

Abstract
The enzyme 5-lipoxygenase (5-LO) catalyzes the conversion of arachidonic acid into the leukotrienes, which are critical regulators of inflammation and inflammatory diseases, such as asthma and arthritis. Although leukotrienes are present in the synovial fluid of Lyme disease patients, their role in the development of Lyme arthritis has not been determined. In the current study, we used a murine model of Lyme arthritis to investigate the role 5-LO products might have in the development of this inflammatory disease. Following infection of Lyme arthritis-susceptible C3H/HeJ mice with B. burgdorferi, mRNA expression of 5-LO and 5-lipoxygenase-activating protein (FLAP) was induced in the joints, and the 5-LO product LTB4 was produced. Utilizing C3H 5-LO-deficient mice, we demonstrated that 5-LO activity was not necessary for the induction of Lyme arthritis, but that its deficiency resulted in earlier joint swelling and an inability to resolve arthritis as demonstrated by sustained arthritis pathology through day 60 post-infection. Although production of anti-Borrelia IgG was decreased in 5-LO-deficient mice, bacterial clearance from the joints was unaffected. Phagocytosis of B. burgdorferi and efferocytosis of apoptotic neutrophils was defective in macrophages from 5-LO-deficient mice, and uptake of opsonized spirochetes by neutrophils was reduced. These results demonstrate that products of the 5-LO metabolic pathway are not required for the development of disease in all models of arthritis, and that caution should be used when targeting 5-LO as therapy for inflammatory diseases.
Introduction
Lyme disease is caused by infection of mammalian hosts with the spirochete, Borrelia burgdorferi (1). It is transferred through the bite of an infected Ixodes tick and is the most common tick-borne disease in the U.S. Recently, the Centers for Disease Control and Prevention reported that in 2009 there were approximately 30,000 confirmed new cases of Lyme disease, with another 8,500 listed as probable (2). If not treated with antibiotics near the time of infection, 60-80% of infected individuals will develop joint pain and sporadic arthritis, which may lead to chronic erosive disease (3). Disease development correlates with the presence of spirochetes in joint tissues, which leads to an inflammatory response that may be excessive in some individuals (4). Most Lyme disease patients respond well to antibiotic therapy and quickly resolve their inflammatory arthritis (5); however, a small minority are refractory to antibiotic therapy and develop a chronic persistent arthritis which is poorly understood (6).
Experimental Lyme arthritis is a murine model that recapitulates many of the arthritis disease parameters seen in human Lyme disease patients (7). Two to three weeks following infection, C3H mice develop a severe arthritis that is mediated by innate immunity and independent of T or B cells (8-10). Disease development requires the recruitment of CXCR2-expressing neutrophils into the infected tissue in response to joint production of the chemokine KC (CXCL1) (11,12); however, the mechanisms by which neutrophils mediate the development of arthritis pathology are incompletely understood. Resolution of arthritis was thought to coincide with antibody-mediated clearance of the spirochetes from joint tissues (13), although recent data challenge this assumption. Infection of mice deficient in TLR2 or MyD88 with B. burgdorferi resulted in exacerbated severity and extended duration of Lyme arthritis concomitant with high numbers and the prolonged presence of spirochetes in tissues despite production of normal levels of anti-Borrelia antibody production (14-16). In addition, B. burgdorferi infection of mice deficient in the cyclooxygenase (COX)-2 enzyme also resulted in development of severe Lyme arthritis with an extended time-course, but antibody production and spirochete clearance appeared similar to that of wild-type mice (17). Thus, the mechanisms of arthritis development and resolution in this model remain unclear.
Experimental Lyme disease is an inflammatory arthritis and, as such, is likely regulated by eicosanoids, products of the arachidonic acid biosynthetic pathway which are known to regulate inflammation (18). Products of the eicosanoid biosynthetic enzyme 5-lipoxygenase (5-LO) have been implicated in the regulation of autoimmune and infectious disease pathology. Leukotrienes (LT), 5-HETE, and pro-resolution molecules such as resolvins and lipoxins, owe their generation wholly or in part to 5-LO activity (19,20). Leukotrienes such as LTB4 have been shown to play critical roles in murine models of rheumatoid arthritis. For example, mice deficient in 5-lipoxygenase–activating protein (FLAP) developed attenuated disease in the collagen-induced arthritis (CIA) model (21). Similarly, blocking the LTB4 receptor (BLT1) with antagonists or using BLT1-/- or BLT1/BLT2 double knockout mice resulted in complete protection from the development of CIA (22-24). In the K/BxN serum transfer model of arthritis, neutrophil-derived LTB4 signaling through either BLT1 or BLT2 is absolutely required for the development of disease (25-27). Signaling of neutrophils through BLT1 has been shown to drive their production of IL-1, which amplifies arthritis severity by inducing the production of neutrophil chemoattractants by synovial cells (28). Thus, 5-LO products play a clear role in arthritis development in autoantibody-mediated arthritis models.
Conversely, 5-LO products play a protective role during the immune response to infectious agents. During infection of mice with the bacterium, Klebsiella pneumoniae, or the parasites Leishmania amazonensis or Toxoplasma gondii, 5-LO deficiency resulted in increased morbidity and mortality. Infection of 5-LO-/- mice with K. pneumoniae or L. amazonensis led to uncontrolled pathogen proliferation and high pathogen tissue loads and death due to decreased phagocytic capacity of macrophages and neutrophils (29-31). Infection of 5-LO-deficient mice with T. gondii caused mice to succumb early due to a marked encephalitis which was linked to high levels of IL-12 and IFNγ and could be prevented via exogenous administration of an analog of lipoxin A4 (32). Thus, 5-LO products play important roles during infection by stimulating macrophage and neutrophil uptake and killing of microbial pathogens and by counter-regulating pro-inflammatory cytokines, thus limiting immunopathology.
The roles of 5-LO products during inflammatory processes are varied, complex, and not well understood. Like many inflammatory mediators, they appear to be detrimental during the development of autoimmune diseases but are critical for regulating the immune response to infectious agents. In the current study, we examined the development of experimental Lyme arthritis in mice deficient in 5-LO. In this study, we found that 5-LO-/- mice developed arthritis similar in severity to wild-type mice but with prolonged duration, suggesting a failure of pro-resolution mechanisms. This failure to resolve joint inflammation was not due to increased spirochete numbers or an inability to clear B. burgdorferi from the joint tissue, despite a profound defect in macrophage phagocytosis in 5-LO-/- mice. Collectively, these results demonstrate that the influence of 5-LO products on inflammatory responses is complex and therapies designed to block 5-LO activity should be used with caution.
Materials and Methods
Animals
Female wild-type C3H/HeJ mice, age 4-6 weeks, were purchased from The Jackson Laboratory (Bar Harbor, ME). 5-lipoxygenase knockout mice (B6;129S2-Alox5tm1Fun/J) were purchased as breeders, from The Jackson Laboratory and were backcrossed onto the C3H/HeJ background for five generations using speed congenics (33). Heterozygous mice were then intercrossed to produce 5-LO knockout and wild-type littermates. Animals were housed in a specific pathogen-free facility and given sterile food and water ad libitum. All studies were conducted in accordance with the Animal Care and Use Committee of the University of Missouri.
Bacteria and infections
A virulent, low-passage, clonal isolate of B. burgdorferi N40 strain was used for all infections. Frozen stocks were placed in 7 ml Barbour, Stoenner, Kelly (BSK) II medium containing 6% rabbit serum (Sigma-Aldrich, St. Louis, MO) and grown to log phase at 32°C. Spirochetes were enumerated using dark field microscopy and a Petroff-Hausser counting chamber (Hausser Scientific, Horsham, PA). Spirochete dilutions were made in sterile BSK II medium and mice were inoculated in both hind footpads with 2.5 × 105 B. burgdorferi organisms in 50 μl of medium, for a final inoculum of 5 × 105 organisms per mouse.
