

Abstract
Thrombospondin repeat (TSR)-like domains are structures involved with cell adhesion. Plasmodium falciparum proteins containing TSR domains play crucial roles in parasite development. In particular, the preerythrocytic P. falciparum circumsporozoite protein is involved in hepatocyte invasion. The importance of these domains in two other malaria proteins, the merozoite-specific thrombospondin-related anonymous protein (MTRAP) and the thrombospondin-related apical membrane protein (PTRAMP), were assessed using near-full-length recombinant proteins composed of the extracellular domains produced in Escherichia coli. MTRAP is thought to be released from invasive organelles identified as micronemes during merozoite invasion to mediate motility and host cell invasion through an interaction with aldolase, an actin binding protein involved in the moving junction. PTRAMP function remains unknown. In this study, the conformation of recombinant MTRAP (rMTRAP) appeared to be a highly extended protein (2 nm by 33 nm, width by length, respectively), whereas rPTRAMP had a less extended structure. Using an erythrocyte binding assay, rMTRAP but not rPTRAMP bound human erythrocytes; rMTRAP binding was mediated through the TSR domain. MTRAP- and in general PTRAMP-specific antibodies failed to inhibit P. falciparum development in vitro. Altogether, MTRAP is a highly extended bifunctional protein that binds to an erythrocyte receptor and the merozoite motor.
INTRODUCTION
Malaria caused by Plasmodium falciparum remains a serious global disease resulting in nearly 800,000 deaths annually, principally in children in sub-Saharan Africa (43). Various control efforts, including insecticide-impregnated bed nets and artemisinin combination therapy, are currently impacting the morbidity and mortality of the disease; however, a long-term, cost-effective strategy requires an effective malaria vaccine (5, 43). Recently, a major step toward this end was reported for an investigative malaria vaccine identified as RTS,S, which is comprised of the carboxyl-terminal half of the circumsporozoite protein (CSP) fused with the hepatitis B surface protein and biophysically presented as a virus-like particle formulated with the adjuvant ASO1 (3). RTS,S in a phase 3 trial reduced the number of clinical episodes by 50.4% compared to the number in a control group for 14 months following the final dose (30). A unique protein motif within P. falciparum CSP (PfCSP) and RTS,S is the presence of a thrombospondin repeat (TSR)-like domain within the carboxyl terminus, which may be involved in cellular adhesion (8–10, 34), similar to other members within the TSR superfamily (42). Analysis of the P. falciparum genome using conserved TSR amino acid motifs identified additional malaria parasite proteins which are also a part of this superfamily (2, 24, 39). Two such members, identified in blood-stage P. falciparum parasites, are the merozoite-specific thrombospondin-related anonymous protein (MTRAP) and the thrombospondin-related apical membrane protein (PTRAMP).
MTRAP is named after the sporozoite thrombospondin-related anonymous protein (TRAP) (2). TRAP and MTRAP are both type 1 membrane proteins with a cytoplasmic domain. TRAP has an essential biological role in sporozoite gliding motility by linking the actin-myosin motor through its cytoplasmic domain while binding to its receptor on the hepatocyte surface through its extracellular domain, which contains a TSR domain and a von Willebrand factor-like A domain (17, 21, 24, 37). TRAP is redistributed from micronemes to the sporozoite surface and subsequently cleaved near the membrane by a rhomboid protease (1, 29). The cytoplasmic domain of TRAP interacts with actin-myosin via aldolase as a bridge (7, 16). Gene disruption of TRAP, as well as mutation analysis of specific amino acid residues within the cytoplasmic domain, impairs sporozoite motility (17, 37). In comparison, no adhesive function for the ectodomain of MTRAP has been identified, although the cytoplasmic domain is reported to interact with aldolase and processing by a rhomboid protease near the membrane surface appears possible (1, 2). Interestingly, MTRAP-specific antibodies have failed to inhibit parasite invasion of erythrocytes in vitro, despite the fact that it has not been possible to knock out this gene by standard genetic techniques, which suggests an essential role for MTRAP in asexual blood-stage development (2, 24).
PTRAMP, another type 1 merozoite protein, is initially localized within micronemes and then may be redistributed onto the merozoite surface (39). PTRAMP contains a TSR domain within its ectodomain and has a cytoplasmic domain which has been shown to weakly interact with aldolase (2). PTRAMP is a conserved protein within the Plasmodium genus and appears to have an essential and conserved unknown biological function considering that the gene that encodes it cannot be disrupted (24, 39).
In order to better understand the functions of MTRAP and PTRAMP and investigate their roles during merozoite invasion of erythrocytes, including susceptibility to antibody blockade, we generated full-length recombinant MTRAP (rMTRAP) and PTRAMP (rPTRAMP) of the extracellular sequence, using Escherichia coli expression and protein refolding. Both rMTRAP and rPTRAMP were extensively biochemically and biophysically characterized and used to produce antigen-specific antisera. The results collated have enabled the identification that MTRAP is a highly extended protein that binds human erythrocytes mediated by the TSR domain. Neither antigen appears to be a robust target for antibody-mediated blockade of erythrocyte invasion.
MATERIALS AND METHODS
Expression and production of recombinant proteins rMTRAP and rPTRAMP.
The amino acid sequence of rPTRAMP (PFL0870w; GenBank accession no. XP_001350582) was used to generate a codon-optimized synthetic gene for expression in E. coli (GeneArt, Regensburg, Germany). The construct, corresponding to amino acids (aa) 25 to 309 of the full-length gene, was subcloned into the E. coli pET-24a+ expression vector downstream of the T7 promoter using the NdeI and XhoI restriction sites (EMD Chemicals, Inc., Gibbstown, NJ). Similarly, the amino acid sequence of rMTRAP (PF10_0281; GenBank protein accession no. XP_001347565.1) was used to generate a codon-optimized synthetic gene for expression in E. coli (GeneArt, Regensburg, Germany). The construct, corresponding to aa 23 to 433 of the full-length gene, was subcloned into the E. coli pET-24a+ expression vector using the NdeI and XhoI restriction sites (EMD Chemicals, Inc., Gibbstown, NJ). The resulting transcribed genes incorporate the additional amino acid sequence LEHHHHHH. Both constructs were transformed into E. coli BL21(DE3) cells (Novagen, San Diego, CA) and used for the expression of rPTRAMP and rMTRAP. Expression of both rPTRAMP and rMTRAP was performed in a 1-liter culture shake flask, using Luria-Bertani medium (KD Medical, Columbia, MD) containing 50 μg/ml kanamycin at 37°C. Once the optical density at 550 nm reached 0.6 to 0.8, the culture was induced with 1 mM isopropyl-1-thio-β-d-1-galactopyranoside (IPTG) for 3 h. Following induction, cells were harvested by centrifugation, and the cell pellet was stored at −80°C.
To break the bacterial cells, the cell pellet was resuspended in 10 volumes of lysis buffer (10 mM Tris-HCl, 10 mM EDTA, 10 mM NaCl, 5 mM dithiothreitol [DTT] [pH 8.0]) and then mechanically disrupted by passage through a Microfluidizer (M110Y; Microfluidics Corp., Newton, MA) five times operating at 19,000 lb/in2. The rPTRAMP and rMTRAP lysates were centrifuged for 45 min at 10,000 × g to separate the soluble fraction from the inclusion bodies and cellular debris. rPTRAMP was subsequently captured from the inclusion body fraction, whereas rMTRAP was captured from the soluble fraction as described below.
