

Abstract
Multiple clinical trials have demonstrated that herpes simplex virus 2 (HSV-2) suppressive therapy using acyclovir (ACV) or valacyclovir in HIV-1/HSV-2-infected persons increased the patient's survival and decreased the HIV-1 load. It has been shown that the incorporation of ACV-monophosphate into the nascent DNA chain instead of dGMP results in the termination of viral DNA elongation and directly inhibits laboratory strains of HIV-1. We evaluated here the anti-HIV activity of ACV against primary HIV-1 isolates of different clades and coreceptor specificity and against viral isolates resistant to currently used drugs, including zidovudine, lamivudine, nevirapine, a combination of nucleoside reverse transcriptase inhibitors (NRTIs), a fusion inhibitor, and two protease inhibitors. We found that, at clinically relevant concentrations, ACV inhibits the replication of these isolates in human tissues infected ex vivo. Moreover, addition of ribavirin, an antiviral capable of depleting the pool of intracellular dGTP, potentiated the ACV-mediated HIV-1 suppression. These data warrant further clinical investigations of the benefits of using inexpensive and safe ACV alone or in combination with other drugs against HIV-1, especially to complement or delay highly active antiretroviral therapy (HAART) initiation in low-resource settings.
INTRODUCTION
Compelling clinical evidence has demonstrated that the treatment of HIV-1-infected persons with acyclovir (ACV) or its prodrug valacyclovir (vACV) reduces HIV-1 load and delays HIV-1 disease progression (20). In particular, ACV/vACV treatments are associated with a reduction of HIV-1 load in plasma, semen, cervico-vaginal secretions, and rectal swabs (3, 9, 11, 20, 27, 45, 46) and in plasma during pregnancy (10). Also, several clinical studies, as well as two meta-analyses of a total of 15 randomized clinical studies, demonstrate that ACV/vACV treatment delays progression to AIDS and prolongs the patient's survival (14, 22).
Understanding the mechanisms of ACV/vACV anti-HIV-1 activity is crucial to assessing its potential clinical use. It is hypothesized that ACV/vACV, an acyclic guanosine analogue, reduces HIV-1 load indirectly by suppressing HSV-2-mediated inflammation (41). However, we and others have shown that upon phosphorylation by kinases expressed by coinfecting human herpesviruses (HHVs) (including, but not limited to, herpes simplex virus 2 [HSV-2]), ACV directly inhibits HIV-1 reverse transcriptase (RT) (21, 25). These findings suggest that the anti-HIV activity of ACV may not be restricted to subjects coinfected with HSV-2, as other HHVs or yet unknown host enzymes (24) are also able to phosphorylate ACV. In consideration of its wide availability, safety, and low cost compared to other antiretroviral agents, ACV/vACV could be utilized to complement or delay highly active antiretroviral therapy (HAART) initiation in low-resource settings.
Here, we further investigate the antiviral activity of ACV against HIV-1 in experiments designed to mimic clinically relevant situations: (i) we evaluated its anti-HIV-1 activity not only against laboratory HIV-1 strains but also against primary HIV-1 clinical isolates of different subtypes and against multidrug-resistant variants; (ii) we evaluated the effect of ribavirin, a drug that is used against hepatitis C virus (HCV), on the anti-HIV-1 activity of ACV, since ribavirin is known to deplete the pool of the intracellular counterpart of ACV-triphosphate (ACV-TP), dGTP.
METHODS AND MATERIALS
Cultures and viral stocks.
Tonsillar tissues obtained from the Children's National Medical Center (Washington, DC), in accordance with an IRB-approved protocol, were dissected and cultured as described elsewhere (13). The MT-4 T-cell line was obtained from ATCC and maintained in RPMI with 10% fetal calf serum (FCS). The analysis of tonsillar tissue samples from 38 different donors showed that all of them contain HHVs of different types (21).
Virus stocks.
HIV-1LAI.04 viral stocks were obtained from the Rush University Virology Quality Assurance Laboratory (Chicago, IL). Primary isolate viruses (HIV-196USNG31, HIV-197USNG30, HIV-196USNN20, and HIV-1ME1), multi-nucleoside reverse transcriptase inhibitor (NRTI)-resistant viruses (HIV-17324-4, HIV-110076-4, HIV-16463-13, HIV-17303-3, HIV-11617-1, and HIV-135764-2), nevirapine-resistant virus (HIV-1N119), fusion inhibitor-resistant virus (HIV-1pNL4-3 gp41(36G)/V38A/N42D), and protease inhibitor-resistant virus (HIV-1L10R/M46I/L63P/V82T/I84V and HIV-1M46I/L63P/V82T/I84V) were obtained through the NIH AIDS Research and Reference Reagent Program (7, 16, 31, 32, 36). Lamivudine-resistant HIV-1 isolates (carrying the M184V mutation in HIV-1 RT) and zidovudine (AZT)-resistant strain 4X (AZT.4X) (carrying D67N, K70R, T215Y, and K219Q mutations in HIV-1 RT) were provided by Raymond Schinazi (1, 18).
Viral infections and drug treatment.
Tonsillar tissue blocks were inoculated with each HIV-1 isolate and treated for the entire culture period with ACV pharmaceutical formulations (Bedford Laboratories, Bedford, OH) and/or ribavirin (Sigma-Aldrich, St. Louis, MO) at the indicated concentrations. Drugs were present during the entire period of culture and were replenished with each medium change (every 3 days). The potentiation effect of ribavirin on the anti-HIV-1 activity of ACV-ProTides was investigated in MT-4 cell cultures: briefly, 105 MT-4 cells infected with HIV-1LAI.04 were cultured for 4 days in the presence of ribavirin at concentrations ranging from 1 to 20 μM and in the presence of ACV-ProTide at concentrations ranging from 0.01 to 50 μM. HIV replication was evaluated by p24gag antigen release in culture medium as previously described (21).