Assessment of arthritis pathology
The progression of arthritis development was monitored by measuring ankle swelling through the thickest craniocaudal portion of the ankle joints using a metric caliper. Joint thickness was determined prior to infection and then at indicated time points throughout the course of infection. Ankle swelling measurements were determined by subtracting the initial baseline measurement from subsequent measurements. Arthritis severity scores were determined as previously described (34). Briefly, formalin-fixed, paraffin-embedded sections of ankle joints were stained with hematoxylin and eosin (H&E), and the sections evaluated by two independent observers for arthritis severity on a scale of 0 to 4. A score of 0 indicated normal tissue and 4 indicated severe inflammation involving greater than 50% of the sample.
Quantitative RT-PCR
Total RNA was extracted with TRIzol reagent (Invitrogen Corp, Carlsbad, CA) according to the manufacturer’s protocol. One-step RT-PCR was performed using the EZ RT-PCR kit (Applied Biosystems, Foster City, CA) and the ABI Prism 7700 Sequence Detection System (Applied Biosystems). The mouse nidogen gene, a single copy gene, was used as an endogenous control as described previously (35). Primer and probe sequences were as follows: 5-LO: 5′ AAC CTG TTC ATC AAC CGC TTC ATG (exon 5), 3′ ACT GGT AGC CAA ACA TGA GGT CTT (exon 6), probe AAC ACT ATA TCT GAG CGA GTC AAG AAC CAC TGG CA (exons 5-6); FLAP: 5′ ATC AAG AGG CTG TGG GCA ACG TT (exon 1), 3′ AGT CCA GAG TAC CAC AAG GAA AGT (exon 3), probe TGC CAA CCA GAA CTG CGT AGA TGC GTA CC (exon 2-3). RT-PCR conditions were: 50°C for 15 min, 60°C for 30 min, 95°C for 10 min, and 45 cycles of 95°C for 30 sec and 60°C for 1 min. Reactions were normalized to copies of nidogen mRNA within the same sample using the −ΔΔCT method. The levels of mRNA are expressed as the fold change in expression level as compared to uninfected controls.
Extraction and measurement of LTB4
The extraction of eicosanoids from ankle joints was performed as described previously (17). Briefly, tissues were snap-frozen in liquid nitrogen, pulverized, and the resulting powder placed in 5 mL ice-cold 100% ethanol, vortexed, weighed, and incubated at −20°C for 72 h. Samples were centrifuged at 3,500 x g for 30 min at 4°C; the clear ethanolic supernatant dried under nitrogen, and resuspended in 50 mM HEPES in 50% methanol. 50 μl of sample was applied to a LUNA C18-2 column (150mm × 2mm, 5μ; Phenomonex, Torrance, CA) equilibrated with Solvent A (methanol/acetonitrile/water/acetic acid; 10:15:100:0.2, v/v, pH 6.0). The column was eluted at a flow rate of 1 mL/min with 55% Solvent A and 45% Solvent B (100% methanol) over 5 min. Solvent B was then increased linearly to 75% over 15 min and maintained at this level for an additional 15 min. 2 mL fractions were collected as determined by elution of a deuterated standard, dried under nitrogen, and resuspended in 250 μl enzyme immunoassay (EIA) buffer. Samples were further diluted in EIA buffer before EIA analysis for leukotriene B4 (LTB4; Cayman Chemical, Ann Arbor, MI).
Anti-Borrelia IgG concentrations
B. burgdorferi-specific IgG levels in the sera of infected animals were determined by ELISA as described previously (36). Immulon 2B ELISA plates (Nalgene, Rochester, NY) were coated with 0.5 μg/mL of B. burgdorferi antigen in coating buffer (0.1 M bicarbonate buffer, pH 9.4). Sera of individual animals were diluted in BSA/PBS and incubated for 2 h at RT. Alkaline phosphatase-conjugated donkey anti-mouse IgG (Jackson ImmunoResearch, West Grove, PA) was applied at 1:1000 dilution. Plates were washed and read at 409 nm after addition of the phosphatase substrate (Sigma 104 tablets, Sigma-Aldrich).
Determination of tissue Borrelia loads
DNA was extracted from ankle joints by homogenization in TRIzol reagent according to manufacturer’s protocol. Multiplex real-time PCR was performed using the TaqMan Universal PCR Master Mix (Applied Biosystems, Foster City, CA) according to manufacturer’s instructions. Real-time PCR reactions for B. burgdorferi flagellin were normalized to copies of nidogen DNA within the same sample (35). Bacterial loads are expressed as copies of B. burgdorferi flagellin per 1000 copies of mouse nidogen.
Cell isolation and culture
Mouse bone marrow macrophages (BMMs) were isolated from mouse femurs using sterile PBS and differentiated on 100 X 15 mm plastic petri dishes in medium containing RPMI 1640 supplemented with 30% L929 cell-conditioned medium, 10% fetal bovine serum (FBS), and 2% penicillin-streptomycin at 37°C in 5% CO2. The media was refreshed every 2 days and the cells were incubated for 5-7 days.
Mouse bone marrow neutrophils (BMPMN) were purified from mouse femurs using a three-layer Percoll gradient centrifugation as described previously (37,38) with some modifications. Bone marrow cells were collected and red blood cells removed by hypotonic lysis. Cells were then washed and laid on top of a discontinuous three-layer Percoll gradient (1.095, 1.085, and 1.070 g/ml). After centrifugation at 500 x g for 30 min at 25° C, the lowest interface (1.085/1.095 g/ml interface) was collected as the PMN fraction. Purity of BMPMNs were >95%, as determined by Quik-Dip differential staining (Mercedes Medical, Inc. Sarasota, FL). In some experiments, BMPMNs were labeled with the fluorescent dye PKH26 according to the manufacturer’s instructions (Sigma). Spontaneous apoptosis was achieved by incubating labeled PMN for 24 h at 37°C in 5% CO2. BMPMN used in phagocytosis assays were 60-70% apoptotic as determined by morphology.
Phagocytosis assay of B. burgdorferi
BMMs were harvested from petri dishes by gentle scraping and plated on glass coverslips at 0.5 × 106 cells per well in 24-well tissue culture plates containing antibiotic-free RPMI 1640 and 30% L929 cell-conditioned media. Live GFP-B. burgdorferi (a kind gift from James Carroll, National Institutes of Health) were added to the cultures at a multiplicity of infection (MOI) of 10. Plates were incubated for 1 or 4 h at 37°C with 5% CO2. Freshly isolated BMPMNs were plated on coverslips in 24-well tissue culture plates in antibiotic-free DMEM media and infected with immune serum-opsonized or nonopsonized live GFP-B. burgdorferi at MOI 10. At indicated time points, both BMMs and BMPMNs were washed three times with PBS to remove extracellular spirochetes and fixed in 4% paraformaldehyde for 15 min at 25°C. Cells were then washed and permeablized with cold methanol for 10 min at 4°C and blocked with 5% BSA in PBS overnight. Anti-mouse CD16/32 was used to block Fc receptors. Lysosome-associated membrane proteins (LAMP) were stained with Alexa Fluor 647-conjugated rat monoclonal anti-mouse LAMP-1(1D4B; Santa Cruz Biotechnology, Inc), and host cell nuclei were stained by Hoechst (1:1000) and examined by fluorescence microscopy. The phagocytic index was calculated by determining the percentage of BMM/BMPMN containing at least one spirochete per 300 cells. Comparable data were obtained in two or more independent experiments. Data are expressed as mean ± SD.
Phagocytosis assay of apoptotic PMN
BMMs were plated in 24-well tissue culture plates as described above. PKH26-labeled apoptotic PMN were added to BMMs at ratio of 10:1. Plates were incubated for 1 or 4 h at 37°C with 5% CO2. Coverslips were then washed with PBS, and fixed in 4% paraformaldehyde for 15 min at 25°C. Following membrane permeablization and blocking with 5% BSA containing Fc blocking antibody, BMMs were stained with Alexa Fluor 488-conjucated monoclonal rat anti-mouse F4/80A (Invitrogen), and coverslips were examined by fluorescence microscopy. A minimum 300 cells were counted in random fields from at least two independent coverslips. Phagocytic indexes were calculated by determining the number of BMM containing at least one PMN per 300 cells. Comparable data were obtained in two or more independent experiments. Data are expressed as mean ± SD.