Capture, refolding, and purification.
rMTRAP was captured from the soluble fraction under denatured and reduced conditions using a nickel Sepharose 6 Fast Flow column (GE Healthcare, Piscataway, NJ). Load material was first dialyzed in a mixture containing 10 mM Tris, 100 mM sodium phosphate, and 14.1 mM β-mercaptoethanol (pH 8.0). Prior to column loading, urea and imidazole were added so that final concentrations of 8 M and 20 mM, respectively, were achieved. A preequilibrated affinity column using a similar buffered solution was loaded, and the bound protein was eluted by an imidazole gradient (20 to 400 mM) in 20 column volumes. Fractions containing rMTRAP were pooled and refolded by rapid dilution at 50 μg/ml protein into a refold buffer containing a 10 mM glutathione oxidized (GSSG) and 2 mM glutathione (GSH) redox system overnight at 10°C. The refolded protein was dialyzed in 50 mM Tris-HCl (pH 7.4), loaded onto a preequilibrated Q Sepharose Fast Flow ion-exchange chromatography column (GE Healthcare), and eluted with an NaCl gradient (0 to 1 M NaCl) in 20 column volumes. The refolded rMTRAP was polished using size exclusion chromatography in a HiLoad 16/60 Superdex 200 prep-grade column (GE Healthcare) equilibrated with phosphate-buffered saline (PBS) at pH 7.4. The final refolded and purified protein was filtered with a 0.22-μm-pore filter and stored at less than −70°C. Similar conditions were used for producing rPTRAMP, with two exceptions. First, the GSH and GSSG concentrations in the refolding buffer were 5 mM and 1 mM, respectively. Second, the final polishing step required a HiLoad 16/60 Superdex 75 prep-grade column (GE Healthcare). Endotoxin levels of the various antigens were measured using Limulus amoebocyte lysate in a 96-well plate with chromogenic reagents and PyroSoft software (Associates of Cape Cod, Inc., East Falmouth, MA).
Amino-terminal sequencing.
Automated Edman sequencing was performed using a Sequenator model Procise 494 HT connected to an online UV detector (Applied Biosystems). Samples were immobilized onto polyvinylidene difluoride (PVDF) using a ProSorb sample preparative cartridge. A 2- by 220-mm C18 microbore column (Applied Biosystems) was used for separation of the phenylthiohydantoin (PTH) amino acids using an acetonitrile–2-propanol gradient elution.
Reversed-phase HPLC.
For reversed-phase high-performance liquid chromatography (HPLC), samples were analyzed on a 2-mm by 250-mm, 5-μm particle size, C4 column (Phenomenex, Torrance, CA). The column was equilibrated in 95% mobile phase A (0.1% [wt/vol] trifluoroacetic acid [TFA] in water) and 5% mobile phase B (0.1% [wt/vol] TFA in acetonitrile), and elution was performed by increasing the percentage of mobile phase B at a constant flow rate (0.2 ml/min). Elution peaks were collected and vacuum dried in a Vacufuge concentrator (Eppendorf, Westbury, NY). Theses samples were submitted to the Research Technology Branch (NIAID/NIH, Rockville, MD) for mass spectrometry (MS) and amino-terminal sequencing.
CD spectroscopy.
Both rMTRAP and rPTRAMP were analyzed as previously described (26). In brief, circular dichroism (CD) spectra of the proteins were recorded on a Jasco J-815 CD spectropolarimeter over the wavelength range of 185 to 260 nm. Data were collected using a data pitch of 0.2 nm, a slit bandwidth of 1.0 nm, and a signal averaging time of 1.0 s in a 1-mm quartz cuvette. Secondary structure content was calculated using the DICHROWEB web server (http://dichroweb.cryst.bbk.ac.uk) and CDPro software.
Analytical SEC-MALS-QELS HPLC.
To evaluate the identity, purity, and solution aggregation state of purified rMTRAP and rPTRAMP, analytical size exclusion chromatography with in-line multiangle light scattering (SEC-MALS) and quasielastic light scatter (QELS) detection was performed. Chromatography was performed on an Alliance HPLC system (Waters, Milford, MA) connected in series to a multiangle DAWN EOS light-scattering detector and a QELS detector (Wyatt Technology, Santa Barbara CA). Samples were prepared and analyzed as described by Tsai et al. (40). In brief, protein separation of rPTRAMP was performed on a TSKgel G2000SWxl column, whereas separation of rMTRAP was accomplished on a TSKgel G3000SWxl column (Tosoh Bioscience, Montgomeryville, PA). The column was equilibrated in the mobile phase consisting of 1.04 mM KH2PO4, 2.97 mM Na2P04 · 7H2O, 308 mM NaCl, and 0.02% azide (pH 7.4), and the samples were run using an isocratic elution at 0.5 ml/min. A gel filtration standard (Bio-Rad, Hercules, CA) was run for size comparison as well as a 25-μl injection of bovine serum albumin (BSA) at 5 mg/ml for configuration of the MALS data.
MS.
Intact mass analyses were performed by electrospray ionization mass spectrometry (ESI-MS). Samples were analyzed by coupling an automated chip-based nanoelectrospray unit, TriVersa Nanomate (Advion BioSciences), to a 4000 Q Trap liquid chromatography-tandem MS (LC-MS/MS) device (Applied Biosystems/Sciex). Sample solutions were first adjusted to low pH (0.1% acetic acid) and low acetonitrile (2%) and then desalted through a microprotein trap cartridge (Michrom BioResources) at 20 ml/min. In LC-MS mode, a Tempo multidimensional liquid chromatography (MDLC) device designed for HPLC applications was used to automate trap desalting of protein samples. Bound protein was step eluted with 60% acetonitrile and 0.1% acetic acid at 800 nl/min into the 4000 Q Trap via a TriVersa capillary coupler.
Polyclonal antibody production.
Rat polyclonal sera were generated at the National Institutes of Health in compliance with guidelines of the National Institutes of Health Institutional Animal Care and Use Committee. In brief, two groups of six rats were immunized with 10 μg of rMTRAP or rPTRAMP formulated in Montanide ISA 720 (Seppic, Inc., Fairfield, NJ) on days 0, 28, and 42 and exsanguinated on day 56. Control rats were immunized with the adjuvant alone. Rabbit polyclonal sera were generated against rMTRAP and rPTRAMP by Spring Valley Laboratories. In brief, 10 healthy female New Zealand White rabbits (Orytolagus cuniculus) were randomly selected into 3 groups and housed individually. Group 1 contained 6 rabbits which were immunized with 20 μg of rPTRAMP formulated in Montanide ISA 720 on day 0 and day 28. The rabbits were prebled on day 0 and then exsanguinated on day 42. Group 2 contained 2 rabbits which were immunized with 20 μg of rMTRAP in the same fashion as rPTRAMP. Group 3 contained 2 rabbits which were immunized with adjuvant alone in the same fashion as rPTRAMP.
IFA and confocal microscopy.
Immunofluorescence assay (IFA) analysis was performed as per the standard protocol (26, 33). In brief, schizont-enriched parasites were smeared on slides and stored at −80°C. Slides were thawed, acetone fixed, and reacted with primary and secondary antibodies diluted in blocking buffer (1× PBS, 3% BSA) at 4°C (25). All primary and secondary antibody incubations were carried out at room temperature and separated by extensive washing with PBS containing 0.05% Tween 20 (Bio-Rad). The samples were mounted under coverslips using Vectashield hard-set mounting medium and stored at 4°C until images were acquired by confocal microscopy as described previously (33). In brief, a Leica SP2 confocal microscope using Leica confocal software was used for image acquisition. All images were collected by using a PL APO ×100/1.4 oil immersion objective and a confocal zoom of ×6. Secondary reagents used Alexa 488 or Alexa 568 species-specific conjugated antibodies (Molecular Probes). All images were collected as three-dimensional (3D) data sets (z-stacks) with a step size of 0.1221 nm between the successive optical sections. Deconvolution of all image stacks was performed using Huygens Essential (Scientific Volume Imaging) to improve the maximum resolution of the data, as well as to minimize background noise. Deconvolved images were saved and analyzed through Imaris image analysis software (version 5.7.2; Bitplane). Confocal images in this study are displayed as maximum projection of the 3D image stacks.
GIA assay.
The standard methodology for the growth inhibition activity (GIA) assay has been described previously (19). The assay was performed with purified IgGs at the indicated concentrations against the 3D7 strain of P. falciparum parasites.
Erythrocyte binding assay.