Statistical analysis.
For each experiment, we compared virus-infected and control tissues obtained from an individual donor in replicates of 27 tissue blocks for each data point. Experiments were repeated with tissues of n donors, where n is specified throughout the text. To average the results of different experiments and to analyze them statistically, we normalized the data as percentages of untreated controls and determined the means ± standard errors of the means (SEM). The data presented in Tables 1 and 2 are means ± standard deviations (SD). We used Student's unpaired t test to evaluate the significance of the differences between the inhibition of each virus (primary isolates AZT.4X and M184V; multi-NRTI-resistant, nevirapine-resistant, protease inhibitor-resistant, and fusion inhibitor-resistant HIV-1) and our laboratory reference strain HIV-1LAI.04.
Table 1.
Inhibition of HIV-1 replication by ACV in tonsillar tissues ex vivoa
| Strainb | HIV replicationc |
% Inhibition by ACVd |
|||
|---|---|---|---|---|---|
| Untreated control | ACV |
3 μM | 30 μM | ||
| 3 μM | 30 μM | ||||
| Primary HIV-1 isolates | |||||
| HIV-1LAI.04 | 13,052 (4,508–19,484) | 7,714 (1,487–12,305) | 749 (267–3,238) | 47 ± 17 | 83 ± 15 |
| HIV-196USNG31 | 4,455 (566–27,716) | 3,836 (608–16,167) | 871 (175–2,031) | 20 ± 18* | 71 ± 13 |
| HIV-197USNG30 | 14,050 (4,373–71,055) | 12,926 (3,568–46,592) | 7,438 (1,404–24,983) | 54 ± 22 | 82 ± 21 |
| HIV-196USNN20 | 5,365 (3,351–10,700) | 4,832 (2,017–10,142) | 2,059 (596–5,403) | 34 ± 26 | 68 ± 18 |
| HIV-1ME1 | 4,462 (2,166–6,919) | 2,263 (1,137–3,287) | 261 (215–336) | 53 ± 11 | 91 ± 6 |
| AZT-resistant variant HIV-1AZT.4X | 18,006 (15,618–30,906) | 10,325 (9,033–15,529) | 6,946 (6,573–9,967) | 57 ± 3 | 74 ± 7 |
| Lamivudine-resistant variant HIV-1M184V | 33,750 (27,305–44,612) | 37,165 (28,921–43,976) | 10,103 (8,118–14,408) | 14 ± 18* | 65 ± 27 |
| Multi-NRTI-resistant variants | |||||
| HIV-17324–4 | 1,904 (752–4,139) | 791 (203–1,782) | 67 (0–598) | 58 ± 17 | 92 ± 12 |
| HIV-110076–4 | 4,664 (513–11,393) | 1,415 (360–13,650) | 29 (13–627) | 57 ± 42 | 90 ± 15 |
| HIV-16463–13 | 793 (577–16,866) | 782 (550–17,295) | 33 (17–3,359) | 4 ± 7* | 90 ± 10 |
| HIV-17303–3 | 3,389 (1,940–10,550) | 2,524 (867–7,047) | 1,054 (253–4,206) | 31 ± 26 | 73 ± 29 |
| HIV-11617–1 | 12,559 (1,693–34,307) | 9,125 (2104–23,525) | 1,861 (216–4,273) | 18 ± 15* | 88 ± 13 |
| HIV-135764–2 | 933 (818–9,311) | 295 (211–3,349) | 98 (91–302) | 55 ± 31 | 91 ± 11 |
| Nevirapine-resistant variant HIV-1N119 | 52,832 (18,027–139,405) | 8,019 (6,935–9,104) | 16,599 (6,152–29,579) | 55 ± 51 | 84 ± 12 |
| Fusion inhibitor-resistant variant HIV-1pNL4–3 gp41(36G)/V38A/N42D | 1,043 (778–2,881) | 619 (600–1,174) | 48 (44–56) | 35 ± 32 | 94 ± 6 |
| Protease inhibitor-resistant HIV-1 variants | |||||
| HIV-1L10R/M46I/L63P/V82T/I84V | 23,863 (18,986–31,508) | 12,590 (11,524–15,546) | 902 (614–1,148) | 40 ± 37 | 96 ± 2 |
| HIV-1M46I/L63P/V82T/I84V | 4,410 (287–10,834) | 3,685 (55–8,507) | 348 (24–1,095) | 47 ± 31 | 86 ± 11 |
For each isolate and each experimental condition, 27 tonsillar tissue blocks were inoculated with each HIV-1 isolate from a multi-NRTI-resistant HIV-1 panel and treated for 12 days with 3 or 30 μM ACV.
Multi-NRTI-resistant clone HIV-17324-4 carried the mutations 41L, 70R, 215F, and 219E on its reverse transcriptase. HIV-110076-4 carried mutations 41L, 215Y, and 184V. HIV-16463-13 carried the mutations 41L, 67N, 210W, 215Y, 184V, and 118I. HIV-17303-3 carried the mutations 41L, 67N, 201W, 215Y, 69D, 44D, and 118I. HIV-11617-1 carried the mutations G70, 184V, 69K, 75I, 77L, 116Y, and 151 M. HIV-135764-2 carried the mutations 75I, 77L, 118Y, and 151 M (11).
