRE reporter cell line was incubated for 6 h with vehicle, dex and/or SGRMs at the concentrations indicated. Luciferase activities are expressed relative to that of cells treated with 10−7 M dex alone. The graph shows averages from three independent experiments ± SEM. ***P < 0.005; **P < 0.01; *P < 0.05 relative to cells treated with 10−7 M dex alone.
Transrepression was then tested following transient transfection of A549 cells with a luciferase reporter construct containing three NF-κB binding sites from the human COX-2 gene upstream of a minimal TATA box (Figure 2B) (Holden et al., 2007). This construct was strongly activated by IL-1β and dose-dependently inhibited by dex with an EC50 of 3.4 × 10−9 M. However, the extent of inhibition did not exceed 67%. Cpd1 and Cpd2 impaired the activation of the NF-κB reporter with similar efficacy (53% and 54%) and potency (EC50 values of 7.8 × 10−9 M and 3.5 × 10−9 M). Dissociation indices were calculated for Cpd1 and Cpd2, based on their potency and efficacy of transactivation and transrepression, relative to the corresponding values for the reference compound dex (see Methods). In either case, a dissociation index greater than 1 indicates selective capacity for transrepression over transactivation. By potency, Cpd1 and Cpd2 had dissociation indices of 3.04 and 30.8, respectively. By efficacy, their dissociation indices were 2.53 and 4.32, respectively. Both compounds therefore appeared to conform to the description of selective GR modulators.
The extent of inhibition of a NF-κB reporter by both dex and the SGRMs was less than anticipated; for example, it was invariably less than the extent of down-regulation of COX-2 mRNA in the same cells (see later, Figure 5). We tested responses of the same NF-κB reporter in a stably transfected A549 cell line; used an alternative reporter based on NF-κB binding sites from the HIV long terminal repeat; or varied concentrations of IL-1β between 0.2 and 10 ng·mL−1. Under no condition was reporter gene expression inhibited by more than 70% (data not shown).
Figure 5.
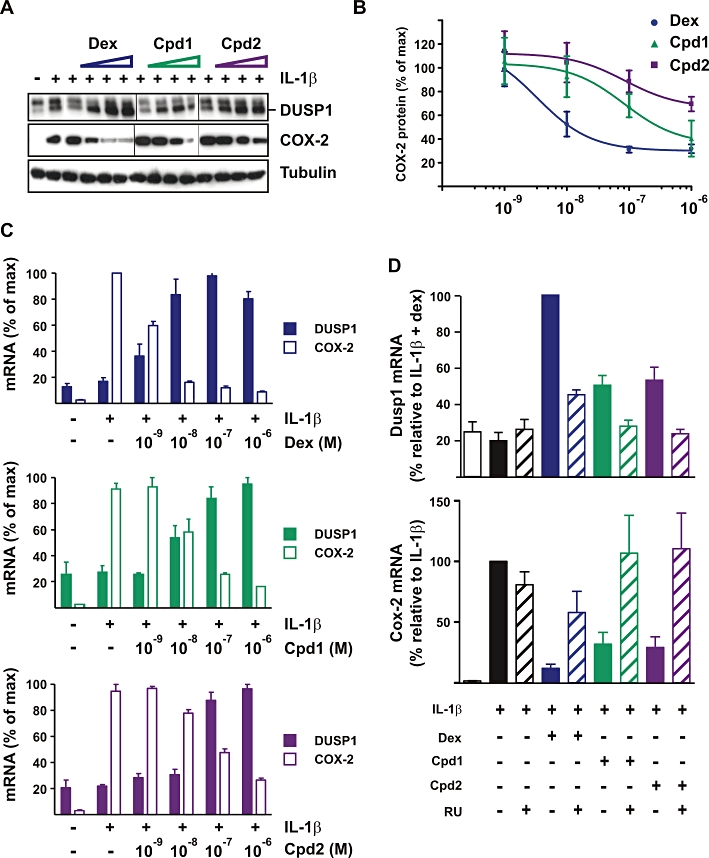
Effects of Cpd1 and Cpd2 on the expression of DUSP1 and COX-2 in A549 cells. (A, B, C) A549 cells were pretreated for 2 h with vehicle (0.1% DMSO) or increasing concentrations (10−9 to 10−6 M) of dex, Cpd1 or Cpd2 then challenged for 4 h with 1 ng·mL−1 of IL-1β. (A) Representative Western blot of DUSP1 and COX-2 protein expression. (B) COX-2 protein expression was quantified by scanning densitometry in three independent experiments. (C) Dusp1 and Cox-2 mRNA were measured by real-time PCR and plotted relative to maximum expression. Graphs show averages ± SEM from three independent experiments. (D) A549 cells were treated with IL-1β (1 ng·mL−1), RU486 (RU), dex, Cpd1, Cpd2 (each 10−6 M) in the combinations indicated. Dusp1 and Cox-2 mRNAs were measured and expressed with respect to cells treated with either IL-1β alone (COX-2) or IL-1β+ dex (Dusp1). Graphs represent averages ± SEM from four independent experiments.
SGRMs are able to induce expression of DUSP1
We and others previously identified two GC responsive regions (GRRs) within the human DUSP1 5′ region, at −1.3 and −4.6 kb with respect to the transcription start site (Johansson-Haque et al., 2008; Shipp et al., 2010; Tchen et al., 2010). Activation of GRR-4.6 by dex was impaired by mutation of the dimerization domain of GR, whereas activation of GRR-1.3 by dex was insensitive to this mutation (Tchen et al., 2010), suggesting that GR may interact differently with the two sites. Having established that SGRMs were capable of transcriptional activation of a stereotypical GRE reporter, we asked whether they could also regulate transcription via GRR-1.3 and -4.6 (Figure 3). The reporter constructs pGL3p-GRR-1.3 and pGL3p-GRR-4.6 (Tchen et al., 2010) are based on the pGL3p vector (Promega), in which firefly luciferase is expressed under the control of an SV40 early promoter. Upstream of the SV40 promoter, pGL3p-GRR-1.3 contains the region of the human Dusp1 locus from −1366 to −1237 with respect to the transcription start site. pGL3p-GRR-4.6 contains the region −4834 to −4369. Cpd1 and Cpd2 significantly activated transcription via both GRR-1.3 and GRR-4.6. The proximal element (GRR-1.3) was slightly less responsive to the two SGRMs than to dex. The distal element (GRR-4.6) was approximately fivefold more responsive to dex than to either of the SGRMs. It was also established by chromatin IP (ChIP), that all three ligands could promote recruitment of GR to both GRR-1.3 and GRR-4.6 in HeLa cells (not shown).