The erythrocyte binding assay was performed as previously described (20), with the following modifications. One hundred microliters of human erythrocytes with a 50% hematocrit was washed three times in incomplete RPMI 1640 medium (Invitrogen) and resuspended in 500 μl of RPMI containing 200 μM rMTRAP, rPTRAMP, PpPfs25 (41), EcMSP142 (32), or BAEBL RII (D. L. Narum, unpublished data) and 10% fetal bovine serum (Invitrogen). The erythrocyte suspension was incubated at 37°C for 1 h with constant shaking and then layered over 300 μl of oil (dibutyl phthalate; Sigma) and centrifuged at 13,000 × g for 30 s. Bound protein was eluted from the erythrocyte pellet by the addition of 100 μl of RPMI with 1 M NaCl. Erythrocyte eluates were analyzed by Western blotting using antigen-specific rabbit antisera produced against the same recombinant antigen and then detected with alkaline phosphatase-conjugated goat anti-rabbit IgG (Kirkegaard & Perry Laboratories, MD).
Analytical ultracentrifugation characterization of rMTRAP.
Boundary sedimentation velocity measurements were made in an Optima XLA analytical ultracentrifuge (Beckman-Coulter Instruments). Sedimentation velocity analysis was performed at 20°C at a rotor speed of 58,000 rpm. The centrifuge cell was filled with 400 μl of protein solution at a concentration of 1.0 mg/ml of rMTRAP. Absorbance scans were obtained at 235 nm. Sedimentation coefficient distribution analysis to deconvolute the boundary velocity data into sedimenting species was performed as previously described (31), using the public domain software Sedfit developed by Peter Schuck (http://www.analyticalultracentrifugation.com/). The result of this computational analysis is a presentation of the distribution of sedimenting species in the form of a c(s) versus s plot. In this computational treatment, the sedimentation boundary velocity data were subjected to maximum entropy regularization statistical analysis for the most parsimonious distribution of sedimenting species (31) that best fits the data. A weight average shape factor (f/fo frictional ratio) is extracted from the boundary spreading, and this allows for the conversion of the c(s) versus s distribution to a c(M) versus M graphical presentation, thereby providing the molar mass of each sedimenting peak. The software program Sednterp developed by Hayes, Laue, and Philo (http://www.bbri.org/RASMB/rasmb.html) was also used to determine the partial specific volume of the rMTRAP and the viscosity (η) and density (ρ) for the PBS solution used in the sedimentation analyses and for the hydrodynamic analysis to be reported below.
AFM.
Samples for atomic force microscopy (AFM) were prepared and analyzed basically as described by Plassmeyer et al. (26) and Tsai et al. (40), with modifications as noted. Briefly, biological AFM imaging of the protein products was carried out under a range of conditions, in both fluid and air, using gentle-tapping-mode AFM, mostly with a PicoForce multimode atomic force microscope (Veeco, CA) consisting of a Nanoscope V controller, a type E scanner head, and a sharpened TESP-SS (Veeco, CA) or similar AFM cantilever (26, 40). For rMTRAP visualization, suitable protein attachment was readily achieved by a 1-min incubation of 7 μl of ∼1 μg/ml solution of rMTRAP in PBS buffer (pH 7.4) on freshly peeled mica substrates, followed by rinsing with ∼1 ml of deionized water and complete drying under an inert gas flow. The sample was then sealed into the instrument compartment dehumidified by Drierite. AFM images were evaluated within the Nanoscope software (version 7.2; Veeco, CA) and exported to Image J version 1.41o (National Institutes of Health, Bethesda, MD) and Mathcad version 14 (Mathsoft, Needham, MA) for further analyses and display.
Computational modeling.
The homology model of the MTRAP TSR domains was constructed using HHPRED (35) and Rosetta (18). Residues 23 to 105 of MTRAP were submitted to the HHPRED server to query the Protein Data Bank (PDB) (6). The crystal structure of human thrombospondin-1 (PDB identification [ID] 1LSL) (38) was identified as the best template (probability, 98.3; E value, 3E−07) and was used for subsequent modeling. For the modeling procedure, the Homo sapiens TSP-1 (HsTSP-1) TSR2 domain spanning residues 434 to 490 was used as the template for both TSR domains of MTRAP (residues 28 to 61 and 62 to 94). Three blocks of residues were restrained to the coordinates of HsTSP-1 residues 434 to 490, with the intervening loop regions modeled using the Rosetta loop relax protocol (27). Specifically, residues 32 to 38 and 66 to 72, 41 to 48 and 75 to 82, and 55 to 61 and 88 to 94 of MTRAP were restrained to residues 441 to 447, 454 to 461, and 479 to 485, respectively, of HsTSP-1. The final alignment was created through iterative modeling and manual adjustments.
Assessment of human antibody responses.
Plasma samples were collected from volunteers (n = 93, 18 to 60 years old) in the village of Kenieroba, Mali. The approval of the human study was obtained from the Ethical Review Committees of the Faculty of Medicine, Pharmacy, and Dentistry at the University of Bamako (Mali) and the National Institute of Allergy and Infectious Diseases (IRB no. 08-I-N120). Individual written informed consent was obtained from all participants.
All 93 of the plasma samples were tested at a 1:500 dilution against MTRAP, PTRAMP, apical membrane antigen 1 (AMA1), and merozoite surface protein 1, 42 kDa (MSP142), enzyme-linked immunosorbent assay (ELISA) plate antigens. The standardized methodology for performing the ELISA has been described previously (22).
RESULTS
Expression, purification, and biological characterization of rMTRAP and rPTRAMP.
In order to investigate the roles of both PfMTRAP and PfPTRAMP during invasion of erythrocytes by Plasmodium falciparum, E. coli codon-optimized genes were produced corresponding to amino acids (aa) 23 to 433 of PfMTRAP and aa 25 to 309 of PfPTRAMP (Fig. 1A). Following expression and purification as described in Materials and Methods, the integrity and purity of the recombinant proteins rMTRAP and rPTRAMP were examined by Coomassie blue-stained SDS-PAGE gel (Fig. 1B). A mobility shift was observed for both recombinant proteins, indicating the presence of disulfide bond formation due to protein refolding. Both rat and rabbit antisera were generated to recombinant proteins rMTRAP and rPTRAMP; as shown in the Western blot using immune polyclonal rat sera for rMTRAP and rPTRAMP (Fig. 1C), additional lower-molecular-weight bands are recognized by the rat antisera which are related fragments or dimers of these two recombinant proteins. Similar results were obtained with the rabbit antisera (data not shown).
Fig 1.

Expression and characterization of purified rMTRAP and rPTRAMP. The diagram in panel A represents the full-length MTRAP and PTRAMP genes, and the solid lines represent recombinant protein boundaries for each ectodomain produced in E. coli. Numerical values represent amino acid positions of native MTRAP and PTRAMP (accession no. XP_001347565.1 and XP_001350582, respectively). Abbreviations in panel A: SS, signal sequence; TSR, thrombospondin repeat-like domain; and TM, transmembrane spanning domain. Panels B and C show purified rMTRAP and rPTRAMP analyzed by Coomassie blue-stained SDS-PAGE and Western blot analyses. Western blots were performed with polyclonal rat antigen-specific sera under nonreduced (NR) and reduced (R) conditions. Panels D and E show the characterization of rMTRAP by reversed-phase HPLC analysis under nonoxidized conditions (D) and the corresponding intact mass of the major peak (E). AU, absorbance units; amu, atomic mass units.