For each viral isolate, the median cumulative p24gag release (pg/ml) and interquartile range (25 to 75%) in culture supernatant of infected tissues were calculated from n independent experiments. n = 3 to 7 for all conditions (except HIV-1AZT.4X replication in the untreated control and HIV-1N119 in the 3 μM ACV-treated condition, where n = 2).
Percentage (mean ± SD) of inhibition of HIV replication by 3 or 30 μM ACV is defined as [1−(RACV/RCtl)] × 100, where RACV and RCtl are the amounts of p24 accumulated in the medium over the 12-day culture period in ACV-treated cultures and in donor-matched untreated cultures, respectively.
, P < 0.05 (inhibition of replication of HIV variants compared to reference strain HIV-1LAI.04).
Table 2.
Potentiation of the ACV anti-HIV-1 activity by ribavirin in human tonsillar tissuesa
| Condition | ACV concn (μM) | HIV-1 replication [median p24gag (IQR)]b | % inhibitionc |
|---|---|---|---|
| Ribavirin (10 μM) | |||
| 0 | 7,379 (4,135–10,737) | NA | |
| 0.3 | 5,020 (3,220–8,837) | 18.3 ± 16.8 | |
| 3 | 2,284 (1,426–4,497) | 65.9 ± 17.0 * | |
| 10 | 1,327 (1,098–1,807) | 91.8 ± 7.5** | |
| No ribavirin | 0 | 8,228 (3,714–12,357) | NA |
| 0.3 | 8,528 (5,844–12,112) | 6.1 ± 11.7 | |
| 3 | 5,869 (3,554–8,257) | 41.3 ± 25.2 | |
| 10 | 3,234 (1,554–5,489) | 65.2 ± 17.6 |
For each condition, 27 tonsillar tissue blocks from each of n donors were inoculated with X4LAI.04 and treated for a 12-day period with 1, 3, or 10 μM ACV in the absence or presence of 10 μM ribavirin.
Median cumulative p24gag release (pg/ml) in culture supernatant of infected tissues. n = 10 for all data points except for 10 μM ACV, where n = 6. IQR, interquartile range.
Percentage (mean ± SD) of inhibition of HIV replication is defined as [1−(RACV/RCtl)] × 100, where RACV and RCtl are the amounts of p24gag accumulated in the medium over a 12-day culture period in ACV-treated cultures and in donor-matched untreated ACV cultures, respectively.
, P value of < 0.05 for HIV-1LAI.04 inhibition in the absence versus in the presence of 10 μM ribavirin at 3 μM ACV.
, P value of < 0.05 for HIV-1LAI.04 inhibition in the absence versus in the presence of 10 μM ribavirin at 30 μM ACV. NA, not available.
RESULTS
ACV suppresses replication of primary HIV-1 isolates.
Previously, we reported that ACV suppresses replication of four laboratory HIV-1 strains, CXCR4-tropic LAI.04 (HIV-1LAI.04), CCR5-tropic BaL, SF162, and AD8, in ex vivo infected human lymphoid tissues (21). Here, we evaluated the susceptibility of four primary isolates to ACV: HIV-196USNG31 and HIV-197USNG30 of clade C, HIV-196USNN20 of clade A, and HIV-1ME1 of clade B. Isolate HIV-196USNG31 uses CCR3, CCR5, and CXCR4; HIV-196USNN20 has a broad coreceptor utilization, including CCR2B, CCR3, CCR4, CCR5, CXCR4, Bob, and Bonzo; HIV-197USNG30 and HIV-1ME1 are exclusively CCR5 tropic.
Human lymphoid tissues were inoculated with each of these isolates and then treated for the entire culture period with ACV at 3 or 30 μM. These concentrations were chosen on the basis of our previous work with this compound (21). The median cumulative release of p24gag in the culture supernatants of tissues infected with each primary HIV-1 isolate with or without ACV is presented in Table 1. ACV suppresses replication of primary isolate HIV-197USNG30, HIV-196USNN20, and HIV-1ME1, and this suppression was not significantly different from that of laboratory isolate HIV-1LAI.04 at both 3 and 30 μM ACV (P > 0.05) (Fig. 1). At 3 μM, the inhibition of HIV-196USNG31 replication was lower than that of the laboratory strain HIV-1LAI.04 (P = 0.03). However, at 30 μM, the ACV inhibitory activity was not statistically different for the two viruses (P > 0.05).
Fig 1.

Inhibition of HIV-1 replication by ACV in human lymphoid tissues. Tissue blocks (27 from each donor) were infected with HIV-197USNG31, HIV-196USNN20, HIV-197USNG30, or HIV-1ME1 and treated with 3 or 30 μM ACV. Replication of HIV-1 was evaluated by p24gag core antigen release in pooled medium bathing 27 tissue blocks using a bead-based assay. Each point represents the measurement of medium pooled from three wells, each of which contained nine tissue blocks. The anti-HIV activity of ACV was evaluated by comparing viral replication in drug-treated tissues with that in untreated donor-matched control tissues. The graph represents the typical replication kinetics of 4 to 6 experiments performed with tissues from different donors. For average replication values, see Table 1.
In summary, we found that primary isolates from clade A and B were inhibited by ACV as efficiently as the laboratory isolate HIV-1LAI.04. However, one of the two clade C HIV-1 subtypes tested had lower susceptibility to ACV compared to HIV-1LAI.04.
ACV suppresses NRTI-resistant HIV-1 isolates.