Statistical Analysis
All experimental groups contained at least 5 mice per group and each experiment was completed at least twice. Group means were analyzed for statistical significance using GraphPad Prism Software. Student’s t-test or ANOVA with the Mann-Whitney U test were used with a = 0.05.
Results
Bb infection activates the 5-LO pathway
Products of the 5-LO pathway are critical regulators of both the development and resolution of inflammatory responses (39,40). Whether products of 5-LO have a role in the generation or resolution of Lyme arthritis, however, is currently unknown. To determine if B. burgdorferi infection could stimulate the 5-LO pathway in joint tissue, Lyme arthritis-susceptible C3H/HeJ mice were infected with B. burgdorferi and the levels of 5-LO and FLAP mRNA in the joint were determined throughout the time course of infection using real-time RT-PCR. Both 5-LO and FLAP mRNA were found to be significantly up-regulated in ankle joints by d7 post-infection (Fig. 1A), a time point prior to the recruitment of inflammatory cells into the joint tissue. In addition, we detected a significant increase in the production of the 5-LO product, LTB4, in the ankle joint at d14 post-infection (Fig. 1B). These data demonstrate that the 5-LO pathway is activated in joint tissues following infection with B. burgdorferi, and could potentially contribute to the anti-Borrelia immune response and development of experimental Lyme arthritis.
Figure 1.

B. burgdorferi infection induces up-regulation of the 5-LO pathway in joints of C3H mice. C3H mice were infected with 1 × 105 B. burgdorferi and sacrificed at indicated time points post-infection. (A) Expression of mRNA for 5-lipoxygenase (5-LO) and FLAP in knee joints was determined by real-time RT-PCR. Values are represented as the increase in expression relative to uninfected controls (day 0). (B) Concentrations of LTB4 in tibiotarsal joints, as determined by EIA. Values expressed are pg LTB4 per mg protein. Bars represent mean values ± SD and are representative of two experiments performed. n = 5 mice/time point. *, p ≤ 0.05 vs. uninfected controls as determined by one-way ANOVA with Dunnett’s multiple comparisons post-test.
5-LO deficiency results in early increased joint swelling
LTB4-mediated recruitment of neutrophils is critical for the subsequent development of arthritis in the K/BxN serum transfer model (25). We have previously shown that recruitment of neutrophils into the infected joint is required for the development of experimental Lyme arthritis (12). To determine the effect of 5-LO deficiency on the development of Lyme arthritis, C3H wild-type (WT) and 5-LO-/- mice were infected with B. burgdorferi and a time course of ankle swelling was determined. In many models of arthritis, including experimental Lyme arthritis, swelling of the joint or paw is considered a reliable indicator of the underlying inflammatory response. Increases in ankle diameter at 3, 7, 14, 21, 28, and 35 days post-infection were measured and compared to uninfected mice. WT mice exhibited a typical ankle joint swelling curve, with a peak increase in ankle diameter occurring at d14-21 post-infection. Unexpectedly, 5-LO-/- mice developed significantly greater (p < 0.01) ankle swelling than the WT control mice, beginning at d7 and continuing past d28 post-infection (Fig 2). This contrasted with results from other models of arthritis such as CIA and K/BxN, in which mice lacking FLAP or 5-LO developed no or significantly less paw swelling (21,25).
Figure 2.
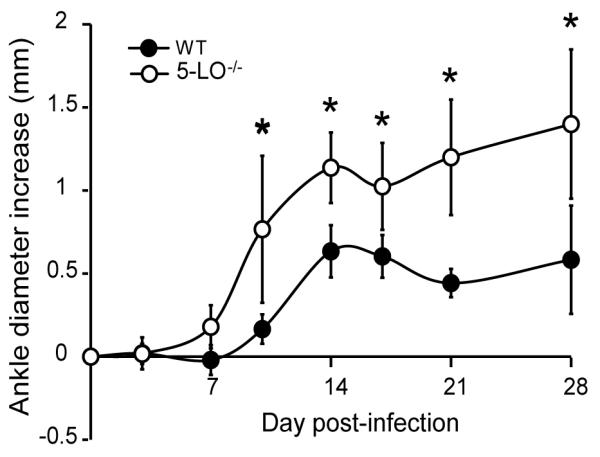
5-LO deficiency results in an early and prolonged increase in ankle swelling. C3H wild-type (WT; filled symbols) and C3H 5-LO-/- (open symbols) mice were infected with 1 × 105 B. burgdorferi and ankle diameter measurements were made at regular intervals post-infection. Ankle swelling was determined by subtracting the ankle measurement pre-infection from the measurement at the indicated time point. Values represent the mean ± SD and are representative of two experiments performed. n = 5 mice/time point. *, p ≤ 0.01 versus WT control, as determined by two-way ANOVA, followed by Bonferroni post-tests.
5-LO deficiency extends the duration of Lyme arthritis
Since eicosanoids are known modulators of vascular leakage and edema, the possibility existed that ankle diameter may be altered while the underlying inflammatory response remained essentially unchanged. Therefore, we ascertained whether the increased ankle diameters in 5-LO-/- mice correlated with increased arthritis severity. C3H WT and 5-LO-/- mice were infected with B. burgdorferi and sacrificed at weekly time points from d7 through d28 post-infection. In Fig. 3A, we show representative histologic sections of ankle joints from WT and 5-LO-/- mice on d14 and 28 post-infection. On d14, severe inflammation is present in both the WT and 5-LO-/- ankle joints characterized by a predominantly neutrophilic inflammatory infiltrate (arrows). By d28 post-infection, inflammation had mostly resolved in the WT mice, with macrophages making up the majority of the remaining inflammatory cells (arrowheads). In contrast, sections from the 5-LO-/- mice still displayed severe acute inflammation with a preponderance of neutrophils, indicating a failure to undergo the spontaneous resolution observed in the WT mice. Interestingly, we also found multinucleate giant cells (MGC) in the joints of 50% of the 5-LO-/- mice at d14-d28 post-infection (white arrow), indicating a change in the inflammatory environment, whereas no MGC were observed in the joints of WT mice. Arthritis severity scores demonstrate that the development of arthritis was similar between C3H WT and 5-LO-/- mice (Fig.3B). However, by day 28 post-infection, the arthritis severity scores for the 5-LO-/- mice were significantly higher than those of the C3H WT control mice (P < 0.001). Together these results indicate normal induction of inflammation in 5-LO-/- mice infected with B. burgdorferi, but a defect in the initiation of resolution of inflammation prolonging the inflammatory response (chronic arthritis).
Figure 3.

5-LO deficiency results in the prolonged duration of experimental Lyme arthritis. (A) Representative H & E sections of tibiotarsal joints from B. burgdorferi-infected C3H wild-type (WT, upper panels) and 5-LO-/- (lower panels) at 14 and 28 days post-infection. The boxed areas in the left panels are shown at a higher magnification in the panels on the right side. Arrows indicate neutrophils, whereas arrowheads indicate macrophages. The white arrow designates a multinucleate giant cell. (B) Arthritis severity scores of C3H WT (filled symbols) and 5-LO-/- (open symbols) mice were determined by examination of H & E sections of ankle joints obtained at weekly time points post-infection, as described in Materials and Methods. Each symbol represents the severity score of an individual animal, and horizontal bars denote the median score of each group. n = 7-15 mice/time point. *, p < 0.01 versus WT control, as determined by two-way ANOVA with Bonferroni post-tests.
We next infected WT and 5-LO-/- mice and allowed the disease to progress for 60 days, a time when most disease parameters in C3H WT mice have returned to homeostatic levels. At this time point, ankle swelling in WT mice had nearly returned to pre-infection levels, and the remaining difference was likely due to the growth of the animals during the experimental period (Fig.4A). Ankle swelling in the 5-LO-/- mice, however, remained significantly greater than in the WT mice, indicating a continued delay in resolution of the inflammatory response. Arthritis severity scores were also higher in the 5-LO-/- mice at this time point (Fig.4B), demonstrating a true extension of the inflammatory response and not simply a defect in the ability to resolve ankle swelling. Histological examination of ankle joints from infected mice revealed the continued presence of a small number of primarily macrophages in WT joints, while joints from 5-LO-/- mice still contained neutrophils - indicative of an ongoing acute inflammatory response (Fig.4C).