Additional biochemical and biophysical characterizations of rMTRAP and rPTRAMP by electrospray ionization mass spectrometry (ESI-MS), N-terminal sequencing, and determination of the endotoxin concentration were performed to establish the integrity, identity, and purity of both recombinant proteins. The primary elution peak obtained from reversed-phase HPLC of rMTRAP (Fig. 1D) was analyzed by ESI-MS. The observed mass of the nonreduced form of rMTRAP was 49,201 Da (Fig. 1E), which is within 1 Da of the nonreduced theoretical mass, 49,199.8 Da, demonstrating excellent integrity of rMTRAP. The identity of rMTRAP was confirmed by N-terminal sequencing. The expected and observed amino acid sequences were in complete agreement for rMTRAP (MISTHDTXDE). Recombinant MTRAP was also analyzed by reduced reversed-phase HPLC. A minor peak was isolated and identified by N-terminal sequencing (MNNSYIY) which corresponded to a truncated form with an amino-terminal deletion of 84 amino acids, which we identified as rMTRAP−84 (see Fig. S1 in the supplemental material). Due to this deletion (aa 105 relative to the native MTRAP sequence), this truncated protein lacks the TSR domain. Additionally, the primary elution peak obtained from reversed-phase HPLC of rPTRAMP was analyzed by ESI-MS and Edman degradation. The observed mass for the nonreduced form was 34,444.2 Da, which is within 1 Da of the nonreduced theoretical mass of 34,443.1 Da, demonstrating excellent integrity of rPTRAMP. The expected and observed N-terminal amino acid sequences were in complete agreement for rPTRAMP (MNDLXSXNLK) as well. The concentrations of endotoxin in the rMTRAP and rPTRAMP stocks were less than 30 and 10 endotoxin units per mg, respectively.
To characterize both the rat and rabbit antisera for biological significance, the anti-rMTRAP and -rPTRAMP polyclonal antibodies were examined for their reactivity to parasitized red blood cells by confocal microscopy. Given the prior localization of MTRAP and PTRAMP to the micronemes (2, 39), we aimed to corroborate the localization to the micronemes and confirm the integrity of the MTRAP- and PTRAMP-specific antibodies. Colocalization of rat anti-rMTRAP polyclonal antibodies with anti-AMA1, -EBA-175, and -RON4 antibodies corroborates that PfMTRAP and PfPTRAMP are located in the micronemes (see Fig. S2A and B in the supplemental material), consistent with the previously published data (2, 39).
The anti-rMTRAP and -rPTRAMP rat and rabbit polyclonal antibodies were further examined for their ability to provide growth inhibition activity (GIA) (see Table S1 in the supplemental material). Only anti-rPTRAMP antibody from two of five rats demonstrated GIA—about 60 or 80% at 10 mg/ml. The inhibition was partially reversible by preincubation with 800 μM rPTRAMP protein. In contrast, no GIA was observed for either rat or rabbit polyclonal antibodies generated to rMTRAP (data not shown).
Biological characterization of rMTRAP and rPTRAMP by erythrocyte binding assay.
Using an erythrocyte binding assay, rMTRAP was observed to bind to human erythrocytes, while no binding was observed for rPTRAMP (Fig. 2). The erythrocyte binding assay was performed using two recombinant negative control proteins, the 25-kDa sexual-stage-specific protein Pfs25 and the 42-kDa fragment of MSP1 and a recombinant positive control protein, BAEBL RII (Fig. 2). A common background band was observed in all five Western blots, as noted by an asterisk in Fig. 2. Further analysis of rMTRAP human erythrocyte binding indicated that only the full-length form bound, while rMTRAP−84, an amino-terminal truncation product which was present in the purified protein sample with an N-terminal 84-aa deletion, did not. (For characterization of the truncated product, see Fig. S1 in the supplemental material.)
Fig 2.
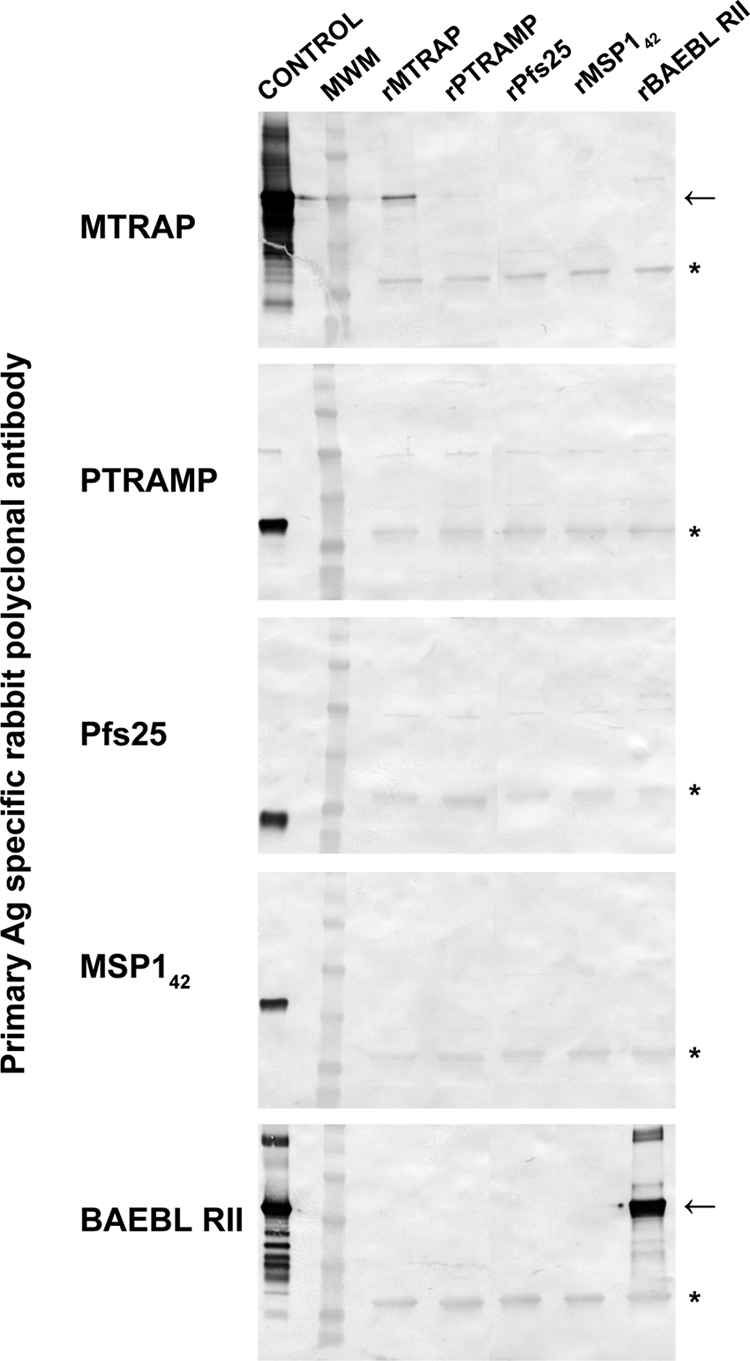
Erythrocyte-binding ability of rMTRAP. Purified recombinant proteins were added to human erythrocytes, and the eluates were separated by SDS-PAGE and tested by Western blotting with antigen (Ag)-specific rabbit antibodies. Arrows indicate expected migration by SDS-PAGE of individual recombinant protein. The asterisks indicate background bands. MWM, molecular weight marker lane.
Biophysical and biochemical characterization by SEC-MALS-QELS HPLC.
rMTRAP and rPTRAMP were examined by analytical SEC-MALS-QELS HPLC to determine their conformation, molar mass and hydrodynamic radius. Analysis of rMTRAP by SEC-HPLC indicated two populations of protein existed in solution: a monomeric population at 94% and an oligomer population at 6%. Similarly, analysis of rPTRAMP by SEC-HPLC indicated two populations of protein existed in solution: a monomeric population at 94% and a dimer population at 6%. Examination of the monomer peaks from SEC-HPLC revealed rMTRAP had a retention time (RT) of 17 to 18 min, similar to the RT of immunoglobulin G (158 kDa) at approximately 17 min (data not shown). In contrast to rMTRAP, rPTRAMP had a longer retention time, approximately 22 min. Further analysis by in-line MALS indicated the weight average molar masses of the monomeric peaks for rMTRAP and rPTRAMP of 53 kDa and 38 kDa corresponded to the theoretical masses of 49.2 kDa and 34.5 kDa, respectively (Fig. 3A; see Fig. S3A in the supplemental material). To further characterize the recombinant proteins, the hydrodynamic radii (Rh) of the monomeric recombinant proteins were examined by QELS. The Rh of rMTRAP was 5.1 nm (Fig. 3B), indicating the monomeric form of rMTRAP (49.2 kDa) is highly extended compared to a typical globular protein like BSA (66.4 kDa), which has an Rh value of ∼3.4 nm (40). In contrast to rMTRAP, the Rh of rPTRAMP was 3.1 nm (see Fig. S3B in the supplemental material), indicating the monomeric form of rPTRAMP is more globular.