We assessed the ACV susceptibility of HIV-1 variants that are resistant to one or more licensed NRTIs. First, we investigated the ACV anti-HIV activity against the AZT-resistant isolate AZT.4X (characterized by the D67N, K70R, T215Y, and K219Q mutations in RT) in ex vivo lymphoid tissues. ACV at concentrations of 3 and 30 μM inhibited AZT.4X replication by 57.2% ± 2.3% and 73.7% ± 4.0%, respectively. No statistical difference between the inhibition of AZT.4X and the inhibition of the laboratory strain HIV-1LAI.04 was observed (P > 0.05) (Table 1).
Similar experiments were performed with a lamivudine-resistant HIV isolate carrying the M184V mutation in RT (M184V HIV-1LAI.04). The replication of M184V virus in tissues treated with 3 μM ACV was suppressed by 13.6% ± 8.9%, while the replication of the parental HIV-1LAI.04 was suppressed by 47.0% ± 7.0% (P = 0.02). At 30 μM ACV, no difference was observed between M184V HIV-1LAI.04 and HIV-1LAI.04 (64.9% ± 15.6% and 83.0% ± 6.0%, respectively [P > 0.05]) (Table 1).
Because drug resistance may often manifest as multidrug resistance, we tested the ACV susceptibility of a panel of six prototypical multi-NRTI-resistant HIV-1 molecular clones (HIV7324–4, HIV10076–4, HIV6463–13, HIV7303–3, HIV1617–1, and HIV35764–2) (Fig. 2). Each of the six molecular clones carries several prototypical mutations that confer resistance to at least two of the following NRTIs: abacavir, zidovudine, stavudine, zalcitabine, didanosine, tenofovir, and lamivudine (12).
Fig 2.
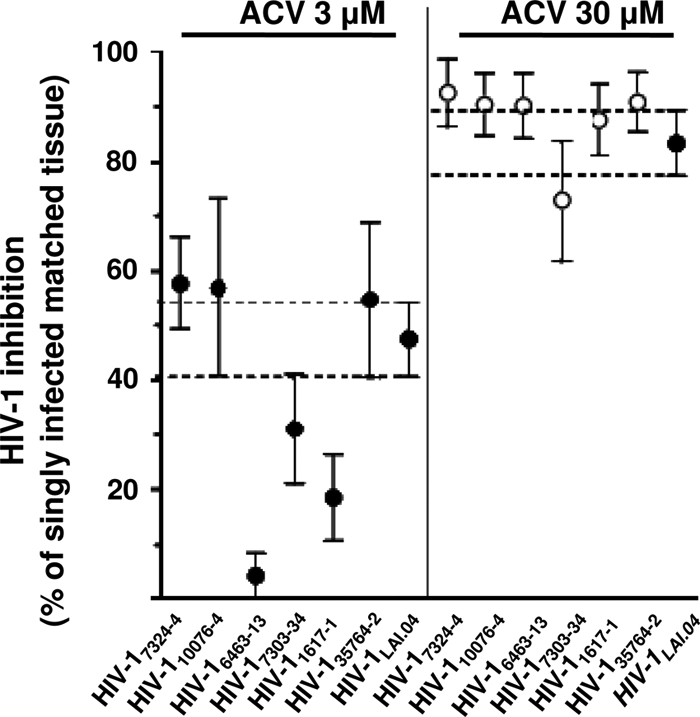
Inhibition of replication of multidrug-resistant HIV-1 clones by ACV in human lymphoid tissues. Donor-matched sets of tissue (27 blocks for each experimental condition from each of three to seven donors) were incubated with ACV (3 or 30 μM) for 12 days or used as untreated controls. Some of these sets were infected with multi-NRTI-resistant clones HIV-17324–4, HIV-110076–4, HIV-17303–3, HIV-16463–13, HIV-11617–1, and HIV-135764–2 or with the laboratory strain HIV-1LAI.04. Replication of HIV-1 was evaluated by p24gag core antigen release in pooled medium bathing 27 tissue blocks using a bead-based assay. The anti-HIV activity of ACV, as a percentage, was evaluated by comparing viral replication in drug-treated tissues with that in untreated donor-matched control tissues. Presented are means ± SEM of HIV-1 inhibition in 27 human tissue blocks from each of three to seven donors, relative to results for matched untreated tissues (n = 3 for HIV-17324–4 and HIV-16463–13; n = 4 for HIV-11617–1; n = 7 for HIV-110076–4, HIV-17303–3, HIV-135764–2, and HIV-1LAI.04).
HIV-1 suppression with 3 μM ACV is summarized in Table 1. The statistical analysis shows that at 3 μM, ACV inhibited the multi-NRTI-resistant HIV7324–4, HIV10076–4, HIV7303–3, and HIV35764–2 as efficiently as HIV-1LAI.04 (P > 0.05). In contrast, replications of HIV6463–13 and HIV1617–1 were significantly less inhibited than that of HIV-1LAI.04 (P = 0.003 and P = 0.02, respectively). At 30 μM, all of the multi-NRTI-resistant viruses were suppressed similarly to HIV-1LAI.04 (P > 0.05) (Fig. 2).
To evaluate whether HIV-1 RT single mutations could interfere with ACV anti-HIV-1 activity, we compared the susceptibilities of the singly RT-mutated viruses HIV-1K65R, HIV-1L74V, HIV-1V75I, and HIV-1Q151M to their HIV-1LAI.04 control at two concentrations of ACV (3 and 30 μM). In five independent experiments performed with tissues from five different donors, we found that all singly mutated viruses were susceptible to ACV at rates equal to that of the HIV-1LAI.04 control (P > 0.05), except HIV-1K65R, which at 30 μM ACV exhibited a ∼2-fold-reduced susceptibility compared to that of HIV-1LAI.04 (48.8% ± 7.7% inhibition versus 80.9% ± 6.6% [P = 0.007]).