Figure 3.
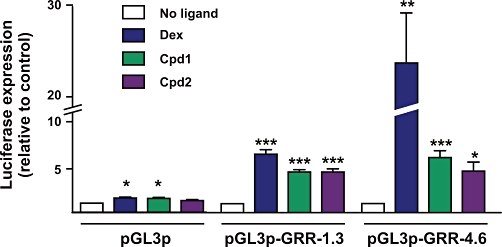
Cpd1 and Cpd2 activate transcription via GC-responsive regions of the human DUSP1 gene. HeLa cells were transiently transfected with pGL3p or derivatives that contain GRR-1.3 or GRR-4.6 from the human DUSP1 gene. A plasmid expressing Renilla luciferase was co-transfected as a control for transfection efficiency. Cells were treated with vehicle (0.1% DMSO) or 10−7 M GR ligands as indicated for 6 h, then lysates were prepared, firefly and Renilla luciferase activities were measured and fold responses to ligand were calculated. The graph represents mean ± SEM from three (pGL3p), six (pGL3p-GRR-1.3) or seven (pGL3p-GRR-4.6) independent experiments. ***P < 0.005; **P < 0.01; *P < 0.05 relative to vehicle-treated cells.
The ability of GR ligands to induce expression of DUSP1 mRNA and protein was next tested in A549, HeLa (human epithelial carcinoma cell line), RAW264.7 (mouse macrophage-like cell line) and BMM (primary mouse bone marrow-derived macrophages) (Figure 4). Note that BMMs displayed anomalous responses to the highest doses of GR ligands (Figure 4D), possibly due to effects on cell viability or proliferation. This phenomenon was not investigated further. The pattern of response of the DUSP1 gene was highly variable between cell types and was not predictable from the response of the GRE reporter, even within a single cell type. For example, in A549 cells Cpd2 was a poor activator of the GRE reporter but activated the endogenous DUSP1 gene with similar efficacy to dex (although with a rather higher EC50). In HeLa cells, the endogenous DUSP1 gene responded similarly to dex and Cpd2. Cpd1 reached the same efficacy (fold activation) but had a higher EC50 (lower potency) than the other two compounds. Compared to dex, both SGRMs had low efficacy and potency in RAW264.7 cells and in primary murine macrophages. Rank orders of efficacy and potency of the three compounds for activation of DUSP1 gene expression were not the same for any two cell lines, illustrating the extreme variability of response.
Figure 4.

Induction of DUSP1 mRNA by Cpd1 and Cpd2. (A) A549 (B) HeLa (C) RAW cells and (D) BMM were treated with increasing concentrations (10−9 to 10−6 M) of dex, Cpd1 and Cpd2 for 1 h. mRNA was harvested and DUSP1 expression quantified using RT-PCR. For each cell type, results were presented as fold induction relative to cells treated with vehicle alone (0.1% DMSO). Graphs indicate means ± SEM from at least three independent experiments.
Anti-inflammatory effects of SGRMs are partially dependent on DUSP1
Having shown that SGRMs are able to induce expression of DUSP1, we investigated the relationship between DUSP1 up-regulation and anti-inflammatory effects of SGRMs in A549 cells (Figure 5) RAW264.7 cells and mouse macrophages (Figure 6). Suppression of COX-2 was selected as a read-out of anti-inflammatory efficacy, because this gene is up-regulated by different pro-inflammatory stimuli in many cell types, and is a well characterized GC target with NF-κB sites in its promoter. In A549 cells, the down-regulation of Cox-2 mRNA and protein mirrored the up-regulation of Dusp1 mRNA and protein (Figure 5A–C). Inhibition of COX-2 protein expression was estimated by scanning densitometry of Western blots. EC50 values for induction of Dusp1 mRNA and inhibition of COX-2 mRNA and COX-2 protein were calculated. EC50 values for induction of DUSP1 protein could not be determined because of non-specific background in the Western blots. For each GR ligand, the calculated EC50 values for DUSP1 induction and COX-2 inhibition were close to one another (Table 1). At the mRNA level, the correspondence between Dusp1 up-regulation and Cox-2 inhibition can be seen in the symmetry of the dose-response curves (Figure 5C). RU486 incompletely blocked the up-regulation of Dusp1 mRNA by dex and incompletely rescued COX-2 from dex-mediated suppression (Figure 5D). RU486 completely prevented the up-regulation of Dusp1 mRNA by Cpd1 or Cpd2 and effectively rescued COX-2 mRNA from inhibition by either of the SGRMs. Like dex, Cpd1 and Cpd2 therefore exert anti-inflammatory effects via GR and not via an off-target mechanism.
Figure 6.
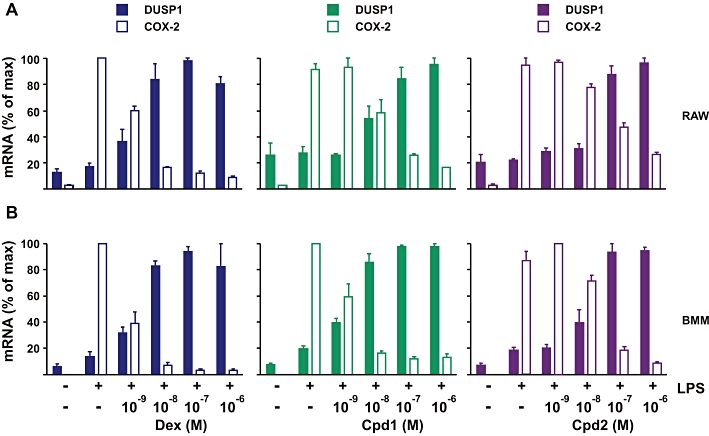
Effects of Cpd1 and Cpd2 on the expression of DUSP1 and COX-2 in RAW264.7 cells and BMMs. RAW264.7 cells (A) or BMMs (B) were pretreated for 2 h with vehicle (0.1% DMSO) or increasing concentrations (10−9 to 10−6 M) of dex, Cpd1 or Cpd2 then challenged for 4 h with 10 ng·mL−1 of LPS. COX-2 and Dusp1 mRNAs were measured and plotted as in Figure 5D.
