

Abstract
CMX001 is a novel, broad-spectrum lipid antiviral conjugate (LAC) that produces high intracellular levels of the active antiviral agent cidofovir diphosphate (CDV-PP). Study CMX001-102 was a randomized, double-blind, placebo-controlled, parallel group, dose-escalating study in healthy volunteers. The objectives of the study were to evaluate the safety and pharmacokinetic parameters of CMX001 after single and multiple doses. Single doses ranging from 0.25 to 2.0 mg/kg of body weight and multiple doses ranging from 0.1 to 1.0 mg/kg (3 total doses, administered every 6 days) were given orally. Safety was assessed using comprehensive clinical and laboratory evaluations, including enhanced monitoring for potential gastrointestinal (GI) effects using wireless capsule endoscopy (WCE). Serial plasma and pooled urine samples were collected to estimate pharmacokinetic parameters for both CMX001 and cidofovir (CDV). No adverse events occurred that prevented dose escalation. No clinically significant drug-related changes in blood chemistry, hematology, renal function, or intraocular pressure were observed. No CMX001-related gastrointestinal mucosal changes were observed by WCE. CMX001 was absorbed rapidly, with maximum plasma concentrations observed 2 to 3 h postdose. Maximum plasma drug concentration and systemic exposure of CMX001 increased approximately in proportion to dose following single and multiple doses; no significant accumulation of CMX001 or CDV was observed following multiple doses. We conclude that CMX001 is orally bioavailable and well tolerated in healthy volunteers at doses up to 2 mg/kg, approximately 140 mg in a typical adult. This is the first demonstration of the use of phospholipid conjugation technology to achieve plasma drug exposures that are expected to result in activity against multiple double-stranded DNA viruses.
INTRODUCTION
There are 5 families of double-stranded DNA (dsDNA) viruses that cause human disease. While a large percentage of the world's population exhibits asymptomatic seropositivity for numerous viruses in these families, active, clinically significant dsDNA viral infections only occur at a substantial rate in immunocompromised populations due to viral escape from host immunity. Viruses of special concern in this category include herpesviruses (e.g., herpes simplex 1 and 2, varicella-zoster virus [VZV], and cytomegalovirus [CMV]), adenovirus (AdV), polyomaviruses (JC virus and BK virus), papillomaviruses, and poxviruses (e.g., vaccinia virus, molluscum contagiosum virus, and the potential for reintroduction of variola/smallpox virus). The specific patient populations at greatest risk of developing serious or life-threatening dsDNA viral diseases are those who are immunosuppressed due to various reasons, including congenital immune deficiencies, HIV infection, intensive cancer chemotherapy, and use of immune-modulating therapies following transplantation. For example, AdV infection is of increasing concern in transplant patients, since more high-risk transplants are being performed with advances in standards of care (11). Previous studies have reported that AdV disease is associated with a mortality rate ranging from 26% to 80% in these patients (2, 9, 10). In addition, an increase in PML (progressive multifocal leukoencephalapathy) resulting from JC virus infection has been reported to occur in subgroups of patients treated with certain biologic drugs (natalizumab and rituximab) that interfere with lymphocytic function (18).
Existing therapeutics used to treat dsDNA virus infections have significant limitations, including dose-limiting toxicities, a low genetic barrier to development of drug-resistant virus, and a narrow spectrum of antiviral activity (7, 12). Hence, the growing number of immune-suppressed patients with life-threatening dsDNA infections has created an urgent medical need for new antiviral agents, preferably with broad antiviral activity, increased potency, and improved resistance and safety profiles.
CMX001 (Fig. 1) is a lipid antiviral conjugate (LAC) comprised of a lipid (1-0-hexadecyl-oxypropyl [HDP]) covalently linked to the acyclic nucleotide analogue cidofovir (CDV). CDV dihydrate, marketed as Vistide (cidofovir injection) for the treatment of CMV retinitis in HIV patients, has shown clinical activity against members of all 5 families of dsDNA viruses that cause human disease (6). However, the clinical utility of CDV dihydrate is limited, as it must be administered by intravenous infusion and is associated with significant nephrotoxicity, necessitating the requirement for hydration and prophylaxis with probenecid prior to infusion of the drug. To create CMX001, the HDP lipid moiety was coupled to CDV, such that the new chemical entity mimicked lysophosphatidylcholine (LPC), enabling the drug to use natural LPC uptake pathways in the small intestine (i.e., passive diffusion and flipases).
Fig 1.
Structure of CMX001 {phosphonic acid, [[(S)-2-(4-amino-2-oxo-1(2H)-pyrimidinyl)-1-(hydroxymethyl) ethoxy]methyl]mono[3-(hexadecyloxy)propyl] ester}.
CMX001 is not a typical prodrug. Unlike a typical prodrug, which is designed to deliver drug to plasma, CMX001 was designed to remain intact in plasma and deliver drug directly to the target cell. This design results in enhanced cellular uptake and high intracellular levels of the active antiviral agent, cidofovir diphosphate (CDV-PP), while producing low plasma drug concentrations of CDV. As a result, CMX001 has the potential for significantly increased antiviral activity relative to that of CDV dihydrate, based on the ability to deliver more of the active antiviral, CDV-PP, to target cells. Additionally, in vitro 50% effective concentrations for representative dsDNA viruses from all 5 families were substantially lower for CMX001 than CDV, in some cases by more than 1,000-fold (8). Finally, CMX001, unlike CDV, is not a substrate for human organic anion transporters, which concentrate CDV in kidney tubules and cause the dose-limiting nephrotoxicity of CDV dihydrate (19). In more than 20 animal toxicology studies conducted to date, there have been no changes in renal function associated with administration of CMX001 (14, 16).
CMX001 is currently in development for the treatment of smallpox under the Animal Efficacy Rule (21 CFR 314) and is being evaluated in phase II clinical trials for the prophylaxis and preemption of CMV infection (study CMX001-201). CMX001 is also being evaluated for treatment of adenovirus infection in stem cell transplant patients (study CMX001-202) and in an open label study that allows for treatment of patients with a wide range of serious and/or life-threatening dsDNA virus diseases (CMX001-350). Reported here are the results from the first study in humans with CMX001, a single- and multiple-dose, placebo-controlled, double-blind, dose escalation, safety, and pharmacokinetic (PK) study of healthy adult volunteers. The results demonstrate the utility of the lipid conjugate technology to create a drug with a favorable pharmacokinetic and safety profile.
MATERIALS AND METHODS
Study CMX001-102 was a randomized, double-blind, placebo-controlled, parallel group, single-dose and multidose, dose escalation study in healthy adult volunteers. The study was conducted in accordance with the principles of the Declaration of Helsinki (2000), International Conference on Harmonization (ICH) E6 Guideline for Good Clinical Practice (GCP), the applicable Code of Federal Regulations (CFR), and all applicable local requirements. Institutional Review Board (IRB) approval and written informed consent were obtained prior to conducting any evaluations or study procedures. All adverse events were coded using the MedDRA dictionary and graded using the Division of AIDS Table for Grading the Severity of Adult and Pediatric Adverse Events (published December 2004).
Study population.