ACV suppresses NNRTI-resistant HIV-1 isolates.
Furthermore, we evaluated the ACV-mediated suppression of HIV-1N119 in human lymphoid tissue ex vivo. HIV-1N119 has the RT mutation Y181C, which confers resistance to nevirapine, as well as to related nonnucleoside reverse transcriptase inhibitors (NNRTIs). HIV-1N119 replication was suppressed by 54.9% ± 35.9% and by 83.9% ± 6.1% at 3 and 30 μM ACV, respectively, rates similar to that of HIV-1LAI.04 (P > 0.05) (21) (Table 1).
ACV suppresses HIV-1 isolates resistant to protease and fusion inhibitors.
We tested the ACV susceptibility of HIV-1 isolates resistant to a fusion inhibitor (HIV-1pNL4-3 gp41(36G)/V38A/N42D) or two protease inhibitors (HIV-1L10R/M46I/L63P/V82T/I84V and HIV-1M46I/L63P/V82T/I84V). HIV-1pNL4-3 gp41(36G)/V38A/N42D is a recombinant virus that contains one or more amino acid substitutions in gp41 which confer resistance to enfuvirtide (T-20). Both HIV-1L10R/M46I/L63P/V82T/I84V and HIV-1M46I/L63P/V82T/I84V are resistant to the structurally diverse protease inhibitors MK-639, XM323, A-80897, Ro31-8959, VX-478, and SC-52151 (7).
We did not observe any difference in ACV-mediated inhibition of HIV-1LAI.04, HIV-1pNL4-3 gp41 (36G) V38A, N42D, HIV-1L10R/M46I/L63P/V82T/I84V, and HIV-1M46I/L63P/V82T/I84V at either 3 or 30 μM ACV (P > 0.05, Table 1). Thus, both fusion and protease inhibitor-resistant HIV-1 variants tested here retained susceptibility to ACV at a level similar to that of the wild-type HIV-1 (HIV-1LAI.04).
Ribavirin potentiates the anti-HIV-1 activity of ACV and of ACV prodrug derivatives.
We investigated in T-cell lines and in ex vivo human lymphoid tissues whether ribavirin, an antiviral compound known to deplete the intracellular dGTP pool, potentiates the anti-HIV activity of a dGTP antagonist, ACV. Control experiments did not show a statistically significant difference between the cumulative p24gag production in HIV-1LAI.04-infected tissues treated and that in tissues not treated with ribavirin (P > 0.05) (Table 2).
Next, in human lymphoid tissue ex vivo, we evaluated the susceptibility of HIV-1 replication to the combination of ribavirin and ACV. Without ribavirin, 3 μM ACV suppressed HIV-1LAI.04 replication by 41.3% ± 8.0%, while in the presence of ribavirin, HIV-1LAI.04 replication was inhibited by 65.9% ± 5.4% (P = 0.008). Similarly, in the absence of ribavirin, 10 μM ACV suppressed HIV-1LAI.04 replication by 65.2% ± 5.6%, while in the presence of ribavirin, HIV-1LAI.04 replication was inhibited by 91.8% ± 2.4% (P = 0.025). Also, 10 μM ribavirin potentiated the anti-HIV-1 activity of 0.3 μM ACV (18.3% ± 5.3% versus 6.1% ± 3.7% inhibition in the absence and presence of ribavirin, respectively) (Fig. 3). However, this potentiation did not reach statistical significance (P > 0.05).
Fig 3.

Potentiation of the anti-HIV-1 activity of ACV by ribavirin in human lymphoid tissue. Donor-matched tissue blocks (27 blocks from each donor) were inoculated with HIV-1LAI.04 and treated with ACV (0.3, 3, or 10 μM) in the absence or presence of 10 μM ribavirin. Replication of HIV-1 was evaluated by p24gag core antigen release in pooled medium bathing 27 tissue blocks using a bead-based assay. The percentage of HIV-1 inhibition was evaluated by comparing viral replication in drug-treated tissues with that in untreated donor-matched control tissues. Presented are means ± SEM of HIV-1LAI.04 inhibition in 27 human tissue blocks from each of 6 to 10 donors, relative to results for matched untreated tissues (n = 6 for experiments performed with 0.3 μM ACV; n = 10 for experiments performed with 3 or 10 μM ACV). An asterisk denotes a statistically significant difference (P < 0.05) in HIV-1LAI.04 inhibition between untreated and ribavirin-treated tissues.
Also, we evaluated the effect of ribavirin on the suppressive activity of ACV in MT-4 cells. Unlike human tissues, which contain various HHVs that activate (phosphorylate) ACV into ACV-MP, MT-4 cells are HHV free; therefore, in these experiments we used ACV-ProTides, which are lipophilic phosphorylated ACV prodrug derivatives (42).
For each experimental condition, 105 MT-4 cells infected with HIV-1LAI.04 were treated with ACV ProTides Cf2648 or Cf2681 at concentrations varying from 0.2 to 50 μM in the absence of ribavirin. When cells were treated with ribavirin at concentrations ranging from 1 to 20 μM, ACV ProTides Cf2648 or Cf2681 was added at concentrations varying from 0.01 to 20 μM. For both ProTides, ribavirin markedly decreased the 50% effective concentration (EC50): ribavirin decreased the EC50 from 1.3 to 0.018 μM for Cf2681 and from 9 to 1.8 μM for Cf2648 (Fig. 4).