Figure 4.

Lyme arthritis in 5-LO-/- mice fails to resolve by 60 days post-infection. C3H wild-type (WT) and 5-LO-/- were infected with B. burgdorferi and disease was allowed to progress for 60 days. (A) Increases in ankle diameter of C3H WT (filled bars) and 5-LO-/- (open bars) mice were determined at 35, 45, and 60 days post-infection with B. burgdorferi. Values are the increase in ankle diameter from day 0 measurements. Bars represent mean values ± SD of 5 mice/genotype. *, p < 0.008 versus WT control at the same time point, as determined by unpaired t-test. (B) Arthritis severity scores of WT (filled symbols) and 5-LO-/- (open symbols) mice at day 60 post-infection were determined by examination of H & E stained sections of ankle joints. Each symbol represents the severity score of an individual animal and horizontal bars represent the median of each group. n = 5 mice/genotype. *, p = 0.02 versus WT controls, as determined by unpaired t-test. (C) Representative H & E sections of ankle joints from WT (left panel) and 5-LO-/- (right panel) mice sacrificed at d60 post-infection. The boxed areas in the upper panels are shown at a higher magnification in the lower panels. Arrows indicate neutrophils.
5-LO deficiency alters the humoral response, but has no effect on spirochete clearance
Our data contrast with results from studies in other arthritis models, in which 5-LO deficiency or inhibition resulted in prevention or amelioration of arthritis pathology (21,25). In the current study, the effect of decreased or absent 5-LO activity during B. burgdorferi infection more closely resembled those observed in models of infectious diseases, rather than those seen in arthritis models. For example, infection of 5-LO-/- mice with K. pneumoniae or T. gondii resulted in increased bacterial or parasite burden accompanied by increased disease pathology (29,32). Thus, it was possible that the failure of arthritis resolution we observed was due to an inhibition or inability to clear spirochetes from the infected joint. Resolution of Lyme arthritis is thought to depend upon the production of anti-Borrelia antibodies and subsequent antibody-mediated clearance of the bacteria from tissues. 5-LO products can affect humoral immunity, as 5-LO is expressed in B cells (41) and exogenous leukotrienes can modify B and T cell activation (42,43). Additionally, in CIA, treatment of mice with MK886, a FLAP inhibitor, resulted in decreased anti-collagen IgG1 and IgG2 production (21).
To determine if the 5-LO deficiency had an effect on the generation of anti-Borrelia antibodies, C3H WT and 5-LO-/- mice were infected with B. burgdorferi and sera collected on days 7, 14, 21, and 28 post-infection. There were no detectable differences in the production of Borrelia-specific IgM levels at any of the time points examined (data not shown); however, by day 28 post-infection, sera from 5-LO-/- mice contained significantly less (P < 0.05) Borrelia-specific IgG than their WT counterparts (Fig.5A). We next investigated whether this resulted in a concomitant decrease in spirochete clearance from the joints. To do this we used quantitative real-time PCR to measure Borrelial loads in the ankle joints of C3H WT and 5-LO-/- mice at time points throughout the infection. Surprisingly, despite the observed decrease in Borrelia-specific IgG antibody production, spirochetes were cleared from the joints of 5-LO-/- mice just as effectively as from the joints of WT animals (Fig.5B). At d28 post-infection, a time point at which significant differences in ankle swelling and joint inflammation were apparent between the WT and 5-LO-/- mice, spirochete DNA was not detected in 50% of joints from either WT or 5-LO-/- mice. By d60 post-infection, a time when WT mice are well into resolution and 5-LO-/- mice exhibit ongoing inflammation, spirochete DNA was not detected in any of the joint tissues from either mouse strain. Thus, despite a decrease in Borrelia-specific IgG antibody production, spirochete clearance from joint tissue appears to be normal, and an increase in bacterial burden could not explain the prolonged arthritis demonstrated in B. burgdorferi-infected 5-LO-/- mice.
Figure 5.
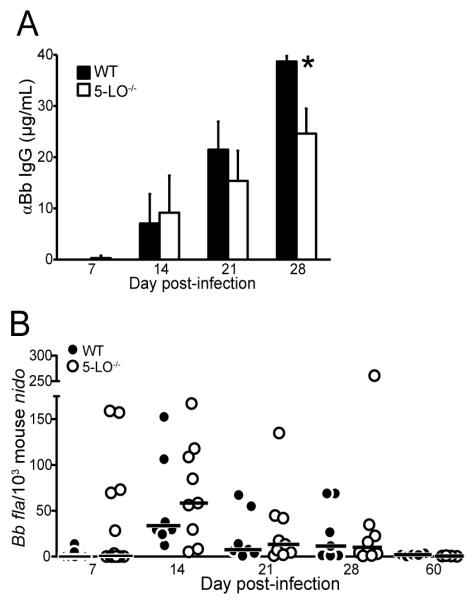
5-LO-deficiency during B. burgdorferi infection has little or no effect on anti-Borrelia antibody production or bacterial clearance. (A) B. burgdorferi-specific IgG concentrations in sera of C3H wild-type (WT; filled bars) and 5-LO-/- (open bars) mice at weekly time points post-B. burgdorferi infection. *, p = 0.004 versus WT controls at the same time point, as determined by two-tailed unpaired t-test. Bars represent mean values ± SD and are representative of two experiments performed. n = 5 mice/genotype. (B) B. burgdorferi loads were determined in the ankle joints of WT (filled symbols) and 5-LO-/- (closed symbols) at 7, 14, 21, 28, and 60 days post-infection. Each symbol represents an individual animal, and values were determined by real-time PCR as described in Materials and Methods and are expressed as copies of B. burgdorferi flagellin (fla) per 103 copies mouse Nidogen (nido). Horizontal bars denote the median value of each group. n = 7-15 mice/genotype/time point.
Macrophage phagocytosis is defective in 5-LO-/- mice
Defects in the clearance of bacteria or apoptotic cells have been cited as instigating chronic inflammatory diseases, such as rheumatoid arthritis and atherosclerosis (44,45). Macrophages and neutrophils from 5-LO-/- mice have been shown to be defective in their uptake of microbial pathogens (29,30,46). Thus, we assessed whether these mechanisms might be involved in the prolonged course of experimental Lyme arthritis seen in 5-LO-deficient mice. Neutrophils were isolated from the bone marrow of C3H wild-type and 5-LO-/- mice and were incubated with B. burgdorferi spirochetes (MOI=10) for 1 or 4h. We found no differences in the uptake of B. burgdorferi spirochetes by neutrophils from wild-type or 5-LO-/- mice at either time point (Fig.6A). The percentage of wild-type neutrophils with at least one internalized spirochete was 31% at 1h, and rose to 44% at 4h and are consistent with other reported results of phagocytosis assays (47). It has been reported that neutrophils are inefficient in phagocytosis of unopsonized B. burgdorferi (48), thus we repeated the experiment using opsonized B. burgdorferi spirochetes. Spirochete opsonization increased the rate of phagocytosis by wild-type neutrophils to 58% at 1h and 62% at 4h. Following opsonization there was a slight but significant (p < 0.01) decrease in the phagocytic index of neutrophils from the 5-LO-/- mice relative to controls (Fig.6B). These results suggest that products of the 5-LO pathway influence FcR-mediated phagocytosis by neutrophils more strongly than other phagocytic mechanisms and supports previous work in this area using other microbial pathogens (30).
Figure 6.

Macrophages from 5-LO-/- mice have reduced phagocytic capacity, but phagocytosis by PMNs is unaffected. Bone marrow-derived PMNs (A & B) or macrophages (Mϕ, C & D) were isolated from wild-type (control) or 5-LO-/- animals and their phagocytic index (% control) determined after 1 or 4 hr of co-culture. PMN were co-cultured with unopsonized (A) or opsonized B. burgdorferi (PMN-OpBb) (B). Macrophages were co-cultured with unopsonized B. burgdorferi (C) or with apoptotic PMN (D) as described in the Materials and Methods. Bars represent means ± SEM and are the results from two pooled experiments. *, p < 0.01 versus WT control cells, as determined by two-way ANOVA, followed by Bonferroni post-tests.