Healthy male and female volunteers 18 to 55 years of age were allowed to participate in the study if they had vital signs within specified parameters and normal laboratory values for urine protein, blood urea nitrogen (BUN), serum creatinine, serum protein, alanine aminotransferase, aspartate aminotransferase, and neutrophil counts, as well as a hematocrit of at least 38% and calculated creatinine clearance of at least 80 ml/min. All subjects were HIV, hepatitis B virus (HBV), and HCV negative. Female subjects were excluded from the study if they were nursing, pregnant, or of childbearing potential. Subjects were excluded from participation if they had clinically relevant medical conditions, including renal or hepatic impairment, gastrointestinal disease, contraindications for wireless capsule endoscopy, cancer, diabetes, autoimmune disease, immunodeficiency, or hypersensitivity to CDV. Potential subjects could not have used products with nephrotoxic potential, over-the-counter medications, or herbal or neutraceutical preparations within 7 days, prescription drugs, illicit drugs, or alcohol within 14 days, aspirin-containing products or nonsteroidal anti-inflammatory drugs within 21 days, or antiviral, corticosteroid, immunosuppressive, anticoagulant, or investigational prescription drugs within 30 days prior to enrollment. Additionally, patients could not have donated blood or plasma within 56 or 30 days, respectively. The first subject was enrolled in the study on 20 September 2006, and the last subject completed dosing on 4 November 2008.
Study design.
Subjects underwent screening procedures 5 to 16 days prior to enrollment. Subjects meeting all inclusion and exclusion criteria were randomized (2:1, CMX001:placebo) and enrolled in cohorts of 6 subjects each. Nine single-dose cohorts and 5 multidose cohorts were enrolled. Study drug was supplied as CMX001 ammonium salt for reconstitution along with matching placebo (crosspovidone). Subjects received an aqueous solution of CMX001 or placebo, administered orally at single doses of 0.025, 0.05, 0.1, 0.2, 0.4, 0.6, 1.0, 1.5, or 2.0 mg/kg or multiple doses of 0.1, 0.2, 0.4, 0.6, or 1.0 mg/kg (3 doses administered on days 0, 6, and 12). The highest dose administered in this study was 191 mg. Prior to dose escalation, safety data from each cohort were evaluated by a Drug Safety Monitoring Board (DSMB) and the FDA.
For each cohort, subjects were admitted to the clinical research facility on day −2. On the morning of day −1, following an overnight fast, subjects underwent wireless capsule endoscopy (WCE; Given Imaging, Duluth, GA) according to the manufacturer's instructions. On day −1, subjects were fed a light lunch (during endoscopy recording) and dinner before fasting overnight. Subjects received blinded study drug on the morning of day 0, and sample collection for safety and pharmacokinetic analyses began. On day 2, subjects in the single-dose cohorts were placed on a full liquid diet after breakfast in preparation for a postdose WCE beginning on the morning of day 3. After verifying capsule excretion, subjects were released from the clinic on day 4. Subjects in the multidose cohorts were released from the clinic on day 2, returning on days 5 and 11 prior to receiving study drug on the mornings of days 6 and 12, respectively. After appropriate fasting, subjects underwent a postdose WCE on the morning of day 14. Subjects were released from the clinic on day 16, following verification of capsule excretion and completion of all required study procedures. All subjects returned to the clinical research unit for follow-up safety visits approximately 2 weeks after their last dose of study drug.
Safety assessments.
Safety was monitored by assessing vital signs and other physical findings, laboratory values for hematology, biochemistry, and urinalysis, fecal occult blood tests, electrocardiograms, WCE studies, and adverse events (AEs). Because CDV is a metabolite of CMX001, volunteers were monitored for events associated with the use of CDV (including nephrotoxicity, neutropenia, and ocular hypotony), even though these were not observed in preclinical studies of CMX001. Changes in intraocular pressure were monitored using standard procedures at the site. Creatinine clearance was calculated using the Cockcroft-Gault equation. As gastrointestinal toxicity was dose-limiting in preclinical studies (13), wireless capsule endoscopy was performed to detect subclinical changes in the small bowel mucosa. Safety analyses included all subjects who received at least one dose of study drug. Safety data were summarized descriptively and are reported by treatment group.
For the single-dose cohorts, blood samples to determine plasma drug concentrations of CMX001 and CDV were obtained at the following time points: for 0.025-mg/kg dose, at predose and 2, 3, 4, 6, 9, 12, 24, 48, and 72 h postdose; for 0.05-, 0.1-, and 0.2-mg/kg doses, predose and 0.5, 1, 1.5, 2, 3, 4, 6, 9, 12, 24, 48, and 72 h postdose; for 0.4-mg/kg dose, predose and 0.5, 1, 1.5, 2, 3, 4, 6, 9, 12, 24, 48, 72, and 96 h postdose; for 0.6-, 1.0-, 1.5-, and 2.0-mg/kg doses, predose and 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 15, 24, 30, 36, 48, 72, and 96 h postdose plus at the day 6/7 visit. Pooled urine samples were collected over the following intervals: 0 to 4, 4 to 8, 8 to 12, 12 to 24, 24 to 36, 36 to 48, 48 to 72, and 72 to 96 h postdose.
For the multidose cohorts, blood samples to determine plasma drug concentrations of CMX001 and CDV were obtained at the following time points for all dose cohorts: day 0, predose and at 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 15, 24, 30, 36, 48, and 72 h after the day 0 dose; day 6, predose; day 12, predose and at 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 15, 24, 30, 36, 48, and 72 h after the day 12 dose; day 16, at the day 19/20 visit and at the day 26/27 visit. Pooled urine samples were collected on days 0 and 12 over the following intervals: 0 to 4, 4 to 8, 8 to 12, and 12 to 24 h postdose, on days 1 and 13 at 24 to 36 h postdose, on day 13 at 36 to 48 h postdose, on day 14 at 48 to 72 h postdose, and on day 15 at 72 to 96 h postdose.
Pharmacokinetic analysis.
Concentrations of CMX001 and CDV were determined in plasma and urine using a validated liquid chromatograph-tandem mass spectrometry assay (Enthalpy Analytical, Durham, NC). Plasma samples were prepared for analysis by protein precipitation using cold acetonitrile followed by solid-phase extraction using Phenomenex Strata X-C (reversed-phase and strong cation exchange) cartridges. Chromatographic separation was performed using an SIELC Primesep B4 column fit with an SIELC Primesep B4 guard column operated under mixed-mode anionic/reversed-phase conditions. The lower limits of quantitation (LLOQ) were 0.1 ng/ml and 0.5 ng/ml for CMX001 and CDV in plasma, respectively. The quantitation limits in urine were 1.0 ng/ml and 5.0 ng/ml for CMX001 and CDV, respectively. A total of 81 analytical runs were performed. Plasma interassay coefficients of variation ranged from 6.5 to 2.2%, while bias ranged from −5.8 to 0.4%. Urine interassay coefficients of variation ranged from 2.8 to 7.0%, while bias ranged from −1.0 to 4.8%. Noncompartmental pharmacokinetic analyses were performed on CMX001 and CDV plasma and urine drug concentration data using WinNonlin (version 5.0.1 or later; Pharsight Corp., Mountain View, CA) and SAS (version 9.1.3; SAS Institute, Cary, NC). All concentrations below the LLOQ were treated as zero for the purpose of PK analysis and presentation of descriptive statistics. Analyses were conducted on all subjects who received study drug and had sufficient data (at least 50% of the required data points presented).