Fig 4.

Potentiation of the anti-HIV-1 activity of ACV ProTides by ribavirin in MT-4 cell culture. MT-4 cells (105) infected with HIV-1LAI.04 were cultured for 4 days in the presence of ribavirin at concentrations ranging from 1 to 20 μM and ACV ProTides at concentrations ranging from 0.01 to 50 μM. HIV replication was evaluated by p24gag antigen release in culture medium at the end of the experiment using a bead-based assay. EC50s were calculated in an experiment in which two ACV ProTides (Cf 2648 and Cf2681) were tested against HIV-1LAI.04 as a function of different ribavirin concentrations.
DISCUSSION
The growing evidence of the important role played by sexually transmitted infections in general and by HSV-2 in particular in HIV transmission and pathogenesis has led to the development of new anti-HIV-1 strategies aimed at suppressing coinfecting viruses. Highly specific and potent anti-herpetic drugs were developed in the mid-1970s, making it possible to test these strategies in randomized clinical trials of HSV-2 suppressive therapy using ACV/vACV. These trials consistently demonstrated that such therapy resulted in increased survival and a decrease in HIV-1 load by 0.3 to 0.5 log10 in plasma, semen, cervico-vaginal secretions, and rectal swabs (3, 9, 11, 20, 27, 45, 46) and by 0.5 log10 in plasma during pregnancy (10). Although this effect is relatively small compared to that produced by the current potent anti-HIV cocktails, it is important to remember that the effect achieved by ACV monotherapy is similar to the monotherapy effect of zidovudine, which is one of the most efficient components of HAART (17). Moreover, two studies have recently reported that HSV-2 suppressive therapy using ACV (400 mg twice daily [b.i.d.]) delayed HIV-1 disease progression by 16% (20) and 27% (30). This delay may seem modest, but given HSV-2 seroprevalence rates as high as 90% in HIV-infected individuals in sub-Saharan Africa and the limited resources in parts of this region, low-cost interventions with well-tolerated drugs which can slow disease progression are an appealing strategy to delay initiation of HAART or the onset of AIDS in coinfected individuals (6, 38). Deciphering the mechanisms by which ACV suppresses HIV-1 is therefore important both for basic virology and for optimization of anti-HIV-1 therapeutic strategies.
Earlier, we and others (21, 25) showed that ACV, upon metabolic activation, is a direct inhibitor of HIV-1. In particular, we found that human tissues, including the tonsil tissues used in these studies, carry HHVs (not necessarily HSV-2) (21). Furthermore, when these tissues are infected ex vivo with HIV-1, ACV is phosphorylated by HHV-encoded kinases (and/or possibly by yet undefined host kinases) (24) and acts as an NRTI, directly inhibiting HIV-1 RT (21).
In the studies referenced above, the anti-HIV-1 activity of ACV was tested for well-defined laboratory-adapted strains of HIV-1, which may be different from primary isolates in several important aspects. In the current work, we tested the anti-HIV activity of ACV against primary isolates and drug-resistant isolates that commonly emerge in treated patients. We performed these tests in an ex vivo system of human tissues that reflects many in vivo tissue features.
We found that ACV inhibited all primary and multidrug-resistant HIV-1 variants tested. Moreover, the understanding of the molecular mechanism of ACV-mediated HIV-1 suppression allowed us to potentiate ACV suppressive activity by adding ribavirin, a known antiviral used in HCV treatment, which is capable of depleting the pool of intracellular dGTP (8).
We first tested the susceptibility of four clinical HIV-1 isolates to two concentrations of ACV, 3 and 30 μM, in ex vivo lymphoid tissue. These concentrations of ACV were chosen because they are in the range of those achieved in patients treated with oral ACV/vACV (23, 34) and they have been reported to inhibit the replication of HIV-1 laboratory isolates by approximately 50 and 90%, respectively (21). ACV suppressed the replication of all tested HIV-1 primary isolates, including two of clade C, which is prevalent in Southern and East Africa and India and is responsible for nearly half of all HIV infections worldwide, one of clade A, mainly present in Western/Central Africa and Russia, and one of clade B, commonly found in Europe, the Americas, and Australia. Also, the inhibition was irrespective of HIV-1 coreceptor specificity (CCR5, CXCR4, CCR3, CCR2B, Bob, Bonzo). In general, there was no difference in suppression of viral replication in ex vivo tonsillar tissues between clinical HIV-1 isolates and laboratory strains, although one of the two clade C isolates (HIV-196USNG31) was not as susceptible to the low concentration of ACV as the laboratory isolate HIV-1LAI.04.
This result should be taken into consideration in the interpretation of the current or recently completed clinical trials on the anti-HIV use of ACV. In fact, although average delays of 16% and 27% of disease progression were observed (20, 30), variations in the response to ACV, which might be explained by differences in susceptibilities of particular HIV-1 isolates, especially to low-dose ACV (i.e., 400 mg b.i.d.), were noted. In this regard, the higher doses of ACV in two ongoing clinical trials (43; NCT01059084 [http://clinicaltrials.gov/ct2/show/NCT01059084″]) (vACV, 500 mg b.i.d. or 1,000 mg thrice daily) may reduce the variability stemming from the intrinsic differences in susceptibility of HIV-1 RT of different isolates, as observed in our experiments in ex vivo tissues treated with a higher concentration of ACV (30 μM). Furthermore, it has been recently reported that, among HIV-1/HSV-2-coinfected persons, vACV suppressive therapy results in greater reduction in plasma HIV-1 levels than standard-dose ACV suppression (26).