We next assayed the ability of primary bone marrow-derived macrophages from 5-LO-deficient mice to phagocytose live B. burgdorferi spirochetes. Phagocytosis of live spirochetes by wild-type macrophages was 16% at 1h of co-incubation, and increased to 31% at 4h. In contrast to results observed in neutrophils, macrophages from 5-LO-/- mice demonstrated significantly decreased (p < 0.01) capacity to phagocytose B. burgdorferi at both the 1 and 4h time points (Fig.6C). To determine if this was a defect only in the uptake of microbes or was a more global defect in macrophage phagocytosis, we next examined the ability of 5-LO-deficient macrophages to take up apoptotic neutrophils. Wild-type macrophage uptake of apoptotic neutrophils was 17% at 1h and 26% at 4h. The ability to phagocytose apoptotic neutrophils was severely impaired in macrophages from 5-LO-/- mice (Fig.6D), as 5-LO-deficient macrophages achieved only 36% of the apoptotic cell engulfment attained by wild-type macrophages at 4h. This is in contrast to a previous report where a 5-LO deficiency had no effect on macrophage uptake of apoptotic thymocytes (49). Thus, a defect in the ability of macrophages to clear away apoptotic neutrophils from the site of infection may play a role in prolonging the inflammatory response and lead to the development of a chronic inflammatory disease.
Discussion
Products of the 5-LO metabolic pathway have been implicated in a number of chronic inflammatory diseases including arthritis, asthma, cardiovascular disease, and cancer, and have once again become targets for therapeutic intervention (50,51). Recent studies using mouse models of arthritis have demonstrated a requirement for LTB4 production by neutrophils, and the involvement of both LTB4 receptors, BLT1 and BLT2, in the development of disease (21-28). In the current study we show that, in contrast to results observed in autoantibody-mediated arthritis models, genetic deletion of 5-LO does not prevent the development of arthritis in response to an infectious agent (B. burgdorferi), but rather exacerbates the development of disease and inhibits resolution of the inflammatory response. Lyme arthritis is caused by infection of the joint by the spirochete, B. burgdorferi, and in mice, disease development is mediated by cells of the innate immune system and is independent of adaptive immunity (8-10). Thus, experimental Lyme arthritis is an infectious disease and is not autoimmune in nature. In contrast, the CIA and K/BxN models of arthritis are dependent upon the induction or spontaneous development of auto-antibodies and are by definition, autoimmune diseases. These models, therefore, occupy opposite ends of the immunological spectrum, demonstrating that arthritis is a complex disease that can manifest via several immunological mechanisms. Therefore, an individual’s disease etiology must be considered when contemplating treatment strategies, as treatment of one type of arthritis may be detrimental for another.
In response to injury or infection, arachidonic acid (AA) is released from membrane phospholipids by phospholipase A2. Once released, AA then serves as a substrate for the generation of a large number of bioactive lipids through the actions of the cyclooxygenase, lipoxygenase, or cytochrome P450 enzymes (52). Substrate availability is the limiting factor in the production of eicosanoids, and under conditions of specific enzyme inhibition or deletion, excess AA can be used by the remaining enzymes to produce increased levels of certain metabolites. This shunting is a possible mechanism for the increased ankle swelling seen in the 5-LO-/- mice in the current study. Due to the genetic deletion of 5-LO, increased free AA may be used by COX-2 to produce higher levels of PGE2, which is known to induce edema (53). Substrate shunting between these two enzymes has been described previously using specific COX-2 inhibitors (54) or mice deficient in microsomal PGE2 synthase (55).
Mice deficient in 5-LO developed severe Lyme arthritis with similar kinetics as wild-type C3H mice. However, as we reported when using COX-2-/- mice (17), the 5-LO-/- mice were unable to resolve their arthritis and still had an acute inflammatory response in their ankle joints 60d post-infection. At this time point, the wild-type mice were well into the resolution phase of disease. Resolution of experimental Lyme arthritis is considered to be mediated by the production of anti-Borrelia antibodies and clearance of spirochetes from the infected joints (13). In the current study, genetic deletion of 5-LO reduced production of Borrelia-specific IgG beginning by day 21 post-infection and became significantly lower (p = 0.004) by day 28. However, spirochete clearance from the infected joint did not appear to be affected by this decrease in specific-antibody production, suggesting the amount of specific-antibody produced was sufficient for normal spirochete clearance. Murine B cells express high levels of 5-LO, but this expression is down-regulated in germinal center B cells or plasma cells (56). Our results are similar to those reported for Schistosoma mansoni infection, where antibody-mediated responses were decreased in 5-LO-/- mice (57). We have recently reported that COX-1 metabolites play a role in antibody class switching via induction of IL-6 and IL-17 production (36), but the role of 5-LO metabolites in antibody production has not been determined.
The inflammatory response in experimental Lyme arthritis is dominated by neutrophils and macrophages (58). These phagocytic cells provide critical host defense through the uptake and killing of microbial pathogens. Defective phagocytosis and killing of microbial pathogens has been demonstrated in macrophages and neutrophils from 5-LO-/- mice (29,30). In the current study, we found that direct uptake of B. burgdorferi was not impaired in neutrophils from 5-LO-/- mice. However, upon spirochete opsonization, we detected a slight but significant decrease in neutrophil phagocytic ability, which was in agreement with previously published results with other pathogens (30). In contrast to the results using neutrophils, phagocytosis of spirochetes by macrophages from 5-LO-/- mice was significantly impaired, with these macrophages demonstrating roughly half of the phagocytic capacity of wild-type macrophages. Again, this is in agreement with previous reports of phagocytic defects in the uptake of K. pneumoniae by macrophages from 5-LO-deficient mice (29). However, despite these defects in phagocyte uptake of Borrelia and decreased Borrelia-specific IgG production, we could not detect a defect in spirochete clearance from the joint. These results suggest that neutrophil phagocytosis of B. burgdorferi is of primary importance for limiting spirochete growth and their eventual elimination from the infected joint. The primary role for macrophages appears to be the uptake and elimination of apoptotic neutrophils, and the subsequent down-regulation of the inflammatory response and return of the tissue to homeostasis. When this function is defective, as seen in the 5-LO-/- mice, the inefficient removal of apoptotic neutrophils could lead to an increase in necrotic neutrophils which are pro-inflammatory. The resolution of inflammation has recently been recognized as an active rather than a passive event, requiring coordinated up-regulation of pro-resolution molecules and down-regulation of pro-inflammatory molecules (59). Phagocytic clearance of apoptotic cells has been recognized as an important component in the resolution of inflammation (60). The ingestion of apoptotic cells (efferocytosis) triggers the release of a number of anti-inflammatory cytokines and lipid mediators, such as TGFβ and PGE2, and inhibits the release of pro-inflammatory mediators, such as TNFα, and IL-1 (61,62). Defects in efferocytosis have been linked to the development of chronic inflammatory diseases and autoimmunity (60,63,64). In the current study, we demonstrate for the first time defective uptake of apoptotic neutrophils by 5-LO-/- macrophages. Thus, it is possible that macrophages in infected joints may not be able to effectively switch to an anti-inflammatory program and thereby continue to promote the inflammatory response beyond clearance of the spirochetes from the joint tissue.
Several lipid mediators have been recently described that have anti-inflammatory or pro-resolution activities. These include the lipoxins from arachidonic acid, protectins and resolvins from DHA and EPA, and the recently described maresins from macrophages (65). We have previously demonstrated that infection of mice with B. burgdorferi induces the production of these pro-resolution lipid metabolites (66). However, lipoxins are produced via transcellular biosynthesis processes that require 5-LO metabolites and are therefore missing in the joints of 5-LO-/- mice (67). Lipoxins are known to stimulate macrophage uptake of apoptotic cells, down-regulate pro-inflammatory mediator production, and inhibit further neutrophil recruitment into the site of inflammation (68,69). Thus, in addition to the mechanisms discussed above, the lack of lipoxins may also contribute to the failure of resolution in 5-LO-/- mice. Indeed, this may be a critical control factor for the initiation of disease resolution in this model. We have previously reported that mice deficient in or treated with an inhibitor of COX-2 have a similar disease phenotype to the 5-LO-/- mice (17). It has been proposed that the production of PGE2 during the initial inflammatory response is critical for the subsequent production of lipoxins (59). Thus, the lack of COX-2 or 5-LO activity may result in a failure of arthritis resolution due to a lack of lipoxins production in the experimental Lyme arthritis model.