Plasma PK parameters determined for both CMX001 and CDV included maximum observed drug concentration (Cmax), time to Cmax (Tmax), half-life of elimination (t½,z), area under the curve (AUC) parameters, mean residence time (MRT∞), apparent total clearance of the drug from plasma after oral administration (CL/F) (or total body clearance of CDV, referred to as CL/fm), and apparent volume of distribution during terminal phase (Vz/F) (or volume of distribution of CDV on the terminal phase, referred to as Vz/fm). Except as noted, the data are presented as the means ± the standard deviation. To assess dose proportionality, PK parameters on days 0 (single and multidose) and 12 (multidose) were compared across doses using one-way analysis of variance (ANOVA) and a power model. For the multidose phase of the study, differences between PK parameters at day 0 versus day 12 were statistically evaluated by ANOVA. Statistical significance was defined as a P value of <0.05.
Data from 10 plasma drug concentration profiles were excluded from the statistical analyses. All subjects who received a single dose of 0.4 mg/kg were excluded from descriptive statistics and dose proportionality analysis for CMX001 t1/2,z, AUC(0–∞), MRT∞, CL/F, and Vz/F, because the CMX001 elimination phase could not be identified with reasonable accuracy in this cohort. All four subjects who received a single dose of 0.025 mg/kg, the lowest dose in this study, and two subjects who received a single dose of 0.050 mg/kg did not have a sufficient CDV plasma concentration data for PK analysis according to prospectively defined criteria rejecting any profile in which fewer than 50% of the samples had quantifiable concentrations of CDV. Although individual Cmax, Tmax, and AUC(0–last) values were reported, the data were excluded from statistical analyses. The CDV t1/2,z could only be estimated in one of the four subjects in each of the cohorts receiving single doses of 0.05 mg/kg and 0.1 mg/kg; thus, data from these cohorts were excluded from descriptive statistics and dose proportionality analysis for CDV t1/2,z, AUC(0–∞), MRT∞, CL/fm, and Vz/fm.
RESULTS
Subject disposition and demographics.
Demographic findings across all cohorts were reflective of a healthy population and were comparable between subjects who received placebo and those who received CMX001 (Table 1). For the first 2 cohorts, for whom testing was conducted at a study site in Miramar, FL, high numbers of Hispanic or Latino subjects (i.e., 75% for the 0.025-mg/kg cohort and 100% for the 0.05-mg/kg cohort) were enrolled. Later cohorts were enrolled at a study site in Morrisville, NC. Females were not excluded from the study; however, all of the subjects who were enrolled into the study were male.
Table 1.
Demographic data for the single-dose and multidose cohorts
| Cohort type and demographic parameter | Placebo (n = 18) | CMX001 dose (mg/kg) (n = 4/cohort) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0.025 | 0.050 | 0.1 | 0.2 | 0.4 | 0.6 | 1.0 | 1.5 | 2.0 | Total (n = 36) | ||
| Single-dose cohorts | |||||||||||
| Mean age (yrs) | 28.8 | 36.5 | 43.0 | 32.8 | 27.8 | 27.8 | 29.0 | 28.8 | 26.8 | 23.8 | 30.7 |
| Gender [n (%)] | |||||||||||
| Male | 18 (100) | 4 (100) | 4 (100) | 4 (100) | 4 (100) | 4 (100) | 4 (100) | 4 (100) | 4 (100) | 4 (100) | 36 (100) |
| Female | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Race [n (%)] | |||||||||||
| Black | 10 (56) | 2 (50) | 0 | 3 (75) | 2 (50) | 2 (50) | 2 (50) | 3 (75) | 2 (50) | 2 (50) | 18 (50) |
| Caucasian | 8 (44) | 2 (50) | 4 (100) | 1 (25) | 2 (50) | 2 (50) | 2 (50) | 1 (25) | 2 (50) | 2 (50) | 18 (50) |
| Ethnicity [n (%)] | |||||||||||
| Hispanic or Latino | 4 (22) | 3 (75) | 4 (100) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 7 (19) |
| Not Hispanic or Latino | 14 (78) | 1 (25) | 0 | 4 (100) | 4 (100) | 4 (100) | 4 (100) | 4 (100) | 3 (75) | 4 (100) | 28 (78) |
| Other | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (25) | 0 | 1 (3) |
| Mean body mass index (kg/m2) | 25.81 | 27.50 | 27.25 | 26.65 | 25.55 | 25.18 | 25.50 | 26.10 | 24.45 | 24.80 | 25.89 |
| Multidose cohorts | |||||||||||
| Mean age (yrs) | 27.8 | 31.8 | 30.8 | 33.0 | 31.3 | 30.5 | 31.5 | ||||
| Gender [n (%)] | |||||||||||
| Male | 10 (100) | 4 (100) | 4 (100) | 4 (100) | 4 (100) | 4 (100) | 20 (100) | ||||
| Female | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||||
| Race [n (%)] | |||||||||||
| Black | 4 (40) | 2 (50) | 4 (100) | 3 (75) | 2 (50) | 2 (50) | 13 (65) | ||||
| Caucasian | 6 (60) | 2 (50) | 0 | 1 (25) | 2 (50) | 2 (50) | 7 (35) | ||||
| Ethnicity [n (%)] | |||||||||||
| Hispanic or Latino | |||||||||||
| Not Hispanic or Latino | 10 (100) | 4 (100) | 4 (100) | 4 (100) | 4 (100) | 4 (100) | 20 (100) | ||||
| Other | |||||||||||
| Mean body mass index (kg/m2) | 25.09 | 26.13 | 25.38 | 26.23 | 25.20 | 24.18 | 25.42 | ||||
A total of 54 subjects enrolled and completed the single-dose phase of the study. Eighteen subjects were randomized to receive placebo, and 36 were randomized to receive active drug (4 subjects at each dose level of CMX001). As shown in Table 1, the mean age was 30.7 years for subjects who received CMX001 and 28.8 years for placebo subjects. Approximately 50% (28 of 54 subjects) were black, and approximately 50% were Caucasian.
Thirty eligible subjects were enrolled sequentially into the multidose phase of the study. Ten subjects were assigned to receive placebo, and 20 subjects were assigned to receive CMX001 on days 0, 6, and 12. Twenty-eight of the 30 subjects (93.3%) completed the multidose phase of the study, while 2 subjects were withdrawn, 1 due to an adverse event and 1 due to a positive urine test for opiates. The mean ages were 31.5 years for subjects who received CMX001 and 27.8 years for subjects who received placebo (Table 1). Fifty-seven percent (17 of 30) of subjects were black, and 43% (13 of 30) were Caucasian.
Safety assessments.
Of all subjects in the single-dose cohorts, 25% who received CMX001 and 39% who received placebo experienced at least one treatment-emergent AE; no dose-related trends were observed (Table 2). The most frequently reported adverse events were coded to the GI MedDRA system organ class (SOC), i.e., 3 out of 36 subjects (8%) who received CMX001 and 2 out of 18 subjects (11%) who received placebo. Preferred terms for reported AEs included abdominal pain, upper abdominal pain, epigastric discomfort, and aphthous stomatitis. All AEs were graded by the principal investigator as mild, except for 2 events graded as moderate (1 increase in creatine kinase and 1 headache). No adverse events were detected based upon physical examinations.
Table 2.