Fig 3.

Biochemical characterization of rMTRAP by SEC-MALS-QELS HPLC and analytical ultracentrifugation. (A) Analysis of rMTRAP by SEC-MALS HPLC provided the molar mass distribution of the main peak (molar mass indicated by thick line) compared to the absorbance at 280 nm. (B) QELS goodness of fit of the autocorrelation function plot at the apex of the peak. (C) Sedimentation coefficient distributions and molar mass obtained from the boundary sedimentation velocity data (not shown) of the rMTRAP sample by the computational analysis described in Materials and Methods. The molar mass distribution c(M) versus M shown in the inset is calculated from the c(s) versus s distribution using the fitted weight average f/fo. The differential c(s) scale is in units of absorbance (AU) per Svedberg unit, and the differential c(M) scale is in units of absorbance per molar mass multiplied by 100,000.
Biophysical and biochemical characterization by boundary sedimentation velocity.
In order to further characterize the recombinant proteins, an orthogonal approach was utilized to determine their mass and hydrodynamic radius. Boundary sedimentation velocity was used to obtain the sedimentation coefficient distribution of rMTRAP and rPTRAMP. The absorbance scans of the sedimenting boundaries versus time (data not shown) were analyzed by the computational program Sedfit to give the sedimentation distribution of protein components shown in Fig. 3C and Fig. S3C in the supplemental material. For rMTRAP, the weight average frictional ratio (f/fo) of 1.93 was extracted in the fitting process to account for boundary diffusion. Integration of the entire c(s) peak gives a weight average uncorrected s value of 2.66S. Correcting for the effects of the density and viscosity of PBS, we obtain an s20,w value of 2.74S. The weight average sedimentation coefficient coupled with the weight average f/fo provides for the conversion of the c(s) distribution into a c(M) versus M distribution, as shown in the inset in Fig. 3C. The weight average molar mass of the major peak is 45,233. This is in good agreement with the sequence molar mass of 49,191.8. Using the latter and the s20,w value of 2.74S, we obtain the following hydrodynamic results: a calculated f/fo of 2.04 and a Stokes radius of 4.9 nm. There is an excellent agreement between the weight average frictional ratio (f/fo) of 1.93 extracted from the boundary fitting process and the f/fo of 2.04 determined from the s20,w value and the sequence molar mass. Based on the large f/fo for MTRAP, it is reasonable to model the shape of this protein as a cylinder. Using this hydrodynamic model with an estimated hydration of 0.3 g of water per g of protein, the MTRAP cylinder or rod would have length and diameter dimensions of 33.4 nm and 2.15 nm, respectively. The AFM results that are presented below support the rod/ribbon shape modeling for MTRAP. A similar analysis was performed for rPTRAMP (see Fig. S3C in the supplemental material). Integration of the entire c(s) peak gives a weight average uncorrected s value of 2.46S. Correcting for the effects of the density and viscosity of PBS, we obtain an s20,w value of 2.54S. A weight average frictional ratio (f/fo) of 1.50 was extracted in the fitting process to account for boundary diffusion. As discussed above, the weight average sedimentation coefficient coupled with the weight average f/fo provides for the conversion of the c(s) distribution into a c(M) versus M distribution in comparable fashion to the rMTRAP results that were shown in the inset in Fig. 3C. The weight average molar mass of the major peak for rPTRAMP is 34,759. This is in excellent agreement with the sequence molar mass of 34,443.1. Using the latter and the s20,w value of 2.54S, we obtain the following hydrodynamic results: a calculated f/fo of 1.49 (in excellent agreement with the fitted f/fo of 1.50) and a Stokes radius of 3.2 nm. Using a prolate model, with an estimated hydration of 0.3 g of water per g of protein, the moderately asymmetric rPTRAMP would have length and diameter dimensions of 16.35 nm and 2.65 nm, respectively.
Biophysical and biochemical characterization of rMTRAP by AFM.
AFM images directly reveal that the recombinant protein rMTRAP is an extended linear and flexible molecule with variable twisted-ribbon-like appearances (Fig. 4A). In agreement with the solution studies, AFM images suggest over 80% of the rMTRAP mass exists in a monomeric state with the measured protein volume of around 50 nm3 (Fig. 4B). However, the balance of the protein mass suggests the presence of a minute dimer population and a very sporadic occurrence of a small oligomer population. The protein particles are varied in shape, although all shapes are consistent with extended twisted-ribbon forms when quantified by the number histogram of the circularity, defined as 4π · area/(perimeter)2 (Fig. 4C). Assuming a straight ribbon shape for a fully extended molecule, the measured circularity values suggest the typical length/width ratio of between 2 and 6. Furthermore, by examination of the more typical rMTRAP monomers with a measured protein volume of between 40 and 60 nm3, the number histogram of the molecular area is tightly distributed and falls within the range of approximately 150 to 220 nm2 (Fig. 4D). On the whole, AFM images of rMTRAP suggest an “average” ribbon shape of the dimensions around 28 nm by 7 nm by 0.3 nm for rMTRAP monomers on mica surfaces. Consequently, the high-resolution AFM images of rMTRAP on a solid surface fully support the interpretations of the structures deduced from the solution state measurements.
Fig 4.

AFM shows rMTRAP as a flexible “rod”-like filamentous protein. (A) Panel of representative images, in three-dimensional plots (50-nm square viewed perpendicular to the scan direction, with a colored scale bar for height up to 0.8 nm bottom left and 30-nm bars representing the x-y scale), showing rMTRAP protein monomer shapes as seen in AFM topographies of uniformly dispersed particles on a mica surface. (B) Mass distribution histogram from ∼3,800 computed particles reveals that over 80% of rMTRAP is seen in a monomeric state (monomer-dimer decompositions illustrated under the blue curve centered at the monomer volume of 50 nm3). (C) A histogram of the circularity of these particles, defined as 4π · area/(perimeter)2, suggests flexible rod-like molecules showing twisted-ribbon-like morphologies with a typical length/width ratio of between 2 and 6. (D) A histogram of the molecular area for 955 rMTRAP monomers, having a more typical measured protein volume between 0.8 and 1.2 of the monomer value in panel B, reveals a distribution range of ∼150 to 220 nm2.
Biophysical and biochemical characterization by CD spectroscopy and thermal stability.
The circular dichroism (CD) spectrum of MTRAP showed a negative peak maxima at 199 nm (Fig. 5A), indicating significant random coil structure within the protein (13). Analysis with the DICHROWEB server and CDPro software gave a composition of 44% ± 15% random coil, 29% ± 7% β-sheet, 19% ± 6% β-turn, and 8% ± 6% α-helix. The spectrum of MTRAP was remarkably similar to that of CSP (Fig. 5A), which has a composition of 49% ± 13% random coil, 23% ± 7% β-sheet, 17% ± 5% β-turn, and 10% ± 4% α-helix (26). Upon heating of MTRAP to 80°C, the peak maxima at 200 nm decreased and ellipticity above 210 nm had a negative shift (see Fig. S4A in the supplemental material). The protein was able to revert back to its original structure upon cooling. The circular dichroism spectrum of PTRAMP had a low negative ellipticity from 205 to 220 nm (see Fig. S4B), indicating significant antiparallel β-pleated sheets (13). Analysis with the DICHROWEB server and CDPro software gave a composition of 41% ± 4% β-sheet, 35% ± 5% random coil, 20% ± 5% β-turn, and 5% ± 2% α-helix. Circular dichroism thermal melt data showed a loss of β-sheet content since the negative molar ellipticity decreased with increasing temperature (see Fig. S4B). The denatured protein was not able to revert to its original folded structure upon cooling.