Based on these data, we propose the following model for the role of 5-LO products in experimental Lyme arthritis (Fig. 7): Upon transmission to the host from an infected tick, B. burgdorferi migrate to the large joints and multiply. This instigates a local inflammatory response, characterized by the production of pro-inflammatory cytokines and lipid mediators before inflammation is histologically evident. Anti-inflammatory molecules are also produced to keep the inflammatory response under control. These likely include some 5-LO products, as edema was significantly increased early in the time course of B. burgdorferi infection in the absence of 5-LO. An anti-B. burgdorferi IgG adaptive immune response is mounted, which is decreased in the absence of 5-LO. Whether this is directly attributable to 5-LO products or is an effect of the altered inflammatory milieu is still unknown. Concomitantly, neutrophils are recruited to the infected joints in a CXCL1-dependent fashion and subsequently phagocytose and kill the spirochetes. Since we found only a minor effect on PMN phagocytosis of B. burgdorferi, this is either largely 5-LO independent, or compensatory mechanisms are invoked in the absence of 5-LO, and thus we see no effect on spirochete numbers in the joints of 5-LO-/- mice. Subsequently, 5-LO products regulating macrophage phagocytosis of apoptotic PMN regulate efferocytosis, encouraging resolution and repair of the damaged joint tissues. A lack of 5-LO, via either genetic or pharmacologic means, would therefore lead to a breakdown in the pathways to resolution, manifested as an ongoing inflammatory response. Studies are currently underway to further define the outcome of 5-LO deficiency with regard to the altered lipid profile, as well as the impact on cytokine and chemokine production. In the future, new data will likely lead to modification of this model. However, we believe this provides an important starting point from which to launch studies that will expand our understanding of how products of 5-LO modulate all phases of the immune response to B. burgdorferi and the generation of Lyme arthritis.
Figure 7.

Proposed model for the effects of 5-LO on Lyme arthritis resolution or the development of chronic inflammation.
Arthritis is a complex disease with many regulatory molecules influencing its development and resolution. Disparate results from animal models suggest caution should be taken when applying treatment modalities, and that treatment successes or failures may apply to subsets within the patient population, rather than globally to all patients with arthritis. More complete characterization of these responder subsets may be required for progress to be made.
Acknowledgments
We thank James Carroll for the GFP Borrelia, and Marc Peters-Golden for helpful discussions.
This work was supported National Institute of Health Grant AR052748.
Abbreviations
- COX
cyclooxygenase
- LTB4
leukotriene B4
- 5-LO
5-lipoxygenase
- FLAP
5-lipoxygenase-activating protein
- RA
rheumatoid arthritis
Reference List
- 1.Burgdorfer W, Barbour AG, Hayes SF, Benach JL, Grunwaldt E, Davis JP. Lyme disease-a tick-borne spirochetosis? Science. 1982;216:1317–1319. doi: 10.1126/science.7043737. [DOI] [PubMed] [Google Scholar]
- 2.CDC 10 A.D. Final 2009 reports of nationally notifiable infectious diseases. :1025–1039. [Google Scholar]
- 3.Steere AC, Schoen RT, Taylor E. The clinical evolution of Lyme arthritis. Ann. Intern. Med. 1987;107:725–731. doi: 10.7326/0003-4819-107-5-725. [DOI] [PubMed] [Google Scholar]
- 4.Nocton JJ, Dressler F, Rutledge BJ, Rys PN, Persing DH, Steere AC. Detection of Borrelia burgdorferi DNA by polymerase chain reaction in synovial fluid from patients with Lyme arthritis. N. Engl. J. Med. 1994;330:229–234. doi: 10.1056/NEJM199401273300401. [DOI] [PubMed] [Google Scholar]
- 5.Wormser GP, Dattwyler RJ, Shapiro ED, Halperin JJ, Steere AC, Klempner MS, Krause PJ, Bakken JS, Strle F, Stanek G, Bockenstedt L, Fish D, Dumler J. Stephen, Nadelman RB. The clinical assessment, treatment, and prevention of Lyme disease, human granulocytic anaplasmosis, and babesiosis: clinical practice guidelines by the Infectious Diseases Society of America. Clin. Infect. Dis. 2006;43:1089–1134. doi: 10.1086/508667. [DOI] [PubMed] [Google Scholar]
- 6.The chronic debate over Lyme disease. Nat. Med. 2008;14:1135–1139. doi: 10.1038/nm1108-1135. [DOI] [PubMed] [Google Scholar]
- 7.Barthold SW, Beck DS, Hansen GM, Terwilliger GA, Moody KD. Lyme borreliosis in selected strains and ages of laboratory mice. J. Infect. Dis. 1990;162:133–138. doi: 10.1093/infdis/162.1.133. [DOI] [PubMed] [Google Scholar]
- 8.Schaible UE, Kramer MD, Museteanu C, Zimmer G, Mossmann H, Simon MM. The severe combined immunodeficiency (scid) mouse. A laboratory model for the analysis of Lyme arthritis and carditis. J. Exp. Med. 1989;170:1427–1432. doi: 10.1084/jem.170.4.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barthold SW, Sidman CL, Smith AL. Lyme borreliosis in genetically resistant and susceptible mice with severe combined immunodeficiency. Am. J. Trop. Med. Hyg. 1992;47:605–613. doi: 10.4269/ajtmh.1992.47.605. [DOI] [PubMed] [Google Scholar]
- 10.Brown CR, Reiner SL. Genetic control of experimental Lyme arthritis in the absence of specific immunity. Infect. Immun. 1999;67:1967–1973. doi: 10.1128/iai.67.4.1967-1973.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ritzman AM, Hughes-Hanks JM, Blaho VA, Wax LE, Mitchell WJ, Brown CR. The chemokine receptor CXCR2 ligand KC (CXCL1) mediates neutrophil recruitment and is critical for development of experimental Lyme arthritis and carditis. Infect. Immun. 2010;78:4593–4600. doi: 10.1128/IAI.00798-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown CR, Blaho VA, Loiacono CM. Susceptibility to experimental Lyme arthritis correlates with KC and monocyte chemoattractant protein-1 production in joints and requires neutrophil recruitment via CXCR2. J. Immunol. 2003;171:893–901. doi: 10.4049/jimmunol.171.2.893. [DOI] [PubMed] [Google Scholar]
- 13.Barthold SW, deSouza M, Feng S. Serum-mediated resolution of Lyme arthritis in mice. Lab. Invest. 1996;74:57–67. [PubMed] [Google Scholar]
- 14.Wooten RM, Ma Y, Yoder RA, Brown JP, Weis JH, Zachary JF, Kirschning CJ, Weis JJ. Toll-like receptor 2 is required for innate, but not acquired, host defense to Borrelia burgdorferi. J. Immunol. 2002;168:348–355. doi: 10.4049/jimmunol.168.1.348. [DOI] [PubMed] [Google Scholar]
- 15.Bolz DD, Sundsbak RS, Ma Y, Akira S, Kirschning CJ, Zachary JF, Weis JH, Weis JJ. MyD88 plays a unique role in host defense but not arthritis development in Lyme disease. J. Immunol. 2004;173:2003–2010. doi: 10.4049/jimmunol.173.3.2003. [DOI] [PubMed] [Google Scholar]