For the future use of ACV or its derivatives in anti-HIV-1 therapy, it is important that ACV remains active against common drug-resistant HIV-1 variants that evolve in the course of regular anti-HIV therapy. Indeed, in the United States, up to 50% of patients receiving antiretroviral therapy are infected with viruses resistant to at least one of the available antiretroviral drugs. Moreover, mutations conferring resistance to one drug may often confer resistance to another drug. Furthermore, a significant proportion of new HIV infections results from the transmission of strains that are already resistant to one or more antiretroviral drugs (33).
Here, we first tested ACV against HIV-1 variants that are resistant to some of the most common anti-HIV NRTIs, zidovudine (AZT) and lamivudine (3TC). We found that ACV efficiently suppressed replication of NRTI-resistant viruses. However, while ACV suppressed replication of AZT-resistant HIV-1 as efficiently as it suppressed the wild-type variant, 3TC resistance, conferred by the M184V mutation, decreased by ∼4-fold the susceptibility to ACV.
Second, to further evaluate the ACV susceptibility of NRTI-resistant HIV-1 variants, we tested a panel of six prototypical infectious multidrug-resistant HIV-1 RT molecular clones. Each of the clones carries several mutations that occur most frequently in HIV-infected individuals treated with NRTIs. ACV (30 μM) suppressed equally well the replication of the six multidrug-resistant clones and of HIV-1LAI.04. However, 3 μM ACV (the EC50 for HIV-1LAI.04) suppressed by 50% the replication of only four of the six multidrug-resistant clones, indicating that some combinations of the common NRTI mutations may provide a reduced susceptibility to low-dose ACV. Finally, none of the tested viruses (except an HIV-1 variant that carries a K65R RT mutation, which showed an ∼2-fold decrease in susceptibility to 30 μM ACV), including one virus carrying the V75I RT mutation, were found to be resistant to ACV. It was reported earlier that this mutation was selected under the pressure of ACV or its prodrug derivative in single-cell cultures of CD4+ lymphoblasts and the MT-4 cell line (25, 39). However, in ex vivo tissues V75I-mutated virus was not resistant to ACV. Also, in vivo ACV does not select for this mutation (2, 19). This further underscores the necessity to evaluate viral resistance in ex vivo tissues, which represent the situation in vivo more adequately than single-cell cultures. Nevertheless, it would be of interest to investigate which particular features determine the difference between tissues and single-cell cultures in viral resistance.
In resource-limited countries, the WHO recommends a first-line regimen of two NRTIs and one NNRTI. Zidovudine and tenofovir disoproxil fumarate are the preferred NRTIs in this regimen in combination with lamivudine or emtricitabine, while the NNRTI is often nevirapine. Currently, inhibitors of viral protease, integrase, or fusion are also important components of anti-HIV cocktails (37). Although cross-resistance generally occurs predominantly between different drugs of the same class, we tested ACV susceptibility to two protease inhibitors and to one fusion inhibitor in HIV-1 variants resistant to nevirapine. We found that these four resistant HIV-1 isolates were as susceptible to ACV as the wild-type reference laboratory isolate HIV-1LAI.04.
Finally, understanding of the molecular mechanism of ACV suppression of HIV-1 allowed us to potentiate its suppressive activity. Indeed, our earlier data demonstrated that termination of HIV-1 DNA elongation results from the incorporation of ACV-TP by HIV-1 RT into the nascent DNA chain instead of its natural counterpart dGTP (21). Therefore, increasing the intracellular ACV-TP/dGTP ratio should increase the probability for RT to incorporate ACV-TP rather than dGTP. This was previously demonstrated in a cell-free system where a 12-fold decrease in the concentration of dGTP resulted in an ∼30% increase in ACV-TP inhibition of HIV-1 RT (21). In the present work, we used ribavirin to decrease the intracellular dGTP concentration and therefore increase the intracellular ACV-TP/dGTP ratio. Ribavirin inhibits IMP dehydrogenase, the enzyme that converts IMP to XMP, a key step in the de novo biosynthesis of GTP and dGTP (8, 35). This strategy was used earlier to potentiate the antiviral activity of ACV, as well as those of ganciclovir and penciclovir, against herpesvirus and hepatitis B virus (8, 28, 44) and also of 2′,3′-dideoxyinosine (ddI) and 2′,3′-dideoxy-2,6-diaminopurine riboside (ddDAPR) against HIV (4, 5).
Here, we used ribavirin at 10 μM, a concentration which per se has no significant effect on HIV replication, and found that ribavirin significantly potentiated the anti-HIV-1 activity of ACV. The ribavirin-mediated potentiation of the ACV anti-HIV activity did not seem to be related to the efficiency of ACV phosphorylation. Indeed, a pronounced potentiation of the anti-HIV activity of ACV by ribavirin was observed both for ACV and for prephosphorylated ACV prodrug derivatives (ACV ProTides). Although both these strategies are based on the assumption that ribavirin acts by decreasing the dGTP pool, other effects of ribavirin cannot be excluded. For example, ribavirin's carboxamide group can make the native nucleoside drug resemble adenosine or guanosine, which therefore can be directly incorporated into DNA. Also, ribavirin is known to modulate host T-cell-mediated immunity against viral infection. Nevertheless, we think that the decrease of dGTP is the main mechanism by which ribavirin potentiates ACV anti-HIV activity. The difference between ex vivo tissues and isolated cells in potentiating these effects may be related to the size of the natural pool of dGTP. It is likely that this pool is larger in highly proliferating immortalized cell lines like MT-4 than in tissue cells.