Overall incidence of treatment-emergent adverse events by dose cohort following each dose of study drug
| CMX001 dose frequency, observation interval, and AE category | No. (%) of subjects who experienced treatment-emergent AE(s) in dose groupa |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Placebob | 0.025 | 0.050 | 0.1 | 0.2 | 0.4 | 0.6 | 1.0 | 1.5 | 2.0 | Totalb | |
| Single dose | |||||||||||
| ≥1 AE | 7/18 (39) | 2 (50) | 2 (50) | 0 | 2 (50) | 0 | 0 | 1 (25) | 0 | 2 (50) | 9/36 (25) |
| AE leading to withdrawal | 0/18 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Multidose | |||||||||||
| Between first and second dose | |||||||||||
| ≥1 AE | 1/10 (10) | 1 (25) | 0 | 3 (75) | 1 (25) | 0 | 5/20 (25) | ||||
| AE leading to withdrawal | 0/10 | 0 | 0 | 0 | 0 | 0 | 0/20 | ||||
| Between second and third dose | |||||||||||
| ≥1 AE | 0/9 | 0 | 2 (50) | 1 (25) | 0 | 0 | 3/20 (15) | ||||
| AE leading to withdrawal | 0/9 | 0 | 1 (25) | 0 | 0 | 0 | 1/20 (5) | ||||
| After third dose until study end | |||||||||||
| ≥1 AE | 2/9 (22) | 1 (25) | 1/3b (33) | 1 (25) | 1 (25) | 1 (25) | 5/19 (26) | ||||
| AE leading to premature discontinuation | 0/9 | 0 | 0 | 0 | 0 | 0 | 0/19 | ||||
| Cumulative AEs throughout multidose study | |||||||||||
| ≥1 AE | 2/10 (20) | 2 (50) | 2 (50) | 3 (75) | 2 (50) | 1 (25) | 10/20 (50) | ||||
| AE leading to premature discontinuation | 0/10 | 0 | 1 (25) | 0 | 0 | 0 | 1/20 (5) | ||||
n = 4 subjects/study group.
Placebo and total groups for each dosage and AE category had varied numbers for each study group, and so the total number of subjects in each group is reported as the denominator of the incidence data.
In the multidose cohorts, CMX001 was well tolerated at all doses assessed in the study (Table 2). Among the 10 subjects who received placebo, 2 (20%) reported at least 1 treatment-emergent AE. Among the 20 subjects who received CMX001, 10 (50%) reported at least 1 treatment-emergent AE. As shown in Table 2, the incidence of events did not increase with additional doses of CMX001. Only one AE was reported that led to discontinuation from the study. One subject randomized to receive 0.2 mg/kg of CMX001 was discontinued after the second dose due to an AE of elevated bilirubin that was considered by the investigator to be possibly related to study drug. The subject, who had a history of hyperbilirubinemia, was released from the research unit after follow-up, and the event resolved without sequelae. The most common events based on system organ class were as follows: gastrointestinal disorders (3 subjects); investigations (2 subjects); nervous system disorders (2 subjects); respiratory, thoracic, and mediastinal disorders (2 subjects); skin and subcutaneous tissue disorders (2 subjects). No individual AE was reported by more than one subject within the same treatment group except for rash, which was reported by 2 subjects who received placebo. One of 10 subjects (10%) who received placebo reported 2 AEs that were assessed by the investigator as possibly related to study drug. Six of 20 subjects (30%) who received CMX001 experienced 17 treatment-emergent AEs that were assessed by the investigator as possibly related to study drug. No dose-effect relationship was established; only 1 subject who received the highest dose of CMX001 (1.0 mg/kg) reported a treatment-emergent AE (constipation). All AEs were judged to be mild in severity, with the exception of 1 headache that was judged to be moderate. There were no severe, life-threatening, or serious adverse events reported during the study.
Gastrointestinal mucosae were evaluated using WCE. Wireless capsule endoscopy data were assessed by two expert gastroenterologists experienced in reviewing endoscopy videos, the first initially scoring the videos and the second reviewing and approving the final PillCam reports. Both reviewers were blinded to treatment as well as timing (i.e., predose versus postdose). All findings judged by the reviewers as clinically significant are displayed in Table 3. Seven placebo-treated subjects, 5 subjects receiving a single dose of CMX001, and 2 subjects in the multidose cohorts (0.1-mg/kg cohort) had clinically significant findings. The one subject who had a clinically significant finding postdose was a placebo subject with a gastric mucosal lesion. In all other cases, the findings were seen at baseline as well as postdose and did not worsen during the course of the study. No subjects had positive fecal occult blood considered possibly or probably related to the treatment.
Table 3.
Clinically significant wireless capsule endoscopy resultsa
| Treatment | Study day | Clinically significant? | Finding(s) |
|---|---|---|---|
| Placebo (pooled across all cohorts; n = 28) | −1 | Yes | Erosions (jejunum) |
| 3 | No | Erosions (jejunum) | |
| −1 | Yes | Retained capsule/delayed gastric emptying (stomach) | |
| 3 | Yes | Delayed gastric emptying (stomach) | |
| −1 | Yes | Gastric ulcer (stomach) | |
| 3 | Yes | Retained capsule (stomach) | |
| 3 | Yes | Gastric ulcer (stomach) | |
| −1 | Yes | Multiple gastric ulcers (stomach) | |
| 3 | Yes | Gastric ulcer (stomach) | |
| −1 | Yes | Delayed gastric emptying (stomach) | |
| 3 | No | Capsule stasis (small bowel) | |
| −1 | No | Focal nodular hyperplasia (small bowel) | |
| 3 | Yes (reported as an AE) | Erosion vs ulcer (stomach) | |
| −1 | Yes | Gastric erosion (stomach) | |
| 3 | Yes | Erythema (dudenum) | |
| 3 | Yes | Erosion (duodenum) | |
| 0.2 mg/kg, single dose | −1 | Yes | Esophagitis (esophagus) |
| 3 | No | Colonic arteriovenous malformation (colon) | |
| 3 | No | Nodular hyperplasia (small bowel) | |
| −1 | Yes | Erosion (small bowel) | |
| 3 | No | Focal nodular hyperplasia (small bowel) | |
| 3 | No | Erythematous spot (small bowel) | |
| 3 | No | Lymphangiectasia (small bowel) | |
| 0.4 mg/kg, single dose | −1 | Yes | Multiple ulcerous erosions (small bowel) |
| 3 | Yes | Erosion vs ulcer (small bowel) | |
| 0.6 mg/kg, single dose | −1 | Yes | Linear gastric erosion (stomach) |
| 3 | No | Hyperemic area (esophagus) | |
| 1.0 mg/kg, single dose | −1 | Yes | Bulb erosion (duodenal bulb) |
| 3 | No | Lymphoid nodular hyperplasia (ileum) | |
| 0.1 mg/kg, multidose | −1 | Yes | Prolonged gastric transit (stomach) |
| 14 | Yes | Prolonged gastric transit (stomach) | |
| −1 | Yes | Prolonged gastric transit (stomach) | |
| −1 | Yes | Subendothelial hemorrhage (stomach) | |
| 14 | Yes | Subendothelial hemorrhage (stomach) | |
All findings judged clinically significant by the reviewing gastroenterologist are listed. Predose and postdose findings for any one subject are grouped together.
Ocular hypotony has been associated with the use of CDV dihydrate in AIDS patients with CMV retinitis, and so intraocular pressure was monitored in all subjects throughout the study. No clinically significant changes from baseline in intraocular pressure were observed in any study subjects.
Nephrotoxicity and neutropenia have been associated with the clinical use of CDV, and so signs and/or symptoms of these events were closely monitored throughout the study (5). No clinically significant changes in BUN, serum creatinine, BUN/creatinine ratio, creatinine clearance, or electrolytes were observed. No clinically significant changes in proteinuria were seen in any treatment group, and no clinically significant, drug-related changes in neutrophil counts (absolute or percentage) or white blood cell counts were noted. One subject (multidose 0.2-mg/kg cohort) exhibited neutropenia, but this was attributed to a concurrent febrile illness.