Table 1.
EC50 values for induction of DUSP1 mRNA, inhibition of COX-2 mRNA and protein expression in A549 cells
| EC50 (M) | |||
|---|---|---|---|
| DUSP1 mRNA | COX-2 mRNA | COX-2 protein | |
| Dex | 7.0 × 10−9 | 6.3 × 10−9 | 5.2 × 10−9 |
| Cpd 1 | 1.52 × 10−8 | 1.01 × 10−8 | 3.35 × 10−8 |
| Cpd 2 | 3.83 × 10−8 | 3.19 × 10−8 | 8.52 × 10−8 |
Values were calculated on the basis of the data illustrated in Figure 5B and C.
In RAW264.7 cells (Figure 6A) or primary mouse macrophages (Figure 6B) stimulated with LPS, the up-regulation of Dusp1 and down-regulation of COX-2 mRNAs by GR ligands displayed similar dose-dependence, again demonstrated by the symmetry of the dose-response curves. In BMM, Cpd1 and Cpd2 up-regulated Dusp1 expression with similar Amax (5 and 5.4, respectively), slightly less than the Amax value of 7.1 for induction of Dusp1 by dex. Cpd1 induced Dusp1 with EC50 of 1.4 × 10−9 M; therefore, Dusp1 expression was near maximal at both 10−8 and 10−6 M. Cpd2 induced Dusp1 with a considerably higher EC50 of 1.9 × 10−8 M; therefore, expression was still increasing in the range 10−8 and 10−6 M. These observations provide context for the following analysis of responses to SGRMs in Dusp1+/+ and Dusp1−/− macrophages.
The above results are consistent with, but do not prove, an important role for DUSP1 in the anti-inflammatory effects of both dex and SGRMs. To investigate this further, we tested effects of GR ligands on the expression of several pro-inflammatory genes in Dusp1+/+ and Dusp1−/− mouse BMMs. As previously reported (Chivers et al., 2006; Hammer et al., 2006), Dusp1−/− macrophages overexpressed IL-6 and TNFα proteins when stimulated with LPS, whilst the expression of IL-12p40 was significantly less than in Dusp1+/+ macrophages (Figure 7). Increased expression of COX-2 mRNA in the knockout macrophages did not reach statistical significance. In agreement with previous observations (Abraham et al., 2006), 10−8 or 10−6 M dex strongly decreased the expression of TNF protein in Dusp1+/+ macrophages but had relatively little effect in Dusp1−/− macrophages (Figure 8). We previously reported that dex down-regulated Il-6 mRNA in a manner largely dependent on DUSP1. Here it is shown that inhibition of IL-6 expression at the protein level is entirely dependent on DUSP1. The same experiments confirm that dex inhibits expression of IL-12p40 protein independently of DUSP1, emphasizing that both DUSP1-dependent and -independent mechanisms contribute to the anti-inflammatory action of GCs (Abraham et al., 2006). Finally, inhibitory effects of dex on COX-2 mRNA were largely DUSP1-dependent as previously shown.
Figure 7.
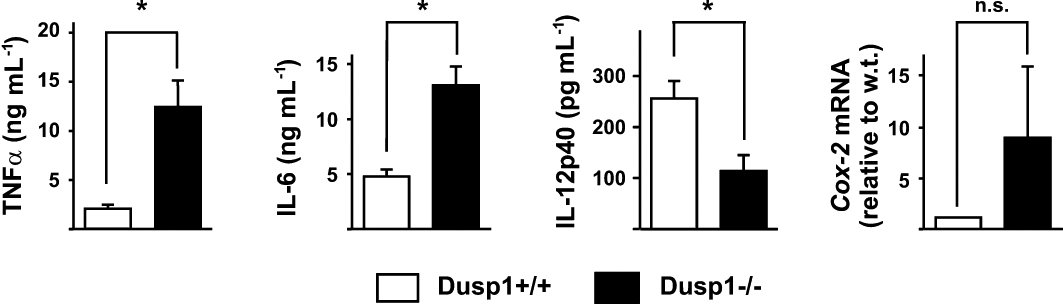
Altered expression of inflammatory mediators in Dusp1−/− macrophages. Dusp1+/+ and Dusp1−/− BMM were stimulated with 10 ng·mL−1 LPS for 4 h, supernatants were harvested and mRNA was isolated. TNFα and IL-6 proteins were quantified by Luminex, IL-12p40 by elisa, and COX-2 mRNA by quantitative PCR. Graphs represent averages from four independent experiments ± SEM. * indicates statistically significant difference between Dusp1+/+ and Dusp1−/− BMM (P < 0.05). n.s., not statistically significant.
Figure 8.