Fig 5.

Biophysical and computational analyses of the structure of rMTRAP. (A) Far-UV analysis of rMTRAP and rCSP by circular dichroism in an aqueous solution. The ellipticity (degrees cm2/dmol) was plotted as a function of wavelength (nm). Raw data measured in millidegrees was converted into ellipticity (degrees cm2/dmol). Spectra were obtained at 20°C. (B) Alignment of MTRAP TSR domains from Plasmodium species with human TSP-1 TSR domains. Regions restrained to the coordinates of HsTSP-1 during homology modeling are indicated by yellow boxes. Conserved residues predicted to participate in the formation of the stacked TSR core are boxed in cyan, and nonconserved arginines in the C-terminal MTRAP TSR domains predicted to participate in stacking are boxed in periwinkle. The glutamic and aspartic acid residues predicted to hydrogen bond to the conserved central stacked arginine are boxed in light red. The secondary structure and disulfide linkages of HsTSP-1 are shown below the alignment. The predicted additional group 2 disulfide linkages of MTRAP TSR are indicated above the alignment. Organism codes and NCBI locus tags are as follows: Pf, Plasmodium falciparum, PF10_0281; Pv, Plasmodium vivax, PVX-111290; Pk, Plasmodium knowlesi, PKH_061300; Pb, Plasmodium berghei, PB000355.03.0; Pc, Plasmodium chabaudi, PC000113.04.0; Hs, Homo sapiens, THBS1. (C) Homology models of the first and second predicted TSR domains of MTRAP and the crystal structure of TSR2 of HsTSP-1. Regions predicted to align between the MTRAP TSRs and the HsTSP-1 TSRs are shown in yellow. Conserved residues participating in the stacked core and the hydrogen-bonded glutamic acid of the TSR domains are shown as cyan sticks. Disulfide linkages are shown in a stick representation.
Computational modeling of TSR domains.
The MTRAP TSR domains are predicted to be considerably more compact than those of the HsTSP-1 (roughly half the length, ∼25 Å) (Fig. 5B and C) with a fold similar to that of the TRAP TSR domain (38). In contrast to HsTSP-1, the MTRAP TSR domains are predicted to have eight fewer amino acids between the domains, which would result in a more compact structure and suggests that the TSR domains may function together as a single unit (Fig. 5B and C). As modeled, the MTRAP TSR folds are predicted to deviate from the TRAP TSR fold in two ways: (i) in the cation-pi stacking core made up of repeating tryptophan and arginine residues, the MTRAP cores contain a single W-R-W stack, as opposed to a W-R-W-R-W stack, and (ii) the cysteine at the N terminus of the A-loop is predicted to form a disulfide with a cysteine at the following strand B (MTRAP TSR residues 28 to 61) or in the B-C loop (MTRAP TSR residues 62 to 94). The latter predicted disulfide linkage would be a variant of the group 2 classification by Tan et al. (38). The MTRAP TSR domains contain only four cysteines, as opposed to the six found in HsTSP-1 and TRAP TSR domains.
Assessment of human antibody responses.
Plasma samples were collected from healthy volunteers, 18 to 60 years old, in the village of Kenieroba, Mali, and were evaluated for their serum reactivity to rMTRAP, rPTRAMP, AMA1, and MSP142. Comparing the relative percentages of individual serum reactivity at a single dilution of 1:500, the levels of antibody recognition of MTRAP and PTRAMP were markedly less than those of AMA1 and MSP142 (Fig. 6). Even though the reactivity of the sera was low, significant correlations were observed between AMA1 and MSP1 titers and MTRAP or PTRAMP titers (Spearman rank test range, P = 0.0156 to P < 0.0001; r2 range, 0.250 to 0.410).
Fig 6.
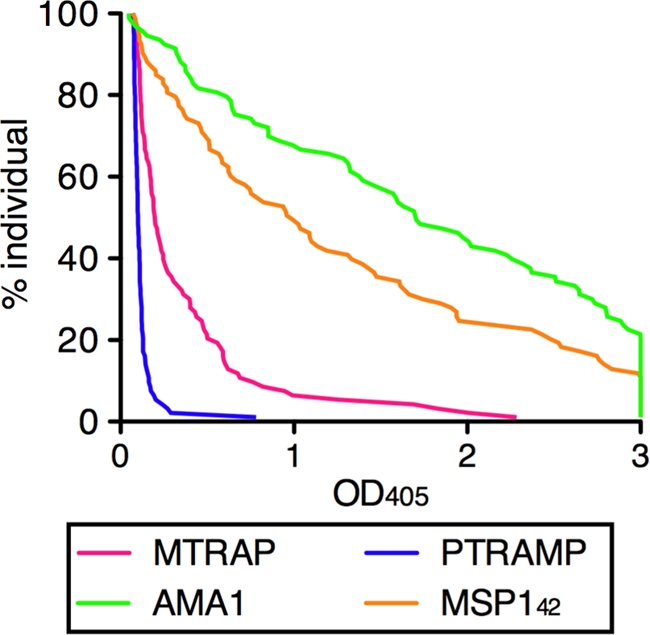
Reverse cumulative distribution plot of antimalarial antibody observed in Malian adults. The antibody levels in 93 Malian adults were determined by ELISA. All samples were tested at a 1:500 dilution, and any optical density at 405 (OD405) values more than 3 were assigned as 3 in this figure. Each OD value (x axis) is plotted against the percentage of subjects who have at least that level of antibody (y axis).
DISCUSSION
A general conservation of the mechanisms and components of a motor complex involved in gliding motility and cell invasion across the Apicomplexan parasites has been previously suggested (4, 11, 14, 17). Recently Baum et al. (2) demonstrated the presence of a conserved motor complex in Plasmodium falciparum blood-stage merozoites. Essential to demonstrating conservation of this motor complex in blood-stage merozoites was the identification of a TRAP-like functional protein, MTRAP, in blood-stage merozoites. However, Baum et al. (2) were unable to demonstrate direct binding of MTRAP to the host cells. To that end, we produced a high-quality recombinant protein (rMTRAP) containing the TSR domain in order to begin to investigate the role in invasion of red blood cells. Furthermore, we also generated an additional TSR-containing protein, PTRAMP, which has demonstrated expression in and on the surface of merozoites and which undergoes proteolytic processing upon invasion of erythrocytes (12, 39). Here we describe the production and biological, biochemical, and biophysical characterization of both MTRAP and PTRAMP.
In order to biologically characterize both rMTRAP and rPTRAMP, antisera were generated to recombinant proteins rMTRAP and rPTRAMP in two different species, rats and rabbits. Confocal microscopy using the antisera generated in both rats and rabbits demonstrated colocation of signal in parasitized red blood cells for the respective recombinant proteins (see Fig. S2 in the supplemental material). Furthermore, localization of MTRAP and PTRAMP to the micronemes was consistent with previously published data (2, 39), indicating the anti-rMTRAP and -rPTRAMP polyclonal antibodies generated in both rats and rabbits specifically recognized PfMTRAP and PfPTRAMP. Next, the anti-rMTRAP and -rPTRAMP polyclonal antibodies were examined for GIA. Only antibodies from two rats generated toward rPTRAMP demonstrated GIA. Differences in GIA between species have previously been observed (23). Additionally, GIA for the anti-rPTRAMP polyclonal antibodies was partially reversed by rPTRAMP, indicating specificity to PTRAMP. Taken altogether, these native proteins do not appear to be effective antimalaria vaccine targets.
Biochemical characterization of purified rMTRAP and rPTRAMP demonstrated that the N-terminal sequence and the mass spectrum directly corresponded to the expected results for each protein. Western blot analysis of rMTRAP was consistent with a good-quality purified recombinant protein. Dimers and multimers of rPTRAMP were observed by Western blotting of nonreduced rPTRAMP but were not observed upon reduction of rPTRAMP (Fig. 1), consistent with the findings of Green et al. (12). The observed dimerization of rPTRAMP is likely due to the presence of an odd number of cysteine residues in the recombinant protein.