Pharmacokinetics.
After a single dose under fasting conditions, CMX001 was readily absorbed, with the time to maximum plasma drug concentration (Tmax) ranging from 2 to 3 h (Table 4; Fig. 2). Maximum plasma drug concentration (Cmax) and systemic exposure (AUC0–∞) increased approximately in proportion to dose over the range of 0.025 to 2.0 mg/kg (Table 4; Fig. 3 and 4). The t1/2,z increased with increasing dose, ranging from 6.15 h at 0.025 mg/kg to 32.7 h at 1.5 mg/kg (Table 4), presumably due to better definition of the elimination phase at higher doses.
Table 4.
Human pharmacokinetics following single doses of CMX001 or CDVa
| Drug and dose level (mg/kg) | Cmax (ng/ml) | Tmax (h) | t1/2,z (h) | AUC0–∞ (h · ng/ml) | MRT∞ (h) | CL/F (ml/h/kg) | Vz/F (liters/kg) |
|---|---|---|---|---|---|---|---|
| CMX001 | |||||||
| 0.025 | 2.58 ± 1.40 | 2.00 (2, 6) | 6.15 ± 0.930 | 21.3 ± 5.71 | 8.58 ± 0.720 | 1,240 ± 382 | 10.8 ± 2.49 |
| 0.05 | 5.28 ± 2.34 | 2.00 (1.5, 3) | 7.83 ± 4.09 | 36.5 ± 10.7 | 8.29 ± 3.10 | 1,480 ± 522 | 15.6 ± 5.75 |
| 0.1 | 10.6 ± 4.63 | 3.00 (3, 3) | 15.0 ± 5.02 | 139 ± 52.6 | 15.8 ± 1.75 | 834 ± 425 | 17.6 ± 7.94 |
| 0.2 | 24.5 ± 4.12 | 2.03 (1.5, 3) | 17.6 ± 4.55 | 235 ± 48.1 | 16.0 ± 1.02 | 877 ± 160 | 22.7 ± 8.38 |
| 0.4 | 68.1 ± 28.0 | 2.00 (1.5, 3) | NC | NC | NC | NC | NC |
| 0.6 | 115 ± 38.3 | 2.96 (1.5, 3) | 25.8 ± 6.31 | 729 ± 253 | 12.1 ± 2.40 | 893 ± 274 | 33.9 ± 15.6 |
| 1.0 | 273 ± 131 | 3.00 (2, 3) | 27.1 ± 2.80 | 1,350 ± 646 | 10.3 ± 1.82 | 909 ± 494 | 34.3 ± 15.2 |
| 1.5 | 371 ± 180 | 3.00 (2, 3) | 32.7 ± 3.46 | 2,340 ± 1,320 | 11.9 ± 0.906 | 792 ± 394 | 36.0 ± 14.8 |
| 2.0 | 350 ± 119 | 3.00 (3, 12) | 24.0 ± 0.705 | 2,650 ± 445 | 12.5 ± 3.00 | 772 ± 129 | 26.7 ± 4.21 |
| CDV | |||||||
| 0.025 | NC | NC | NC | NC | NC | NC | NC |
| 0.05 | 1.76 ± 0.481 | 9.00 (9, 9) | NC | NC | NC | NC | NC |
| 0.1 | 3.44 ± 0.603 | 12.0 (12, 12) | NC | NC | NC | NC | NC |
| 0.2 | 5.41 ± 0.998 | 9.15 (9, 12) | 24.2 ± 1.83 | 195 ± 31.4 | 38.0 ± 4.19 | 520 ± 73.9 | 18.2 ± 3.07 |
| 0.4 | 10.4 ± 1.55 | 9.00 (9, 12) | 45.5 ± 23.2 | 491 ± 83.1 | 63.1 ± 20.2 | 415 ± 73.8 | 26.1 ± 9.77 |
| 0.6 | 12.2 ± 2.39 | 15.0 (12, 15) | 55.6 ± 4.04 | 565 ± 60.3 | 64.5 ± 5.76 | 532 ± 51.5 | 42.6 ± 3.36 |
| 1.0 | 18.3 ± 5.87 | 7.00 (6, 15) | 64.5 ± 12.4 | 1,030 ± 317 | 76.5 ± 7.22 | 522 ± 170 | 48.5 ± 17.9 |
| 1.5 | 25.8 ± 2.90 | 10.0 (8, 12) | 56.8 ± 4.18 | 1,510 ± 290 | 70.0 ± 3.68 | 512 ± 121 | 42.2 ± 11.6 |
| 2.0 | 31.1 ± 7.00 | 11.5 (6, 15) | 63.0 ± 11.0 | 1,740 ± 409 | 76.4 ± 13.7 | 596 ± 143 | 52.7 ± 6.04 |
n = 4 subjects/dose group. NC, not calculated. All PK parameters are means ± standard deviations, except Tmax, which is presented as the median (with minimum, maximum in parentheses).
Fig 2.

Semilog plot of the mean plasma CMX001 concentration versus time profiles for each single-dose cohort. Four subjects in each cohort received CMX001.
Fig 3.
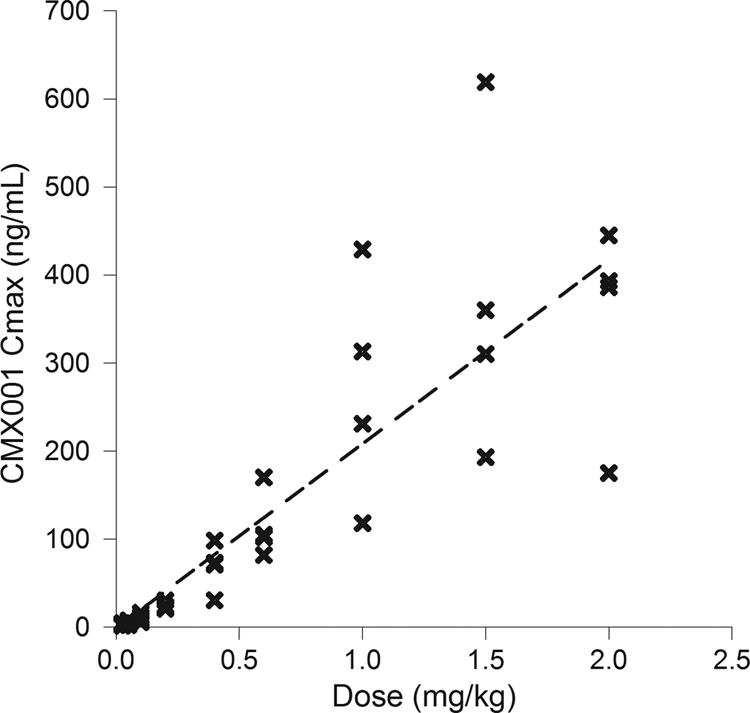
CMX001 Cmax versus dose (single-dose cohorts). Each x represents the data for one subject at that dose. Cohorts of 4 patients each received doses of 0.025, 0.05, 0.1, 0.2, 0.4, 0.6, 1.0, 1.5, or 2.0 mg/kg. The linear regression had an R2 value of 0.737.
Fig 4.