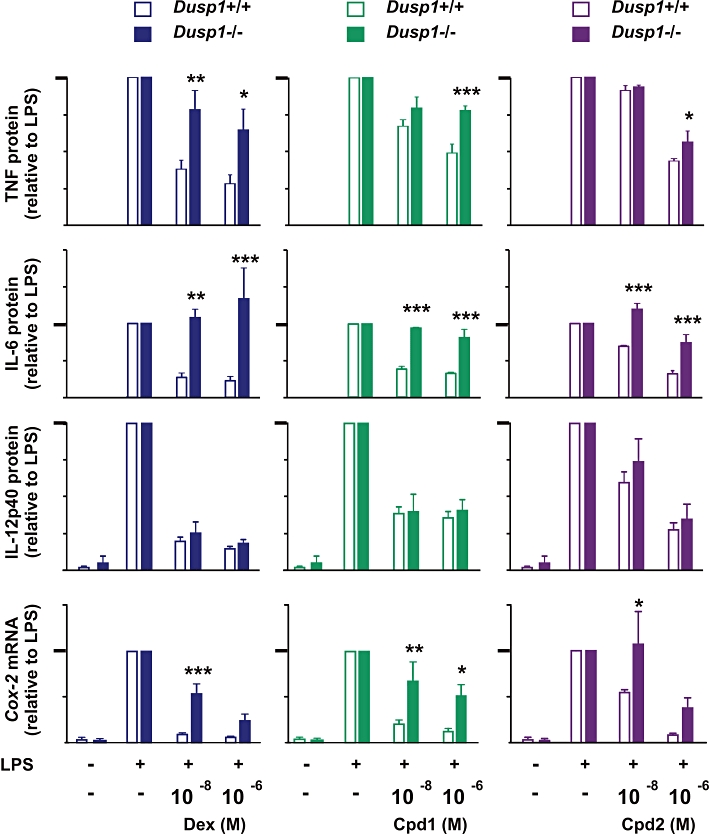
Effects of Cpd1 and Cpd2 on expression of pro-inflammatory mediators in Dusp1+/+ and Dusp1−/− macrophages. BMM from Dusp1+/+ mice and Dusp1−/− mice were pretreated for 2 h with vehicle (0.1% DMSO), 10−8 or 10−6 M dex, Cpd1 or Cpd2, then challenged with 10 ng·mL−1 of LPS for another 4 h. TNF, IL-6 and IL-12p40 protein expression were measured by Luminex or elisa and COX-2 mRNA by real-time PCR. Results are presented as percentages of the response in cells treated with LPS and vehicle. The y-axes differ in scale. In each case, the 100% level is represented by a heavy tick mark. Graphs indicate mean ± SEM from at least three independent experiments. Statistical analysis is shown for Dusp1−/− versus Dusp1+/+ under the same conditions. ***P < 0.005; **P < 0.01; *P < 0.05.
In terms of its anti-inflammatory effects, Cpd1 generally behaved like a version of dex with lower efficacy but similar potency. It inhibited IL-6 less effectively than dex but again in an entirely DUSP1-dependent manner. Its inhibitory effects on COX-2 mRNA were less strong than those of dex but also largely dependent on DUSP1. In all of these cases, inhibitory effects were similar at 10−8 and 10−6 M. Cpd1-mediated inhibition of TNF expression was also clearly dependent on DUSP1, although in this case the difference between Dusp1+/+ and Dusp1−/− macrophages became significant only at 10−6 M. IL-12p40 expression was equally inhibited by Cpd1 in Dusp1+/+ and Dusp1−/− macrophages. These observations suggest that, like dex, Cpd1 exerts anti-inflammatory effects in part via DUSP1.
Cpd1 and Cpd2 had similar inhibitory effects on pro-inflammatory genes at 10−6 M concentration in wild-type macrophages. At 10−8 M concentration, Cpd2 was invariably less effective than Cpd1. Nevertheless, some of the anti-inflammatory effects of Cpd2 were clearly dependent on DUSP1. For example, expression of IL-6 was significantly decreased by either 10−8 or 10−6 M Cpd2 in Dusp1+/+ macrophages but not in Dusp1−/− macrophages. In the cases of TNF protein or COX-2 mRNA, responses to Cpd2 significantly differed between Dusp1+/+ and Dusp1−/− macrophages at only one of the two concentrations tested. Inhibition of IL-12p40 expression was independent of DUSP1 as expected. The overall picture is of a compound that depends upon DUSP1 for some of its anti-inflammatory effects but induces DUSP1 expression with relatively low potency.
Discussion
Novel compounds that preferentially mediate transrepression have been predicted to cause fewer side effects than classical GCs (Newton and Holden, 2007; Schacke et al., 2007; Berlin, 2010; De Bosscher et al., 2010). The notional mechanistic uncoupling of therapeutic and harmful consequences of GR activation suggested a straightforward course of action. Safer GR ligands might be discovered through screening strategies based on constructs that contain multimerized binding sites for GR itself (as a reporter for transactivating function) or for NF-κB or AP-1 (as reporters for transrepressing function). However, this idea has its roots in relatively simplistic and now outdated conceptions of how GR controls gene expression. Transcriptional activation by GR is now known to be a remarkably diverse process. Binding sites for GR can extensively vary from the idealized consensus AGAACAnnnTGTTCT, only five or six positions within this sequence being strongly constrained (So et al., 2007; Reddy et al., 2009; John et al., 2011). Single nucleotide variations in binding site sequence can have profound effects on the conformation adopted by GR and the downstream consequences (Meijsing et al., 2009). Only about 0.4% of possible binding sites are recognized by GR in one cell type, this repertoire being dictated by cell type-specific modulation of chromatin accessibility (John et al., 2011). GR also co-operates with a large number of other transcription factors to control transcription (Clark, 2007; Kassel and Herrlich, 2007). Some of these accessory factors are likely to be required for the establishment of domains of open chromatin structure within which GR can bind to DNA. At individual GC-regulated genes, and probably at individual cis-acting elements of one gene, GR displays different requirements for transcriptional cofactors (Chen et al., 2006; Galliher-Beckley et al., 2008; John et al., 2011). Finally, GR is extensively post-translationally modified, and GC-responsive elements may display differential requirements for different GR modifications (Beck et al., 2009; Galliher-Beckley and Cidlowski, 2009).
To stand as representative of all GR-mediated transcriptional activation events is therefore an unreasonably large burden for one highly simplified reporter, or even for one or two endogenous genes (Clark, 2007). Some of the problems of extrapolating from simple reporters are well illustrated by the present study. Even within one cell type (A549), the dose-dependence of induction of Dusp1 gene expression by the two SGRMs did not resemble the dose dependence of activation of the GRE reporter (compare Figures 2A and 4A). The predictive value of the reporter became even poorer when other cell types were considered (Figure 4B–D). Individual response elements of the Dusp1 locus were not necessarily better predictors of the behaviour of the endogenous gene. For example, a previous study identified a powerfully GC-responsive region located 4.6 kb upstream of the Dusp1 transcription start site (Tchen et al., 2010). In HeLa cells, this element was quite weakly activated by Cpd1 and Cpd2 compared with dex (Figure 3), whereas the endogenous gene was similarly activated by all three GR ligands at the relevant dose of 10−8 M (Figure 4B). At least some of this variation in response of reporter constructs and endogenous GC-regulated genes is probably explained by differential cofactor requirements and variable expression of cofactors in different cell types.