- 16.Liu N, Montgomery RR, Barthold SW, Bockenstedt LK. Myeloid differentiation antigen 88 deficiency impairs pathogen clearance but does not alter inflammation in Borrelia burgdorferi-infected mice. Infect. Immun. 2004;72:3195–3203. doi: 10.1128/IAI.72.6.3195-3203.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blaho VA, Mitchell WJ, Brown CR. Arthritis develops but fails to resolve during inhibition of cyclooxygenase 2 in a murine model of Lyme disease. Arthritis Rheum. 2008;58:1485–1495. doi: 10.1002/art.23371. [DOI] [PubMed] [Google Scholar]
- 18.Harizi H, Corcuff JB, Gualde N. Arachidonic-acid-derived eicosanoids: roles in biology and immunopathology. Trends Mol. Med. 2008;14:461–469. doi: 10.1016/j.molmed.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 19.Chen XS, Sheller JR, Johnson EN, Funk CD. Role of leukotrienes revealed by targeted disruption of the 5-lipoxygenase gene. Nature. 1994;372:179–182. doi: 10.1038/372179a0. [DOI] [PubMed] [Google Scholar]
- 20.Serhan CN, Brain SD, Buckley CD, Gilroy DW, Haslett C, O’Neill LAJ, Perretti M, Rossi AG, Wallace JL. Resolution of inflammation: state of the art, definitions and terms. FASEB J. 2006;21:1–8. doi: 10.1096/fj.06-7227rev. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Griffiths RJ, Smith M, Roach ML, Stock JL, Stam EJ, Milici AJ, Scampoli DN, Eskra JD, Byrum RS, Koller BH, McNeish JD. Collagen-induced arthritis is reduced in 5-lipoxygenase-activating protein-deficient mice. J. Exp. Med. 1997;185:1123–1130. doi: 10.1084/jem.185.6.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Griffiths RJ, Pettipher ER, Koch K, Farrell CA, Breslow R, Conklyn MJ, Smith MA, Hackman BC, Wimberly DJ, Milici AJ, Scampoli DN, Cheng JB, Pillar JS, Pazoles CJ, Doherty NS, Melvin LS, Reiter LA, Biggars MS, Falkner FC, Mitchell DY, Liston TE, Showell HJ. Leukotriene B4 plays a critical role in the progression of collagen-induced arthritis. Proc. Nat. Acad. Sci. , USA. 1995;92:517–521. doi: 10.1073/pnas.92.2.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuwabara K, Yasui K, Jyoyama H, Maruyama T, Fleisch JH, Hori Y. Effects of the second-generation leukotriene B4 receptor antagonist, LY293111Na, on leukocyte infiltration and collagen-induced arthritis in mice. Eur. J. Pharmacol. 2000;402:275–285. doi: 10.1016/s0014-2999(00)00518-5. [DOI] [PubMed] [Google Scholar]
- 24.Shao WH, Del Prete A, Bock CB, Haribabu B. Targeted disruption of leukotriene B4 receptors BLT1 and BLT2: A critical role for BLT1 in collagen-induced arthritis in mice. J. Immunol. 2006;176:6254–6261. doi: 10.4049/jimmunol.176.10.6254. [DOI] [PubMed] [Google Scholar]
- 25.Chen M, Lam BK, Kanaoka Y, Nigrovic PA, Audoly LP, Austen KF, Lee DM. Neutrophil-derived leukotriene B4 is required for inflammatory arthritis. J. Exp. Med. 2006;203:837–842. doi: 10.1084/jem.20052371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim ND, Chou RC, Seung E, Tager AM, Luster AD. A unique requirement for the leukotriene B4 receptor BLT1 for neutrophil recruitment in inflammatory arthritis. J. Exp. Med. 2006;203:829–835. doi: 10.1084/jem.20052349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mathis SP, Jala VR, Lee DM, Haribabu B. Nonredundant Roles for Leukotriene B4 Receptors BLT1 and BLT2 in Inflammatory Arthritis. J. Immunol. 2010;185:3049–3056. doi: 10.4049/jimmunol.1001031. [DOI] [PubMed] [Google Scholar]
- 28.Chou RC, Kim ND, Sadik CD, Seung E, Lan Y, Byrne MH, Haribabu B, Iwakura Y, Luster AD. Lipid-cytokine-chemokine cascade drives neutrophil recruitment in a murine model of inflammatory arthritis. Immunity. 2010;33:266–278. doi: 10.1016/j.immuni.2010.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bailie MB, Standiford TJ, Laichalk LL, Coffey MJ, Strieter R, Peters-Golden M. Leukotriene-deficient mice manifest enhanced lethality from Klebsiella pneumonia in association with decreased alveolar macrophage phagocytic and bactericidal activities. J. Immunol. 1996;157:5221–5224. [PubMed] [Google Scholar]
- 30.Mancuso P, Nana-Sinkam P, Peters-Golden M. Leukotriene B4 augments neutrophil phagocytosis of Klebsiella pneumoniae. Infect. Immun. 2001;69:2011–2016. doi: 10.1128/IAI.69.4.2011-2016.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Serezani CH, Perrela JH, Russo M, Peters-Golden M, Jancar S. Leukotrienes are essential for the control of Leishmania amazonensis infection and contribute to strain variation in susceptibility. J. Immunol. 2006;177:3201–3208. doi: 10.4049/jimmunol.177.5.3201. [DOI] [PubMed] [Google Scholar]
- 32.Aliberti J, Serhan C, Sher A. Parasite-induced lipoxin A4 is an endogenous regulator of IL-12 production and immunopathology in Toxoplasma gondii infection. J. Exp. Med. 2002;196:1253–1262. doi: 10.1084/jem.20021183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wakeland E, Morel L, Achey K, Yui M, Longmate J. Speed congenics: a classic technique in the fast lane (relatively speaking) Immunol. Today. 1997;18:472–477. doi: 10.1016/s0167-5699(97)01126-2. [DOI] [PubMed] [Google Scholar]
- 34.Brown CR, Lai AYC, Callen ST, Blaho VA, Hughes JM, Mitchell WJ. Adenoviral delivery of interleukin-10 fails to attenuate experimental Lyme disease. Infect. Immun. 2008;76:5500–5507. doi: 10.1128/IAI.00808-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morrison TB, Ma Y, Weis JH, Weis JJ. Rapid and sensitive quantification of Borrelia burgdorferi-infected mouse tissues by continuous fluorescent monitoring of PCR. J. Clin. Microbiol. 1999;37:987–992. doi: 10.1128/jcm.37.4.987-992.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Blaho VA, Buczynski MW, Dennis EA, Brown CR. Cyclooxygenase-1 orchestrates germinal center formation and antibody class-switch via regulation of IL-17. J. Immunol. 2009;183:5644–5653. doi: 10.4049/jimmunol.0901499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boxio R, Bossenmeyer-Pourie C, Steinckwich N, Dournon C, Nusse O. Mouse bone marrow contains large numbers of functionally competent neutrophils. J. Leukoc. Biol. 2003;75:604–611. doi: 10.1189/jlb.0703340. [DOI] [PubMed] [Google Scholar]
- 38.Maderna P, Yona S, Perretti M, Godson C. Modulation of phagocytosis of apoptotic neutrophils by supernatant from dexamethasone-treated macrophages and annexin-derived peptide Ac2-26. J. Immunol. 2005;174:3727–3733. doi: 10.4049/jimmunol.174.6.3727. [DOI] [PubMed] [Google Scholar]
- 39.Takano T, Fiore S, Maddox JF, Brady HR, Petasis NA, Serhan CN. Aspirin-triggered 15-epi-lipoxin A4 (LXA4) and LXA4 stable analogues are potent inhibitors of acute inflammation: evidence for anti-inflammatory receptors. J. Exp. Med. 1997;185:1693–1704. doi: 10.1084/jem.185.9.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Samuelsson B, Dahlen SE, Lindgren JA, Rouzer CA, Serhan CN. Leukotrienes and lipoxins: structures, biosynthesis, and biological effects. Science. 1987;237:1171–1176. doi: 10.1126/science.2820055. [DOI] [PubMed] [Google Scholar]
- 41.Jakobsson PJ, Steinhilber D, Odlander B, Radmark O, Claesson HE, Samuelsson B. On the expression and regulation of 5-lipoxygenase in human lymphocytes. Proc. Nat. Acad. Sci. , USA. 1992;89:3521–3525. doi: 10.1073/pnas.89.8.3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim DC, Hsu FI, Barrett NA, Friend DS, Grenningloh R, Ho IC, Al-Garawi A, Lora JM, Lam BK, Austen KF, Kanaoka Y. Cysteinyl leukotrienes regulate Th2 cell-dependent pulmonary inflammation. J. Immunol. 2006;176:4440–4448. doi: 10.4049/jimmunol.176.7.4440. [DOI] [PubMed] [Google Scholar]