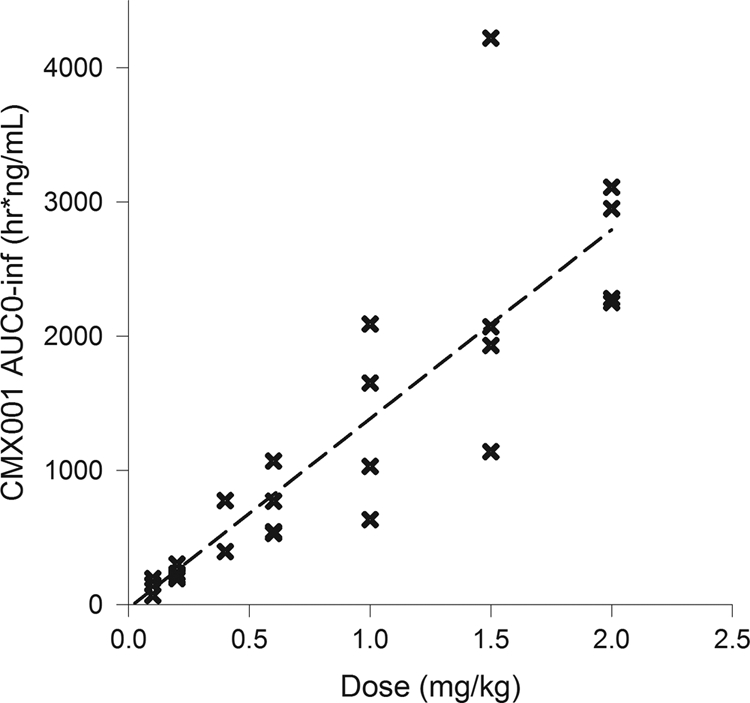
CMX001 AUC0–∞ versus dose (single-dose cohorts). Each x represents the data for one subject at that dose. Cohorts of 4 patients each received doses of 0.025, 0.05, 0.1, 0.2, 0.4, 0.6, 1.0, 1.5, or 2.0 mg/kg. The linear regression had an R2 value of 0.798.
When assessed by ANOVA, dose-proportional increases in the AUC were observed over the entire dose range from 0.025 to 2 mg/kg. Analysis by ANOVA also confirmed dose-proportional increases in Cmax values over the dose ranges of 0.025 to 0.4 mg/kg and 0.4 to 2.0 mg/kg, with the apparent break at 0.4 mg/kg due to exclusion of this cohort from the analysis (see Materials and Methods). Results of the power model generally confirmed the results of ANOVA for CMX001; dose proportionality for AUC parameters was demonstrated over the dose range of 0.1 to 2.0 mg/kg. The lack of dose proportionality at the 0.025- and 0.05-mg/kg doses was attributed to low drug concentrations (just above the limit of quantitation), which resulted in poor definition of the elimination phases at these doses.
After a single dose of CMX001, the rate of appearance of CDV in plasma was dependent on the absorption rate of CMX001 and the rate of conversion of CMX001 to CDV. Hence, Tmax of CDV ranged from 7 to 15 h (Table 4). Cmax of CDV increased somewhat less than in proportion to dose; however, the AUC increased approximately in proportion to dose. The t1/2,z increased from 24.2 h at 0.2 mg/kg to 63.0 h at 2 mg/kg (Table 4), again presumably due to better definition of the elimination phase at higher doses. CDV exposure (AUC0–last) was dose proportional over the dose range of 0.1 to 2.0 mg/kg as assessed by ANOVA and over the dose range of 0.5 to 2.0 mg/kg as assessed by the power model.
CMX001 was not detected in urine samples, indicating that urinary excretion is not a significant elimination pathway of CMX001. CDV was detected in urine as an elimination product of CMX001.
Quantifiable concentrations of CMX001 were measured in the plasma of all multidose subjects who were randomized to active study drug. A dose-proportional increase in plasma PK parameters (Cmax and AUC) was generally observed following administration of CMX001 over the dose range of 0.1 to 1.0 mg/kg on both days 0 and 12 (Table 5; Fig. 5). Statistical analyses indicated dose-proportional increases over the range of doses of the AUC0–last, as assessed by both ANOVA and the power model on day 0, but not by ANOVA on day 12. Statistical analyses of Cmax indicated dose-proportional increases on both day 0 and day 12, as assessed by both ANOVA and the power model.
Table 5.
Human pharmacokinetics, by day, following multiple doses of CMX001 or CDVa
| Drug and dose level (mg/kg) | Day postdose | Cmax (ng/ml) | Tmax (h) | AUC0–last (h · ng/ml) | AUC0–∞ (h · ng/ml) | t1/2,z (h) | MRT∞ (h) | CL/F or CLss/F (ml/h/kg) | Vz/F (liters/kg) |
|---|---|---|---|---|---|---|---|---|---|
| CMX001 | |||||||||
| 0.1 | 0 | 13.1 ± 4.70 | 1.50 (1.5, 1.5) | 94.1 ± 31.7 | 96.2 ± 32.5 | 11.1 ± 5.18 | 9.01 ± 1.73 | 1,130 ± 344 | 17.5 ± 8.73 |
| 12 | 13.3 ± 6.63 | 1.76 (1.5, 3) | 96.7 ± 38.4 | 98.7 ± 38.8 | 11.1 ± 6.55 | 10.7 ± 3.73 | 1,120 ± 387 | 17.2 ± 9.74 | |
| 0.2 | 0 | 24.2 ± 4.91 | 1.50 (1, 3) | 173 ± 30.5 | 182 ± 37.4 | 33.1 ± 9.86 | 14.5 ± 3.99 | 1,130 ± 193 | 51.9 ± 4.44 |
| 12 | 29.1 ± 16.4 | 1.50 (1.5, 1.5) | 200 ± 57.2 | 205 ± 57.8 | 30.8 ± 9.32 | 12.7 ± 1.20 | 1,050 ± 349 | 46.0 ± 16.9 | |
| 0.4 | 0 | 80.0 ± 19.7 | 3.00 (1.5, 3) | 579 ± 149 | 611 ± 169 | 29.0 ± 2.87 | 14.0 ± 0.813 | 696 ± 223 | 28.5 ± 6.24 |
| 12 | 97.3 ± 49.2 | 1.50 (1.5, 3) | 610 ± 251 | 621 ± 253 | 27.7 ± 8.96 | 11.4 ± 2.23 | 717 ± 229 | 28.5 ± 11.7 | |
| 0.6 | 0 | 81.8 ± 20.2 | 2.50 (1.5, 3) | 547 ± 92.5 | 574 ± 105 | 13.7 ± 7.01 | 10.8 ± 4.00 | 1,070 ± 211 | 19.9 ± 7.43 |
| 12 | 67.3 ± 19.6 | 2.00 (1.5, 3) | 475 ± 123 | 531 ± 149 | 23.3 ± 22.0 | 15.7 ± 13.1 | 1,240 ± 416 | 36.0 ± 29.7 | |
| 1.0 | 0 | 176 ± 42.1 | 3.00 (3, 3) | 1,300 ± 472 | 1,360 ± 514 | 17.0 ± 8.81 | 13.0 ± 6.54 | 829 ± 331 | 18.1 ± 4.97 |
| 12 | 185 ± 10.4 | 3.00 (1.5, 3) | 1,430 ± 254 | 1,480 ± 265 | 19.9 ± 7.10 | 17.3 ± 8.15 | 694 ± 123 | 19.7 ± 6.30 | |
| CDV | |||||||||
| 0.1 | 0 | 2.64 ± 0.468 | 15.0 (15, 15) | 82.8 ± 27.2 | 107 ± 46.1 | 20.1 ± 11.1 | 37.4 ± 14.8 | 518 ± 167 | 13.2 ± 2.51 |
| 12 | 2.79 ± 0.594 | 13.5 (6, 15) | 94.5 ± 34.3 | 122 ± 40.3 | 34.6 ± 13.1 | 49.6 ± 14.0 | 474 ± 157 | 21.9 ± 7.02 | |
| 0.2 | 0 | 6.35 ± 0.696 | 15.0 (8, 15) | 200 ± 20.1 | 242 ± 31.3 | 26.3 ± 3.06 | 41.5 ± 5.02 | 415 ± 53.0 | 15.7 ± 1.48 |
| 12 | 5.67 ± 2.07 | 12.0 (8, 15) | 224 ± 52.0 | 295 ± 63.9 | 52.5 ± 5.33 | 67.2 ± 9.55 | 397 ± 78.2 | 30.1 ± 6.77 | |
| 0.4 | 0 | 11.2 ± 2.32 | 10.0 (8, 12) | 475 ± 88.4 | 542 ± 91.0 | 53.0 ± 5.75 | 65.1 ± 10.6 | 375 ± 62.7 | 28.8 ± 6.93 |
| 12 | 11.1 ± 1.20 | 12.0 (12, 12) | 545 ± 115 | 623 ± 133 | 57.7 ± 15.1 | 73.2 ± 17.4 | 388 ± 54.8 | 31.9 ± 7.46 | |
| 0.6 | 0 | 13.4 ± 1.55 | 10.0 (10, 10) | 534 ± 71.7 | 585 ± 85.5 | 44.2 ± 7.32 | 53.7 ± 4.38 | 519 ± 86.5 | 32.8 ± 5.23 |
| 12 | 13.0 ± 2.39 | 13.5 (10, 15) | 588 ± 146 | 646 ± 146 | 50.2 ± 12.3 | 64.1 ± 9.95 | 544 ± 103 | 38.4 ± 7.76 | |
| 1.0 | 0 | 18.5 ± 5.24 | 10.0 (8, 15) | 859 ± 272 | 996 ± 351 | 53.8 ± 9.36 | 67.0 ± 11.6 | 538 ± 150 | 40.5 ± 8.06 |
| 12 | 19.0 ± 4.37 | 12.0 (10, 12) | 1,090 ± 400 | 1,210 ± 442 | 58.2 ± 3.36 | 77.7 ± 6.98 | 541 ± 184 | 45.9 ± 18.0 |
n = 4 subjects per dose group. All PK parameters are summarized as means ± standard deviations, except for Tmax, which is reported as the median (with minimum, maximum in parentheses).
Fig 5.

Mean (± standard deviation) plasma drug Cmax and AUC0–∞ for CMX001 and cidofovir on day 0 and day 12 following oral administration of CMX001 on days 0, 6, and 12 (n = 4/cohort). There was no significant accumulation of CMX001 or CDV on day 12 compared with day 0.