It is hard to escape the conclusion that idealized reporters containing tandem GR binding sites are of little practical help when trying to determine the transactivating properties (and hence dissociated nature) of GR ligands. RU24858, an earlier example of a supposedly dissociated GR ligand, was later found to be capable of up-regulating a subset of GR-regulated genes (Chivers et al., 2006; Janka-Junttila et al., 2006). LGD-5552, a near-relative of Cpd2, was also found to be capable of transcriptional activation but differed from the classical GC prednisolone in the profile of genes activated (Lopez et al., 2008). When a compound is described as being dissociated or as having poor capacity to activate transcription, it should therefore be asked exactly what this means and how it has been demonstrated. It is unclear whether genuine and consistent separation between transactivation and transrepression properties of GR can be demonstrated using simple reporters, or whether it can be achieved. We and other investigators have questioned whether such separation is even desirable, given that GCs up-regulate a number of anti-inflammatory factors, and depend on these factors for at least some of their anti-inflammatory effects (Smoak and Cidlowski, 2006; Clark, 2007; Newton and Holden, 2007).
Both Cpd1 and Cpd2 have emerged from deliberate efforts to selectively promote the transrepression rather than transactivation function of GR. With respect to transcriptional activation of GRE reporters and endogenous Dusp1 genes, both compounds behaved like partial agonists of GR. However, Cpd1 did not significantly block transcriptional activation by the full agonist dex (Figure 2D). This surprising finding is reminiscent of the first generation SGRM RU24858, which was also found not to inhibit transcriptional activation by the full agonist dex (Vayssiere et al., 1997). A possible conclusion is that dex and Cpd1 recognize different surfaces of GR and do not bind the receptor in a mutually exclusive manner. This appears unlikely because of the ability of RU486 to antagonize transcriptional activation by either compound (Figure 2C). A second possibility is that Cpd1 has relatively low affinity for GR in intact cells and is therefore not an effective competitor for binding. It should also be pointed out that demonstration of the expected partial antagonism is quite challenging in the case of Cpd1. At the respective concentrations of 10−7 and 10−6 M, dex and Cpd1 differ by only about 40% in transactivation of the GRE reporter (Figure 2D). This is a relatively small window in which to demonstrate competitive inhibition by Cpd1 of the response to dex. We cannot conclude that partial antagonism does not occur, only that we have been unable to demonstrate it. Cpd2 being a weaker activator of transcription, partial antagonistic behaviour was more straightforward to demonstrate (Figure 2D). To fully understand the partial agonist/antagonist properties of the two SGRMs requires biochemical and crystallographic studies that are beyond the scope of the present study. In any case, this issue does not effect our major conclusions.
Most importantly, the anti-inflammatory effects of the SGRMs were in direct proportion to their capacity to induce DUSP1 expression in a number of cell types and demonstrably dependent on DUSP1 in mouse macrophages. Cpd2, which had the higher dissociation indices, was the weaker inducer of DUSP1 and the poorer anti-inflammatory agent in all experimental settings used. The importance of DUSP1 as a mediator effect of GR was particularly clearly demonstrated in the case of IL-6. Neither dex, Cpd1 nor Cpd2 was capable of decreasing expression of IL-6 protein in Dusp1−/− macrophages. It is ironic that IL-6 is a well-characterized NF-κB target, whose inhibition by GCs has usually been interpreted in terms of transrepression.
Cpd1 is closely related to ZK 245186 (otherwise known as BOL-303242-X or Mapracorat), which is being tested as a novel drug for dermatological or opthalmological indications such as atopic dermatitis, dry-eye syndrome and postoperative eye inflammation (Schacke et al., 2009; Zhang et al., 2009; Cavet et al., 2010; Pfeffer et al., 2010; Shafiee et al., 2010). ZK 245186 was found to inhibit JNK and p38 MAPK phosphorylation in corneal epithelial cells subjected to hyperosmolar stress (Cavet et al., 2010). Inhibition of the MAPKs is thought to contribute to the therapeutic effect of the SGRM and may be mediated by up-regulation of DUSP1. The classical GC dex induces expression of Dusp1 in primary human lens epithelium (Gupta et al., 2005). In the present study, we were not able to assess responses to ZK 245186 itself, and we acknowledge the danger of extrapolating too far from in vitro studies. Nevertheless, we consider it quite possible that the anti-inflammatory efficacy and safety of SGRMs like ZK 245186 have little to do with whether or not these compounds are ‘dissociated’. This raises important questions about how the properties of the current generation of SGRMs should be interpreted and how the next generation of SGRMs might best be identified.
Acknowledgments
The work of AC's group is supported by Arthritis Research UK. We are grateful to Bristol-Myers Squibb for provision of the original Dusp1−/− mouse strain, and Roche Palo Alto for provision of SGRMs. We sincerely thank Robert Newton for the gift of reporter constructs and A549 cell lines stably transfected with these constructs.
Glossary
- AP-1
activating protein 1
- BMM
bone marrow-derived macrophage
- CXCL1
chemokine (CXC motif) ligand 1
- dex
dexamethasone
- DUSP1
dual specificity phosphatase 1
- GC
glucocorticoid
- GR
GC receptor
- GRE
GC response element
- GRR
GC responsive region
- SEGRA
selective GR agonist
- SGRM
selective GR modulator
Conflicts of interest
The authors have no conflict of interest.