The biophysical studies of rMTRAP and rPTRAMP by SEC-MALS and QELS indicated a predominant monomeric conformation for both proteins. The weight average molar mass for both rMTRAP and rPTRAMP is similar to the theoretical mass of our designed product within the error of the assay. Furthermore, QELS data indicated rMTRAP was an extended molecule based on the observed hydrodynamic radii (Fig. 3B), which was corroborated by sedimentation velocity (Fig. 3C). Consequently, two orthogonal approaches, QELS and sedimentation velocity analysis, support the concept that MTRAP acquires a nonglobular highly extended structure. To further investigate and begin to resolve the structural features of MTRAP, we employed an additional orthogonal technique, high-resolution AFM, which permitted visualization of individual rMTRAP molecules. The AFM images and the subsequent measurements via direct visualizations on mica substrate indicate that rMTRAP is an extended linear, rod-like flexible molecule with measurements centered around 33 nm by 2 nm. In contrast, QELS data indicated rPTRAMP was a less-extended molecule (∼16 nm by 2.6 nm) based on the observed hydrodynamic radii (see Fig. S3B in the supplemental material), which was corroborated by sedimentation velocity (see Fig. S3C). Consequently, two orthogonal approaches, QELS and sedimentation velocity analysis, support the concept that PTRAMP acquires a less-extended structure.
The TSR domains, when modeled end to end, could at most account for approximately 6 of the 33 nm of rMTRAP, leaving roughly 330 amino acids to account for the remaining length and width. Interpreting the far-UV spectrum within the constraints of those dimensions, the ∼330 amino acids are likely ordered with significant β-strand content. MTRAP, like CSP, contains a domain with internal repeats (KNDDD in MTRAP, residues ∼124 to 168; NANP in CSP); however, the length of the domain is shorter by about a factor of 3. These observations, taken together with the similarity in the far-UV spectra between MTRAP and CSP, suggest that the extracellular domain of MTRAP may be composed of a domain with regularly repeating units of mixed sheet-coil character along with a larger domain (residues ∼169 to 360) with less internal symmetry of similar secondary structure content.
The predicted disulfide linkage between loop A and strand B in the MTRAP TSR-like domains puts these domains in a new group (group 3), as they do not fit into either group defined by Tan et al. (38). The model presented here is consistent with substitutions seen within the N- and C-terminal domains and between homologs of these TSR-like domains (see Fig S5 in the supplemental material). For example, in a homologous domain from a hypothetical protein in the Plasmodium-related organism Theileria annulata, in the N-terminal domain (residues 22 to 55), both cysteines in this A-B linkage are replaced by small amino acids. In the C-terminal domain of this homolog, residues in the cation-pi stacking interaction and the proposed Arg-Glu hydrogen bonding interaction are all replaced by hydrophobic resides. Experimental evidence will be necessary to confirm the modeling predictions.
These data are the first direct evidence using orthogonal techniques with good agreement demonstrating the relative shapes of both MTRAP and PTRAMP. Both proteins have different, extended, and multidomain shapes: MTRAP has a long flexible rod-like shape versus PTRAMP, which has a more globular structure. Moreover, determination of the relative structure notwithstanding, we desired to examine binding of both proteins to erythrocytes. Using an erythrocyte binding assay, we were unable to demonstrate direct binding of PTRAMP to erythrocytes; however, there may be an indirect interaction between an erythrocyte receptor via interactions with other parasite proteins. Indirect interactions with host cell receptors via multiprotein complexes are common biological occurrences. Such an interaction for a TRAP homolog is not unprecedented given that the Toxoplasma TRAP homolog, Toxoplasma gondii MIC2 (TgMIC2), interacts with TgM2AP (28). Furthermore, disruption of this association, TgMIC2-M2AP, adversely affects invasion (15). Given the fact that only two of the rats immunized with rPTRAMP produced polyclonal antibodies capable of GIA, in addition to incomplete GIA reversal by rPTRAMP, it is plausible that these two polyclonal antibodies contain antibody which recognizes a low-immunogenicity site involved in protein complex formation.
Conversely, while we were unable to demonstrate direct binding of PTRAMP to erythrocytes, we were able to provide the first evidence demonstrating the direct binding of rMTRAP to erythrocytes (Fig. 2). The eluted rMTRAP from the erythrocytes corresponded to the full-length rMTRAP, not the truncated version rMTRAP−84, in which the TSR domain is missing, localizing the functional region to the TSR domain. As demonstrated by Western blotting, there is an obvious discrepancy between the quantity of rMTRAP incubated with the erythrocytes and the quantity of rMTRAP eluted by the high-salt elution buffer. This could be due to a low number of host receptors or possibly due to a low affinity between the host cells' receptor and rMTRAP. While we were able to demonstrate direct binding to erythrocytes, we were unable to show GIA with either rat or rabbit anti-rMTRAP polyclonal antibodies, consistent with the findings of Baum et al. (2). Given the lack of GIA and MTRAP's apparent lack of positive selection along with the absence of an excess of nonsynonymous polymorphisms within PfMTRAP (2), it is feasible that MTRAP is concealed from the host immune response by its sequestration within the tight junction. MTRAP and PTRAMP are not markedly immunogenic since the prevalence of naturally induced human antibodies is low compared to those of other merozoite antigens, such as AMA1 and MSP142.
MTRAP appears to be a bifunctional molecule that is involved in merozoite invasion. It is interesting to speculate that due to the highly extended structure of MTRAP, with an adhesive TSR domain located at the amino terminus, it is involved in early attachment and reorientation through the interaction with aldolase and the motor complex. There are two challenges to this model, which are that antibodies against rMTRAP do not block merozoite invasion, as reported here and in a previous work (2), and that the protein sequence of MTRAP appears conserved, indicating no immune selection. The amino acid sequence conservation suggests a biological function that occurs later than AMA1, as an example, since AMA1 is subject to immune pressure. However, other adhesion molecules, such as the EBLs (EBA-175, and BAEBL) are relatively conserved and function prior to AMA1. Finally, it is easier to understand the necessity of a long filamentous protein of 25 nm in length before a moving junction has formed between AMA1 and RON2 (36) since the molecular distance inside the junction is likely to be quite small (<10 nm). While others have demonstrated MTRAP is expressed on the surface of the merozoite following secretion from the micronemes (2), the ability of MTRAP to interact with aldolase in vitro (2), and the possibility of PfROM4 cleavage of MTRAP (1), we have provided evidence that MTRAP is a long flexible rod-like protein capable of binding erythrocytes and consequently tethering the motor complex to an erythrocyte receptor.
Supplementary Material
ACKNOWLEDGMENTS
We are thankful to Matthew Plassmeyer for expertise in recombinant protein production and confocal microscopy and Carl Hammer and Mark Garfield for mass spectrometry and protein sequencing, respectively. We appreciate the contributions of Mahamadou Diakite and Rick Fairhurst for the Malian samples, Ababacar Diouf and Hong Zhou for the GIA and ELISA data, and the villagers of Kenieroba, Mali, for their participation. We appreciate helpful discussions with Prakash Srinivasan regarding the function of these proteins during merozoite invasion.
This research was supported by the Intramural Research Program of the NIH, including NIAID and NIBIB, and the GIA Reference Center is supported by the PATH Malaria Vaccine Initiative.
Footnotes
Published ahead of print 30 March 2012
Supplemental material for this article may be found at http://ec.asm.org/.