- 43.Chen H, Qin J, Wei P, Zhang J, Li Q, Fu L, Li S, Ma C, Cong B. Effects of leukotriene B4 and prostaglandin E2 on the differentiation of murine Foxp3+ T regulatory cells and Th17 cells. Prostaglandins Leukot. Essent. Fatty Acids. 2009;80:195–200. doi: 10.1016/j.plefa.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 44.Firestein GS, Yeo M, Zvaifler NJ. Apoptosis in rheumatoid arthritis synovium. J. Clin. Invest. 1995;96:1631–1638. doi: 10.1172/JCI118202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schrijvers DM, De Meyer GRY, Kockx MM, Herman AG, Martinet W. Phagocytosis of apoptotic cells by macrophages is impaired in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2005;25:1256–1261. doi: 10.1161/01.ATV.0000166517.18801.a7. [DOI] [PubMed] [Google Scholar]
- 46.Mancuso P, Standiford TJ, Marshall T, Peters-Golden M. 5-Lipoxygenase reaction products modulate alveolar macrophage phagocytosis of Klebsiella pneumoniae. Infect. Immun. 1998;66:5140–5146. doi: 10.1128/iai.66.11.5140-5146.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ravichandran KS. Find-me and eat-me signals in apoptotic cell clearance: progress and conundrums. J. Exp. Med. 2010;207:1807–1817. doi: 10.1084/jem.20101157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Montgomery RR, Lusitani D, de Boisfleury Chevance A, Malawista SE. Human phagocytic cells in the early innate immune response to Borrelia burgdorferi. J. Infect. Dis. 2002;185:1773–1779. doi: 10.1086/340826. [DOI] [PubMed] [Google Scholar]
- 49.Canetti C, Hu B, Curtis JL, Peters-Golden M. Syk activation is a leukotriene B4-regulated event involved in macrophage phagocytosis of IgG-coated targets but not apoptotic cells. Blood. 2003;102:1877–1883. doi: 10.1182/blood-2003-02-0534. [DOI] [PubMed] [Google Scholar]
- 50.Fourie AM. Modulation of inflammatory disease by inhibitors of leukotriene A4 hydrolase. Curr. Opin. Investig. Drugs. 2009;10:1173–1182. [PubMed] [Google Scholar]
- 51.Sampson AP. FLAP inhibitors for the treatment of inflammatory diseases. Curr. Opin. Investig. Drugs. 2009;10:1163–1172. [PubMed] [Google Scholar]
- 52.Buczynski MW, Dumlao DS, Dennis EA. Thematic review series: Proteomics. An integrated omics analysis of eicosanoid biology. J. Lipid Res. 2009;50:1015–1038. doi: 10.1194/jlr.R900004-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Alencar NM, Oliveira RS, Figueiredo JG, Cavalcante IJ, Matos MP, Cunha FQ, Nunes JV, Bomfim LR, Ramos MV. An anti-inflammatory lectin from Luetzelburgia auriculata seeds inhibits adhesion and rolling of leukocytes and modulates histamine and PGE2 action in acute inflammation models. Inflamm. Res. 2010;59:245–254. doi: 10.1007/s00011-009-0092-9. [DOI] [PubMed] [Google Scholar]
- 54.Mao JT, Tsu IH, Dubinett SM, Adams B, Sarafian T, Baratelli F, Roth MD, Serio KJ. Modulation of pulmonary leukotriene B4 production by cyclooxygenase-2 inhibitors and lipopolysaccharide. Clin. Cancer Res. 2004;10:6872–6878. doi: 10.1158/1078-0432.CCR-04-0945. [DOI] [PubMed] [Google Scholar]
- 55.Kapoor M, Kojima F, Qian M, Yang L, Crofford LJ. Shunting of prostanoid biosynthesis in microsomal prostaglandin E synthase-1 null embryo fibroblasts: regulatory effects on inducible nitric oxide synthase expression and nitrite synthesis. FASEB J. 2006;20:2387–2389. doi: 10.1096/fj.06-6366fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mahshid Y, Lisy MR, Wang X, Spanbroek R, Flygare J, Christensson B, Bjorkholm M, Sander B, Habenicht A, Claesson HE. High expression of 5-lipoxygenase in normal and malignant mantle zone B lymphocytes. BMC Immunology. 2009;10:1–12. doi: 10.1186/1471-2172-10-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Secor WE, Powell MR, Morgan J, Wynn TA, Funk CD. Mice deficient for 5-lipoxygenase, but not leukocyte-type 12-lipoxygenase, display altered immune responses during infection with Schistosoma mansoni. Prostaglandins Other Lipid Mediat. 1998;56:291–304. doi: 10.1016/s0090-6980(98)00059-8. [DOI] [PubMed] [Google Scholar]
- 58.Weis JJ. Host-pathogen interactions and the pathogenesis of murine Lyme disease. Curr. Opin. Rheumatol. 2002;14:399–403. doi: 10.1097/00002281-200207000-00011. [DOI] [PubMed] [Google Scholar]
- 59.Levy BD, Clish CB, Schmidt B, Gronert K, Serhan CN. Lipid mediator class switching during acute inflammation: signals in resolution. Nat. Immunol. 2001;2:612–619. doi: 10.1038/89759. [DOI] [PubMed] [Google Scholar]
- 60.Vandivier RW, Henson PM, Douglas IS. Burying the dead. The impact of failed apoptotic cell removal (efferocytosis) on chronic inflammatory lung disease. Chest. 2006;129:1673–1682. doi: 10.1378/chest.129.6.1673. [DOI] [PubMed] [Google Scholar]
- 61.Voll RE, Herrmann M, Roth EA, Stach C, Kalden JR, Girkontaite I. Immunosuppressive effects of apoptotic cells. Nature. 1997;390:350–351. doi: 10.1038/37022. [DOI] [PubMed] [Google Scholar]
- 62.Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-β, PGE2, and PAF. J. Clin. Invest. 1998;101:890–898. doi: 10.1172/JCI1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Huynh ML, Malcolm KC, Kotaru C, Tilstra JA, Westcott JY, Fadok VA, Wenzel SE. Defective apoptotic cell phagocytosis attenuates prostaglandin E2 and 15-hydroxyeicosatetraenoic acid in severe asthma alveolar macrophages. Am. J. Respir. Crit. Care Med. 2005;172:972–979. doi: 10.1164/rccm.200501-035OC. [DOI] [PubMed] [Google Scholar]
- 64.Munoz LE, Gaipl US, Franz S, Sheriff A, Voll RE, Kalden JR, Herrmann M. SLE–a disease of clearance deficiency? Rheumatology. 2005;44:1101–1107. doi: 10.1093/rheumatology/keh693. [DOI] [PubMed] [Google Scholar]
- 65.Serhan CN. Novel Lipid Mediators and Resolution Mechanisms in Acute Inflammation: To Resolve or Not? Am. J. Pathol. 2010;177:1576–1591. doi: 10.2353/ajpath.2010.100322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Blaho VA, Buczynski MW, Brown CR, Dennis EA. Lipidomic analysis of dynamic eicosanoid responses during the induction and resolution of Lyme arthritis. J. Biol. Chem. 2009;284:21599–21612. doi: 10.1074/jbc.M109.003822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Serhan CN, Yacoubian S, Yang R. Anti-inflammatory and proresolving lipid mediators. Ann. Rev. Pathol. 2008;3:279–312. doi: 10.1146/annurev.pathmechdis.3.121806.151409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Godson C, Mitchell S, Harvey K, Petasis NA, Hogg N, Brady HR. Cutting Edge: Lipoxins rapidly stimulate nonphlogistic phagocytosis of apoptotic neutrophils by monocyte-derived macrophages. J. Immunol. 2000;164:1663–1667. doi: 10.4049/jimmunol.164.4.1663. [DOI] [PubMed] [Google Scholar]
- 69.Serhan CN. Lipoxins and aspirin-triggered 15-epi-lipoxins are endogenous components of antiinflammation: emergence of the counterregulatory side. Arch. Immunol. Ther. Exp. (Warsz. ) 2001;49:177–188. [PubMed] [Google Scholar]