References
- Abraham SM, Clark AR. Dual-specificity phosphatase 1: a critical regulator of innate immune responses. Biochem Soc Trans. 2006;34:1018–1023. doi: 10.1042/BST0341018. [DOI] [PubMed] [Google Scholar]
- Abraham SM, Lawrence T, Kleiman A, Warden P, Medghalchi M, Tuckermann J, et al. Antiinflammatory effects of dexamethasone are partly dependent on induction of dual specificity phosphatase 1. J Exp Med. 2006;203:1883–1889. doi: 10.1084/jem.20060336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes PJ. Corticosteroids: the drugs to beat. Eur J Pharmacol. 2006;533:2–14. doi: 10.1016/j.ejphar.2005.12.052. [DOI] [PubMed] [Google Scholar]
- Beck IM, Vanden Berghe W, Vermeulen L, Yamamoto KR, Haegeman G, De Bosscher K. Crosstalk in inflammation: the interplay of glucocorticoid receptor-based mechanisms and kinases and phosphatases. Endocr Rev. 2009;30:830–882. doi: 10.1210/er.2009-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlin M. Recent advances in the development of novel glucocorticoid receptor modulators. Expert Opin Ther Pat. 2010;20:855–873. doi: 10.1517/13543776.2010.493876. [DOI] [PubMed] [Google Scholar]
- Bladh LG, Johansson-Haque K, Rafter I, Nilsson S, Okret S. Inhibition of extracellular signal-regulated kinase (ERK) signaling participates in repression of nuclear factor (NF)-kappaB activity by glucocorticoids. Biochim Biophys Acta. 2009;1793:439–446. doi: 10.1016/j.bbamcr.2008.11.013. [DOI] [PubMed] [Google Scholar]
- Cavet ME, Harrington KL, Ward KW, Zhang JZ. Mapracorat, a novel selective glucocorticoid receptor agonist, inhibits hyperosmolar-induced cytokine release and MAPK pathways in human corneal epithelial cells. Mol Vis. 2010;16:1791–1800. [PMC free article] [PubMed] [Google Scholar]
- Chen W, Rogatsky I, Garabedian MJ. MEDI14 and MED1 differentially regulate target-specific gene activation by the glucocorticoid receptor. Mol Endoncrinol. 2006;20:560–572. doi: 10.1210/me.2005-0318. [DOI] [PubMed] [Google Scholar]
- Chivers JE, Gong W, King EM, Seybold J, Mak JC, Donnelly LE, et al. Analysis of the dissociated steroid RU24858 does not exclude a role for inducible genes in the anti-inflammatory actions of glucocorticoids. Mol Pharmacol. 2006;70:2084–2095. doi: 10.1124/mol.106.025841. [DOI] [PubMed] [Google Scholar]
- Cho IJ, Kim SG. A novel mitogen-activated protein kinase phosphatase-1 and glucocorticoid receptor (GR) interacting protein-1-dependent combinatorial mechanism of gene transrepression by GR. Mol Endocrinol. 2009;23:86–99. doi: 10.1210/me.2008-0257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AR. Anti-inflammatory functions of glucocorticoid-induced genes. Mol Cell Endocrinol. 2007;275:79–97. doi: 10.1016/j.mce.2007.04.013. [DOI] [PubMed] [Google Scholar]
- Coghlan MJ, Elmore SW, Kort ME, Kym PR, Moore JL, Pratt JK, et al. 1999. Preparation of glucocorticoid-selective anti-inflammatory agents. Patent WO99041256.
- De Bosscher K, Haegeman G, Elewaut D. Targeting inflammation using selective glucocorticoid receptor modulators. Curr Opin Pharmacol. 2010;10:497–504. doi: 10.1016/j.coph.2010.04.007. [DOI] [PubMed] [Google Scholar]
- Diefenbacher M, Sekula S, Heilbock C, Maier JV, Litfin M, van Dam H, et al. Restriction to Fos family members of Trip6-dependent coactivation and glucocorticoid receptor-dependent trans-repression of activator protein-1. Mol Endocrinol. 2008;22:1767–1780. doi: 10.1210/me.2007-0574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorfman K, Carrasco D, Gruda M, Ryan C, Lira SA, Bravo R. Disruption of the erp/mkp-1 gene does not affect mouse development: normal MAP kinase activity in ERP/MKP-1-deficient fibroblasts. Oncogene. 1996;13:925–931. [PubMed] [Google Scholar]
- Frijters R, Fleuren W, Toonen EJ, Tuckermann JP, Reichardt HM, Van der Maaden H, et al. Prednisolone-induced differential gene expression in mouse liver carrying wild type or a dimerization-defective glucocorticoid receptor. BMC Genomics. 2010;11:359. doi: 10.1186/1471-2164-11-359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furst R, Schroeder T, Eilken HM, Bubik MF, Kiemer AK, Zahler S, et al. MAPK phosphatase-1 represents a novel anti-inflammatory target of glucocorticoids in the human endothelium. FASEB J. 2007;21:74–80. doi: 10.1096/fj.06-6752com. [DOI] [PubMed] [Google Scholar]
- Galliher-Beckley AJ, Cidlowski JA. Emerging roles of glucocorticoid receptor phosphorylation in modulating glucocorticoid hormone action in health and disease. IUBMB Life. 2009;61:979–986. doi: 10.1002/iub.245. [DOI] [PubMed] [Google Scholar]
- Galliher-Beckley AJ, Williams JG, Collins JB, Cidlowski JA. Glycogen synthase kinase 3β-mediated serine phosphorylation of the human glucocorticoid receptor redirects gene expression profiles. Mol Cell Biol. 2008;28:7309–7322. doi: 10.1128/MCB.00808-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass CK, Saijo K. Nuclear receptor transrepression pathways that regulate inflammation in macrophages and T cells. Nat Rev Immunol. 2010;10:365–376. doi: 10.1038/nri2748. [DOI] [PubMed] [Google Scholar]
- Gupta V, Galante A, Soteropoulos P, Guo S, Wagner BJ. Global gene profiling reveals novel glucocorticoid induced changes in gene expression of human lens epithelial cells. Mol Vis. 2005;11:1018–1040. [PubMed] [Google Scholar]
- Hammer M, Mages J, Dietrich H, Servatius A, Howells N, Cato A, et al. Dual specificity phosphatase 1 (DUSP1) regulates a subset of LPS-induced genes and protects mice from lethal endotoxin shock. J Exp Med. 2006;203:15–20. doi: 10.1084/jem.20051753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillier SG. Diamonds are forever: the cortisone legacy. J Endocrinol. 2007;195:1–6. doi: 10.1677/JOE-07-0309. [DOI] [PubMed] [Google Scholar]
- Holden NS, Gong W, King EM, Kaur M, Giembycz MA, Newton R. Potentiation of NF-κB-dependent transcription and inflammatory mediator release by histamine in human airway epitheial cells. Br J Pharmacol. 2007;152:891–902. doi: 10.1038/sj.bjp.0707457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson AR, Roach SL, Higuchi RI. Recent developments in the discovery of selective glucocorticoid receptor modulators (SGRMs) Curr Top Med Chem. 2008;8:750–765. doi: 10.2174/156802608784535048. [DOI] [PubMed] [Google Scholar]
- Humphrey EL, Williams JH, Davie MW, Marshall MJ. Effects of dissociated glucocorticoids on OPG and RANKL in osteoblastic cells. Bone. 2006;38:652–661. doi: 10.1016/j.bone.2005.10.004. [DOI] [PubMed] [Google Scholar]
- Issa R, Xie S, Khorasani N, Sukkar M, Adcock IM, Lee KY, et al. Corticosteroid inhibition of growth-related oncogene protein-alpha via mitogen-activated kinase phosphatase-1 in airway smooth muscle cells. J Immunol. 2007;178:7366–7375. doi: 10.4049/jimmunol.178.11.7366. [DOI] [PubMed] [Google Scholar]
- Janka-Junttila M, Moilanen E, Hasala H, Zhang X, Adcock I, Kankaanranta H. The glucocorticoid RU24858 does not distinguish between transrepression and transactivation in primary human eosinophils. J Inflamm (Lond) 2006;3:1–10. doi: 10.1186/1476-9255-3-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaroch S, Lehmann M, Schmees N, Buchmann B, Rehwinkel H, Droescher P, et al. 2002. Preparation of amido-benzoxazinone/phthalides as non-steroidal inflammation inhibitors. Patent WO02010143.