On day 12, following the third dose of CMX001, the mean accumulation index for steady state (indexac) was not greater than 1.05, indicating that there was no significant accumulation of CMX001 following repeat dosing at 6-day intervals (Fig. 5). ANOVA of natural log-transformed Cmax and AUC0–last values on day 0 versus day 12 demonstrated general dose proportionality with only one minor variation, which was observed for Cmax at the 0.6-mg/kg dose. The value on day 12 was lower than the value on day 0, further supporting the lack of accumulation of CMX001 following repeat dosing at 6-day intervals.
As with CMX001, dose-proportional kinetics for CDV AUC values were observed on both day 0 and day 12 (Table 5; Fig. 5). Plasma CDV Cmax values increased in a dose-proportional manner over the 0.1- to 0.6-mg/kg dose range and a less-than-dose-proportional increase was observed at the 1.0-mg/kg dose level. The mean indexac was 1.07 at 0.10 mg/kg and 1.22 at 0.4 mg/kg and 1.0 mg/kg. ANOVA of natural log-transformed Cmax and AUC0–last values on day 0 versus day 12 indicated one significant difference, which was observed for AUC0–last at the 0.4-mg/kg dose. The value for AUC0–last at the 0.4-mg/kg dose was higher on day 12 than on day 0. CDV but not CMX001 was detected in the urine of subjects who received CMX001 during the multidose phase of the study.
DISCUSSION
Ten drugs have been approved for the treatment and/or prophylaxis of herpesvirus infections due to herpes simplex virus 1 (HSV-1), HSV-2, VZV, and CMV. Six of these drugs are approved specifically for the treatment and/or prophylaxis of CMV, the single most clinically important viral pathogen in the transplant population (17). Valganciclovir, ganciclovir, and foscarnet are the most frequently used antivirals for CMV, with cidofovir used as second-line therapy. However, there are important limitations on the use of these drugs, including neutropenia, nephrotoxicity, and the development of resistance (12, 15). Consequently, there is a pressing need for new antiviral drugs, or preferably a single broadly active drug, with increased potency, a better resistance profile, and an improved safety profile to address the growing problem of dsDNA virus infections.
The study results presented in this report demonstrate that CMX001 is well-tolerated when administered as a single dose of up to 2 mg/kg or as 3 doses of up to 1 mg/kg given once every 6 days. Specifically, the incidence of treatment-emergent AEs did not exhibit dose dependence in either the single-dose or multidose cohorts. In the multidose cohorts, the incidence of AEs seen following a single dose of study drug was comparable to the incidence of AEs seen following the second dose and third dose of study drug. No dose-effect relationship was demonstrated following administration of 3 doses of study drug. For example, only one subject who received the highest multiple dose of CMX001 (1 mg/kg) reported a treatment-emergent AE (constipation). No serious adverse events (SAEs) were reported during the study, and all AEs were judged to be mild in intensity by the principal investigator, except for one creatine kinase elevation and one headache, both judged to be moderate. There were no AEs that prevented dose escalation. A maximum tolerated dose was not identified in this trial, and no “expected adverse events” were identified as a result of this study. In studies conducted in animals, the dose-limiting toxicity was gastrointestinal, characterized by diarrhea, decreased body weight, and gastro/enteropathy. The GI toxicity occurred at doses >10-fold higher than those studied here and was quickly and fully reversible.
Pharmacokinetic analysis demonstrated that maximum plasma concentrations and systemic exposures (AUC) for both CMX001 and CDV increased approximately in proportion to dose over the entire dose range. Only CDV was detected in urine, indicating that CMX001 is not eliminated unchanged by urinary excretion. There was no accumulation of either CMX001 or CDV in the multidose phase of the study in which CMX001 was administered once every 6 days for a total of 3 doses.
Following intravenous administration of CDV dihydrate (Vistide), CDV is distributed in total body water and is rapidly eliminated by a combination of filtration and active secretion and is excreted unchanged in urine (5). The dose-limiting toxicity of CDV dihydrate is a Fanconi-like syndrome that occurs when proximal tubule cells in the kidney accumulate CDV, a process that is mediated by organic anion transporter 1 (OAT1) (3, 4, 13, 20). When administered concurrently, probenecid inhibits OAT-mediated CDV uptake, reducing accumulation of CDV and the associated risk of CDV dihydrate nephrotoxicity (4). Unlike cidofovir, CMX001 is not a substrate for OAT1 (19). Peak plasma drug concentrations of CDV produced as a metabolite of CMX001 remain low. For example, following administration of the 2-mg/kg dose of CMX001, the highest dose used in this study, the Cmax for CDV was more than 100-fold lower than that observed following administration of a 3-mg/kg dose of CDV dihydrate (without probenicid). It is hypothesized that the lower peak CDV concentrations in plasma following an oral dose of CMX001 result in lower CDV concentrations in kidney tubules, which do not exceed the capacity for efflux from the cell. This is supported by tissue distribution studies of mice showing minimal concentrations of CMX001-derived radioactivity in the kidney, as well as toxicity studies in both rodent and non-rodent species in which no nephrotoxicity was observed at high doses of CMX001 (16).