REFERENCES
- 1. Baker RP, Wijetilaka R, Urban S. 2006. Two Plasmodium rhomboid proteases preferentially cleave different adhesins implicated in all invasive stages of malaria. PLoS Pathog. 2:e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Baum J, et al. 2006. A conserved molecular motor drives cell invasion and gliding motility across malaria life cycle stages and other apicomplexan parasites. J. Biol. Chem. 281:5197–5208 [DOI] [PubMed] [Google Scholar]
- 3. Bejon P, et al. 2008. Efficacy of RTS,S/AS01E vaccine against malaria in children 5 to 17 months of age. N. Engl. J. Med. 359:2521–2532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bergman LW, et al. 2003. Myosin A tail domain interacting protein (MTIP) localizes to the inner membrane complex of Plasmodium sporozoites. J. Cell Sci. 116:39–49 [DOI] [PubMed] [Google Scholar]
- 5. Bhattarai A, et al. 2007. Impact of artemisinin-based combination therapy and insecticide-treated nets on malaria burden in Zanzibar. PLoS Med. 4:e309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bourne PE, et al. 2004. The distribution and query systems of the RCSB Protein Data Bank. Nucleic Acids Res. 32:D223–D225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Buscaglia CA, Coppens I, Hol WG, Nussenzweig V. 2003. Sites of interaction between aldolase and thrombospondin-related anonymous protein in Plasmodium. Mol. Biol. Cell 14:4947–4957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cerami C, et al. 1992. The basolateral domain of the hepatocyte plasma membrane bears receptors for the circumsporozoite protein of Plasmodium falciparum sporozoites. Cell 70:1021–1033 [DOI] [PubMed] [Google Scholar]
- 9. Coppi A, et al. 2011. The malaria circumsporozoite protein has two functional domains, each with distinct roles as sporozoites journey from mosquito to mammalian host. J. Exp. Med. 208:341–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Frevert U, et al. 1993. Malaria circumsporozoite protein binds to heparan sulfate proteoglycans associated with the surface membrane of hepatocytes. J. Exp. Med. 177:1287–1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gaskins E, et al. 2004. Identification of the membrane receptor of a class XIV myosin in Toxoplasma gondii. J. Cell Biol. 165:383–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Green JL, Hinds L, Grainger M, Knuepfer E, Holder AA. 2006. Plasmodium thrombospondin related apical merozoite protein (PTRAMP) is shed from the surface of merozoites by PfSUB2 upon invasion of erythrocytes. Mol. Biochem. Parasitol. 150:114–117 [DOI] [PubMed] [Google Scholar]
- 13. Greenfield NJ. 2006. Using circular dichroism spectra to estimate protein secondary structure. Nat. Protoc. 1:2876–2890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Herm-Gotz A, et al. 2002. Toxoplasma gondii myosin A and its light chain: a fast, single-headed, plus-end-directed motor. EMBO J. 21:2149–2158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Huynh MH, et al. 2003. Rapid invasion of host cells by Toxoplasma requires secretion of the MIC2-M2AP adhesive protein complex. EMBO J. 22:2082–2090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jewett TJ, Sibley LD. 2003. Aldolase forms a bridge between cell surface adhesins and the actin cytoskeleton in apicomplexan parasites. Mol. Cell 11:885–894 [DOI] [PubMed] [Google Scholar]
- 17. Kappe S, et al. 1999. Conservation of a gliding motility and cell invasion machinery in Apicomplexan parasites. J. Cell Biol. 147:937–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Leaver-Fay A, et al. 2011. ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 487:545–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Malkin EM, et al. 2005. Phase 1 clinical trial of apical membrane antigen 1: an asexual blood-stage vaccine for Plasmodium falciparum malaria. Infect. Immun. 73:3677–3685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mayer DC, et al. 2006. The glycophorin C N-linked glycan is a critical component of the ligand for the Plasmodium falciparum erythrocyte receptor BAEBL. Proc. Natl. Acad. Sci. U. S. A. 103:2358–2362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Menard R. 2000. The journey of the malaria sporozoite through its hosts: two parasite proteins lead the way. Microbes Infect. 2:633–642 [DOI] [PubMed] [Google Scholar]
- 22. Miura K, et al. 2008. Development and characterization of a standardized ELISA including a reference serum on each plate to detect antibodies induced by experimental malaria vaccines. Vaccine 26:193–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Miura K, et al. 2007. In immunization with Plasmodium falciparum apical membrane antigen 1, the specificity of antibodies depends on the species immunized. Infect. Immun. 75:5827–5836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Morahan BJ, Wang L, Coppel RL. 2009. No TRAP, no invasion. Trends Parasitol. 25:77–84 [DOI] [PubMed] [Google Scholar]
- 25. Ozaki LS, Gwadz RW, Godson GN. 1984. Simple centrifugation method for rapid separation of sporozoites from mosquitoes. J. Parasitol. 70:831–833 [PubMed] [Google Scholar]
- 26. Plassmeyer ML, et al. 2009. Structure of the Plasmodium falciparum circumsporozoite protein, a leading malaria vaccine candidate. J. Biol. Chem. 284:26951–26963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Qian B, et al. 2007. High-resolution structure prediction and the crystallographic phase problem. Nature 450:259–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rabenau KE, et al. 2001. TgM2AP participates in Toxoplasma gondii invasion of host cells and is tightly associated with the adhesive protein TgMIC2. Mol. Microbiol. 41:537–547 [DOI] [PubMed] [Google Scholar]
- 29. Rogers WO, et al. 1992. Characterization of Plasmodium falciparum sporozoite surface protein 2. Proc. Natl. Acad. Sci. U. S. A. 89:9176–9180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. RTS,S Clinical Trials Partnership 2011. First results of phase 3 trial of RTS,S/AS01 malaria vaccine in African children. N. Engl. J. Med. 365:1863–1875 [DOI] [PubMed] [Google Scholar]
- 31. Schuck P. 2000. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys. J. 78:1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shimp RL, Jr, et al. 2006. Production and characterization of clinical grade Escherichia coli derived Plasmodium falciparum 42 kDa merozoite surface protein 1 (MSP1(42)) in the absence of an affinity tag. Prot. Expr. Purif. 50:58–67 [DOI] [PubMed] [Google Scholar]
- 33. Singh S, Plassmeyer M, Gaur D, Miller LH. 2007. Mononeme: a new secretory organelle in Plasmodium falciparum merozoites identified by localization of rhomboid-1 protease. Proc. Natl. Acad. Sci. U. S. A. 104:20043–20048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sinnis P, et al. 1994. Structural and functional properties of region II-plus of the malaria circumsporozoite protein. J. Exp. Med. 180:297–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Soding J. 2005. Protein homology detection by HMM-HMM comparison. Bioinformatics 21:951–960 [DOI] [PubMed] [Google Scholar]
- 36. Srinivasan P, et al. 2011. Binding of Plasmodium merozoite proteins RON2 and AMA1 triggers commitment to invasion. Proc. Natl. Acad. Sci. U. S. A. 108:13275–13280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sultan AA, et al. 1997. TRAP is necessary for gliding motility and infectivity of Plasmodium sporozoites. Cell 90:511–522 [DOI] [PubMed] [Google Scholar]
- 38. Tan K, et al. 2002. Crystal structure of the TSP-1 type 1 repeats: a novel layered fold and its biological implication. J. Cell Biol. 159:373–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Thompson J, et al. 2004. PTRAMP; a conserved Plasmodium thrombospondin-related apical merozoite protein. Mol. Biochem. Parasitol. 134:225–232 [DOI] [PubMed] [Google Scholar]
- 40. Tsai CW, et al. 2009. Characterization of a protective Escherichia coli-expressed Plasmodium falciparum merozoite surface protein 3 indicates a non-linear, multi-domain structure. Mol. Biochem. Parasitol. 164:45–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tsai CW, Duggan PF, Shimp RL, Jr, Miller LH, Narum DL. 2006. Overproduction of Pichia pastoris or Plasmodium falciparum protein disulfide isomerase affects expression, folding and O-linked glycosylation of a malaria vaccine candidate expressed in P. pastoris. J. Biotechnol. 121:458–470 [DOI] [PubMed] [Google Scholar]
- 42. Tucker RP. 2004. The thrombospondin type 1 repeat superfamily. Int. J. Biochem. Cell Biol. 36:969–974 [DOI] [PubMed] [Google Scholar]
- 43. World Health Organization 2010. World malaria report. World Health Organization Press, Geneva, Switzerland [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.