- Johansson-Haque K, Palanichamy E, Okret S. Stimulation of MAPK-phospatase 1 (MKP-1) gene expression by glucocorticoids occurs through a tethering mechanism involving C/EBP. J Mol Endocrinol. 2008;41:239–249. doi: 10.1677/JME-08-0015. [DOI] [PubMed] [Google Scholar]
- John S, Sabo PJ, Thurman RE, Sung MH, Biddie SC, Johnson TA, et al. Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nat Genet. 2011;43:264–268. doi: 10.1038/ng.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang BN, Jude JA, Panettieri RA, Jr, Walseth TF, Kannan MS. Glucocorticoid regulation of CD38 expression in human airway smooth muscle cells: role of dual specificity phosphatase 1. Am J Physiol Lung Cell Mol Physiol. 2008;295:L186–L193. doi: 10.1152/ajplung.00352.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassel O, Herrlich P. Crosstalk between the glucocorticoid receptor and other transcription factors: Molecular aspects. Mol Cell Endocrinol. 2007;275:13–29. doi: 10.1016/j.mce.2007.07.003. [DOI] [PubMed] [Google Scholar]
- King EM, Holden NS, Gong W, Rider CF, Newton R. Inhibition of NF-kappaB-dependent transcription by MKP-1: transcriptional repression by glucocorticoids occurring via p38 MAPK. J Biol Chem. 2009;284:26803–26815. doi: 10.1074/jbc.M109.028381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiman A, Tuckermann JP. Glucocorticoid receptor action in beneficial and side effects of steroid therapy: lessons from conditional knockout mice. Mol Cell Endocrinol. 2007;275:98–108. doi: 10.1016/j.mce.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Kym PR, Kort ME, Coghlan MJ, Moore JL, Tang R, Ratajczyk JD, et al. Nonsteroidal selective glucocorticoid modulators: the effect of C-10 substitution on receptor selectivity and functional potency of 5-allyl-2,5-dihydro-2,2,4-trimethyl-1H-[1]benzopyrano[3,4-f]quinolines. J Med Chem. 2003;46:1016–1030. doi: 10.1021/jm020335m. [DOI] [PubMed] [Google Scholar]
- Lari R, Fleetwood AJ, Kitchener PD, Cook AD, Pavasovic D, Hertzog PJ, et al. Macrophage lineage phenotypes and osteoclastogenesis – complexity in the control by GM-CSF and TGFβ. Bone. 2007;40:323–326. doi: 10.1016/j.bone.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Lasa M, Brook M, Saklatvala J, Clark AR. Dexamethasone destabilizes cyclooxygenase 2 mRNA by inhibiting mitogen-activated protein kinase p38. Mol Cell Biol. 2001;21:771–780. doi: 10.1128/MCB.21.3.771-780.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasa M, Abraham SM, Boucheron C, Saklatvala J, Clark AR. Dexamethasone causes sustained expression of mitogen-activated protein kinase (MAPK) phosphatase 1 and phosphatase-mediated inhibition of MAPK p38. Mol Cell Biol. 2002;22:7802–7811. doi: 10.1128/MCB.22.22.7802-7811.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Zhang M, Hussain F, Triantaphyllopoulos K, Clark AR, Bhavsar PK, et al. Inhibition of p38-MAPK-dependent bronchial contraction after ozone by corticosteroids. Eur Respir J. 2010;37:933–942. doi: 10.1183/09031936.00021110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez FJ, Ardecky RJ, Bebo B, Benbatoul K, De Grandpre L, Liu S, et al. LGD-5552, an antiinflammatory glucocorticoid receptor ligand with reduced side effects, in vivo. Endocrinology. 2008;149:2080–2089. doi: 10.1210/en.2007-1353. [DOI] [PubMed] [Google Scholar]
- Meijsing SH, Pufall MA, So AY, Bates DL, Chen L, Yamamoto KR. DNA binding site sequence directs glucocorticoid receptor structure and activity. Science. 2009;324:407–410. doi: 10.1126/science.1164265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miner JN, Ardecky B, Benbatoul K, Griffiths K, Larson CJ, Mais DE, et al. Antiinflammatory glucocorticoid receptor ligand with reduced side effects exhibits an altered protein-protein interaction profile. Proc Natl Acad Sci USA. 2007;104:19244–19249. doi: 10.1073/pnas.0705517104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton R. Molecular mechanisms of glucocorticoid action: what is important? Thorax. 2000;55:603–613. doi: 10.1136/thorax.55.7.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton R, Holden NS. Separating transrepression and transactivation: a distressing divorce for the glucocorticoid receptor? Mol Pharmacol. 2007;72:799–809. doi: 10.1124/mol.107.038794. [DOI] [PubMed] [Google Scholar]
- Newton R, Leigh R, Giembycz MA. Pharmacological strategies for improving the efficacy and therapeutic ratio of glucocorticoids in inflammatory lung diseases. Pharmacol Ther. 2010;125:286–327. doi: 10.1016/j.pharmthera.2009.11.003. [DOI] [PubMed] [Google Scholar]
- Owens DM, Keyse SM. Differential regulation of MAP kinase signalling by dual-specificity protein phosphatases. Oncogene. 2007;26:3203–3213. doi: 10.1038/sj.onc.1210412. [DOI] [PubMed] [Google Scholar]
- Pfeffer BA, DeWitt CA, Salvador-Silva M, Cavet ME, Lopez FJ, Ward KW. Reduced myocilin expression in cultured monkey trabecular meshwork cells induced by a selective glucocorticoid receptor agonist: comparison with steroids. Invest Ophthalmol Vis Sci. 2010;51:437–446. doi: 10.1167/iovs.09-4202. [DOI] [PubMed] [Google Scholar]