The doses of CMX001 used in this study were designed to escalate to clinically relevant doses. When concentrations of 1, 3, and 10 μM CDV or CMX001 were incubated with MRC-5 human lung fibroblasts in vitro, intracellular concentrations of CDV-PP that are anticipated to have antiviral activity were observed (1). The maximum concentration of CMX001 in plasma in this study ranged from 2.58 ng/ml (4.6 nM) to 350 ng/ml (623.1 nM), and the maximum concentration of CDV ranged from 1.76 ng/ml (6.3 nM) to 31.1 ng/ml (111.4 nM) over the dose range of 0.05 to 2.0 mg/kg CMX001. Peak plasma drug concentrations achieved at doses of 1.0 mg/kg and higher were within the range of median effective concentrations previously reported against herpesvirus (0.001 μM for human CMV to 0.06 μM for HSV), adenovirus (<0.009 to 0.3 μM), and variola virus (0.1 μM) isolates as assessed in plaque reduction assays using human fibroblast cell lines in vitro (8, 14), suggesting that the doses used in the current study are expected to produce an efficacious clinical response in humans. To specifically define clinical response, CMX001 antiviral activity will be assessed in randomized clinical trials in dsDNA virus-infected patients.
In conclusion, CMX001 was well tolerated at all doses, with no dose-limiting toxicity. In particular, there was no evidence of nephrotoxicity, which was the dose-limiting toxicity of CDV dihydrate (Vistide), or myelotoxicity, which is a toxicity that is potentially dose-limiting following administration of other nucleoside analogues, including ganciclovir. CMX001 was readily absorbed following oral administration under fasting conditions, exhibiting linear, dose-proportional pharmacokinetics. Based on comparison to results of in vitro studies, the doses administered in this study produced plasma drug concentrations of CMX001 that are projected to be efficacious in humans.
The ability to dose orally, the high in vitro potency, and the potentially improved safety profile of CMX001 make it a promising candidate for development as a broad-spectrum antiviral drug.
ACKNOWLEDGMENTS
This study was supported by grant 1-UOI-AI057233 from the National Institute of Allergies and Infectious Diseases Biodefense Partnership Program.
Footnotes
Published ahead of print 5 March 2012
REFERENCES
- 1. Aldern KA, Ciesla SL, Winegarden KL, Hostetler KY. 2003. Increased antiviral activity of 1-0-hexadeyloxypropyl-[2-14C] cidofovir in MRC-5 human lung fibroblasts is explained by unique cellular uptake and metabolism. Mol. Pharmacol. 63:678–681 [DOI] [PubMed] [Google Scholar]
- 2. Bruno B, et al. 2003. Adenovirus infection in hematopoietic stem cell transplantation: effect of ganciclovir and impact on survival. Biol. Blood Marrow Transplant. 9:341–352 [DOI] [PubMed] [Google Scholar]
- 3. Cihlar T, et al. 1999. The antiviral nucleotide analogs cidofovir and adefovir are novel substrates for human and rat renal organic anion transporter 1. Mol. Pharmacol. 56:570–580 [DOI] [PubMed] [Google Scholar]
- 4. Cihlar T, Ho ES, Lin DC, Mulato AS. 2001. Human renal organic anion transporter 1 (hOAT1) and its role in the nephrotoxicity of antiviral nucleotide analogs. Nucleosides Nucleotides Nucleic Acids 20:641–648 [DOI] [PubMed] [Google Scholar]
- 5. Cundy KC, et al. 1995. Clinical pharmacokinetics of cidofovir in human immunodeficiency virus-infected patients. Antimicrob. Agents Chemother. 39:1247–1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. De Clercq E. 2003. Clinical potential of the acyclic nucleoside phosphonates cidofovir, adefovir, and tenofovir in treatment of DNA virus and retrovirus infections. Clin. Microbiol. Rev. 16:569–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Emery VC. 2001. Progress in understanding cytomegalovirus drug resistance. J. Clin. Virol. 21:223–228 [DOI] [PubMed] [Google Scholar]
- 8. Hostetler KY. 2009. Alkoxyalkyl prodrugs of acyclic nucleoside phosphonates enhance oral antiviral activity and reduce toxicity: current state of the art. Antiviral Res. 82:A84–A98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ison MG, Hayden FG. 2002. Viral infections in immunocompromised patients: what's new with respiratory viruses? Curr. Opin. Infect. Dis. 15:355–367 [DOI] [PubMed] [Google Scholar]
- 10. Ison MG. 2005. Respiratory viral infections in transplant recipients. Curr. Opin. Organ Transplant. 10:312–319 [Google Scholar]
- 11. Ison MG. 2006. Adenovirus infections in transplant recipients. Clin. Infect. Dis. 43:331–339 [DOI] [PubMed] [Google Scholar]
- 12. Jacobsen T, Sifontis N. 2010. Drug interactions and toxicities associated with the antiviral management of cytomegalovirus infection. Am. J. Health Syst. Pharm. 67:1417–1425 [DOI] [PubMed] [Google Scholar]
- 13. Lalezari JP, et al. 1995. (S)-1-[3-hydroxy-2-(phosphonylmethoxy)propyl]cytosine (cidofovir): results of a phase I/II study of a novel antiviral nucleotide analogue. J. Infect. Dis. 171:788–796 [DOI] [PubMed] [Google Scholar]
- 14. Lanier R, et al. 2010. Development of CMX001 for the treatment of poxvirus infections. Viruses 2:2740–2762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lurain NS, Chou S. 2010. Antiviral drug resistance of human cytomegalovirus. Clin. Microbiol. Rev. 23:689–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Painter RG, et al. 2008. CMX001: anti-smallpox agent, anti-cytomegalovirus agent, viral polymerase inhibitor. Drugs Future 33:655 [Google Scholar]
- 17. Razonable RR, Emery VC. 2004. Management of CMV infection and disease in transplant patients. Herpes 11:77–86 [PubMed] [Google Scholar]
- 18. Tan CS, Koralnik IJ. 2010. Progressive multifocal leukoencephalopathy and other disorders caused by JC virus: clinical features and pathogenesis. Lancet Neurol. 9:425–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tippin TK, Lampert BM, Painter GP, Lanier R. 2010. Lipid conjugates of cidofovir and tenofovir, CMX001 and CMX157, are not substrates of human organic anion transporters OAT1 and OAT3, poster T3396. Am. Assoc. Pharm. Sci. Annu. Meet., New Orleans, LA, 14 to 18 November 2010 American Association of Pharmaceutical Scientists, Arlington, VA [Google Scholar]
- 20. Uwai Y, Ida H, Tsuji Y, Katsura T, Inui K-I. 2007. Renal transport of adefovir, cidofovir, and tenofovir by SLC22A family members (hOAT1, hOAT3, and hOCT2). Pharm. Res. 24:811–815 [DOI] [PubMed] [Google Scholar]