- Quante T, Ng YC, Ramsay EE, Henness S, Allen JC, Parmentier J, et al. Corticosteroids reduce IL-6 in ASM cells via up-regulation of MKP-1. Am J Respir Cell Mol Biol. 2008;39:208–217. doi: 10.1165/rcmb.2007-0014OC. [DOI] [PubMed] [Google Scholar]
- Rauch A, Seitz S, Baschant U, Schilling AF, Illing A, Stride B, et al. Glucocorticoids suppress bone formation by attenuating osteoblast differentiation via the monomeric glucocorticoid receptor. Cell Metab. 2010;11:517–531. doi: 10.1016/j.cmet.2010.05.005. [DOI] [PubMed] [Google Scholar]
- Reddy TE, Pauli F, Sprouse RO, Neff NF, Newberry KM, Garabedian MJ, et al. Genomic determination of the glucocorticoid response reveals unexpected mechanisms of gene regulation. Genome Res. 2009;19:2163–2171. doi: 10.1101/gr.097022.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schacke H, Docke WD, Asadullah K. Mechanisms involved in the side effects of glucocorticoids. Pharmacol Ther. 2002;96:23–43. doi: 10.1016/s0163-7258(02)00297-8. [DOI] [PubMed] [Google Scholar]
- Schacke H, Schottelius A, Docke WD, Strehlke P, Jaroch S, Schmees N, et al. Dissociation of transactivation from transrepression by a selective glucocorticoid receptor agonist leads to separation of therapeutic effects from side effects. Proc Natl Acad Sci USA. 2004;101:227–232. doi: 10.1073/pnas.0300372101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schacke H, Berger M, Rehwinkel H, Asadullah K. Selective glucocorticoid receptor agonists (SEGRAs): novel ligands with an improved therapeutic index. Mol Cell Endocrinol. 2007;275:109–117. doi: 10.1016/j.mce.2007.05.014. [DOI] [PubMed] [Google Scholar]
- Schacke H, Zollner TM, Docke WD, Rehwinkel H, Jaroch S, Skuballa W, et al. Characterization of ZK 245186, a novel, selective glucocorticoid receptor agonist for the topical treatment of inflammatory skin diseases. Br J Pharmacol. 2009;158:1088–1103. doi: 10.1111/j.1476-5381.2009.00238.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafiee A, Bucolo C, Budzynski E, Ward KW, Lopez FJ. In vivo ocular efficacy profile of BOL-303242-X, a novel selective glucocorticoid receptor agonist, in rabbit models of ocular disease. Invest Ophthalmol Vis Sci. 2010;52:1422–1430. doi: 10.1167/iovs.10-5598. [DOI] [PubMed] [Google Scholar]
- Shipp LE, Lee JV, Yu CY, Pufall M, Zhang P, Scott DK, et al. Transcriptional regulation of human dual specificity protein phosphatase 1 (DUSP1) gene by glucocorticoids. PLoS ONE. 2010;5:e13754. doi: 10.1371/journal.pone.0013754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smoak K, Cidlowski JA. Glucocorticoids regulate tristetraprolin synthesis and posttranscriptionally regulate tumor necrosis factor alpha inflammatory signaling. Mol Cell Biol. 2006;26:9126–9135. doi: 10.1128/MCB.00679-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- So AY, Chaivorapol C, Bolton EC, Li H, Yamamoto KR. Determinants of cell- and gene-specific transcriptional regulation by the glucocorticoid receptor. PLoS Genet. 2007;3:e94. doi: 10.1371/journal.pgen.0030094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchen CR, Martins JR, Paktiawal N, Perelli R, Saklatvala J, Clark AR. Glucocorticoid regulation of mouse and human dual specificity phosphatase 1 (DUSP1) genes: unusual cis-acting elements and unexpected evolutionary divergence. J Biol Chem. 2010;285:2642–2652. doi: 10.1074/jbc.M109.037309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuckermann JP, Reichardt HM, Arribas R, Richter KH, Schutz G, Angel P. The DNA binding-independent function of the glucocorticoid receptor mediates repression of AP-1-dependent genes in skin. J Cell Biol. 1999;147:1365–1370. doi: 10.1083/jcb.147.7.1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuckermann JP, Kleiman A, McPherson KG, Reichardt HM. Molecular mechanisms of glucocorticoids in the control of inflammation and lymphocyte apoptosis. Crit Rev Clin Lab Sci. 2005;42:71–104. doi: 10.1080/10408360590888983. [DOI] [PubMed] [Google Scholar]
- Vayssiere BM, Dupont S, Choquart A, Petit F, Garcia T, Marchandeau C, et al. Synthetic glucocorticoids that dissociate transactivation and AP-1 transrepression exhibit antiinflammatory activity in vivo. Mol Endocrinol. 1997;11:1245–1255. doi: 10.1210/mend.11.9.9979. [DOI] [PubMed] [Google Scholar]
- Wang X, Nelin LD, Kuhlman JR, Meng X, Welty SE, Liu Y. The role of MAP kinase phosphatase-1 in the protective mechanism of dexamethasone against endotoxemia. Life Sci. 2008;83:671–680. doi: 10.1016/j.lfs.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JZ, Cavet ME, VanderMeid KR, Salvador-Silva M, Lopez FJ, Ward KW. BOL-303242-X, a novel selective glucocorticoid receptor agonist, with full anti-inflammatory properties in human ocular cells. Mol Vis. 2009;15:2606–2616. [PMC free article] [PubMed] [Google Scholar]