In general, the fact that a drug that reduces the dGTP pool potentiates the anti-HIV activity of ACV emphasizes that ACV-TP and its natural counterpart dGTP compete for incorporation during DNA elongation by HIV-1 RT and confirms once again the existence of a direct effect of the activated ACV metabolite on HIV-1 RT.
The strategy of potentiating ACV anti-HIV activity may be clinically relevant, as the concentration of 10 μM ribavirin required for potentiation in our experiments is attained in human plasma upon oral dosing of ribavirin in HCV-infected patients (15). Our finding that ribavirin, an HCV drug, potentiates acyclovir, an anti-HSV drug recently reported to have anti-HIV-1 activity, is important in light of frequent coinfection with these three viruses: about 50% of HIV-1-infected patients are coinfected with HSV-2 in the United States (29), and about one quarter of the people infected with HIV are also infected with HCV (40). Our results show that in combination with ACV, ribavirin could have a dual-targeted action in coinfected individuals by inhibiting both HCV and HIV-1. Although both drugs are already used for therapy, only further studies may show whether this strategy could be pursued in vivo.
In summary, we found that in human lymphoid tissue, ACV suppresses the replication of HIV-1 of different clades and of different coreceptor tropisms as well as HIV-1 variants with mutations conferring resistance to the currently used NRTIs, NNRTIs, and inhibitors of viral protease, integrase, and fusion. Together with the reported absence of emergence of HIV variants resistant to ACV in vivo (2, 19) and the well-documented beneficial effect of ACV on disease progression (20), these data warrant further clinical investigation of the use of ACV alone or in combination with anti-HIV drugs against this virus. Moreover, this inexpensive and safe drug may be used to slow disease progression and delay HAART in low-income countries where access to HAART is limited. The combination of ACV with ribavirin may be envisioned also in topical microbicide applications where both HSV-2 and HIV-1 might be suppressed with this drug combination regimen. Further studies are needed to address the feasibility, the potential benefits, and the cost-effectiveness of novel anti-HIV-1 strategies based on ACV and its derivatives.
ACKNOWLEDGMENTS
This work was supported by the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, and the KU Leuven (GOA no. 10/14). Also, this work was supported in part by NIH grant 5P30-AI-50409 (R.F.S.) and by the Department of Veterans Affairs (R.F.S.).
We thank Dana Ashley Hill and the staff of the Department of Pathology of Children's National Medical Center for their generous assistance in obtaining human tonsillar tissues.
We declare no conflict of interest.
Footnotes
Published ahead of print 6 February 2012
REFERENCES
- 1. Arion D, Kaushik N, McCormick S, Borkow G, Parniak MA. 1998. Phenotypic mechanism of HIV-1 resistance to 3′-azido-3′-deoxythymidine (AZT): increased polymerization processivity and enhanced sensitivity to pyrophosphate of the mutant viral reverse transcriptase. Biochemistry 37:15908–15917 [DOI] [PubMed] [Google Scholar]
- 2. Baeten JM, et al. 2011. Herpes simplex virus type 2 suppressive therapy with acyclovir or valacyclovir does not select for specific HIV-1 resistance in HIV-1/HSV-2 dually infected persons. J. Infect. Dis. 203:117–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Baeten JM, et al. 2008. Herpes simplex virus (HSV)-suppressive therapy decreases plasma and genital HIV-1 levels in HSV-2/HIV-1 coinfected women: a randomized, placebo-controlled, cross-over trial. J. Infect. Dis. 198:1804–1808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Balzarini J, Lee CK, Herdewijn P, De Clercq E. 1991. Mechanism of the potentiating effect of ribavirin on the activity of 2′,3′-dideoxyinosine against human immunodeficiency virus. J. Biol. Chem. 266:21509–21514 [PubMed] [Google Scholar]
- 5. Balzarini J, Naesens L, Robins MJ, De Clercq E. 1990. Potentiating effect of ribavirin on the in vitro and in vivo antiretrovirus activities of 2′,3′-dideoxyinosine and 2′,3′-dideoxy-2,6-diaminopurine riboside. J. Acquir. Immune Defic. Syndr. 3:1140–1147 [PubMed] [Google Scholar]
- 6. Buve A, Lynen L. 2010. Treating HIV infection with drugs for HSV-2 infection? Lancet 375:782–784 [DOI] [PubMed] [Google Scholar]
- 7. Condra JH, et al. 1995. In vivo emergence of HIV-1 variants resistant to multiple protease inhibitors. Nature 374:569–571 [DOI] [PubMed] [Google Scholar]
- 8. De Clercq E. 2009. Another ten stories in antiviral drug discovery (part C): “old” and “new” antivirals, strategies, and perspectives. Med. Res. Rev. 29:611–645 [DOI] [PubMed] [Google Scholar]
- 9. Delany S, et al. 2009. Impact of aciclovir on genital and plasma HIV-1 RNA in HSV-2/HIV-1 co-infected women: a randomized placebo-controlled trial in South Africa. AIDS 23:461–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Drake A, et al. 2012. Valacyclovir suppressive therapy reduces plasma and breast milk HIV-1 RNA levels during pregnancy and postpartum: a randomized trial. J. Infect. Dis. 205:366–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dunne EF, et al. 2008. Suppressive acyclovir therapy reduces HIV cervicovaginal shedding in HIV- and HSV-2-infected women, Chiang Rai, Thailand. J. Acquir. Immune Defic. Syndr. 49:77–83 [DOI] [PubMed] [Google Scholar]
- 12. Dupnik K, Gonzales MJ, Shafer RW. 2001. Most multidrug-resistant HIV-1 reverse transcriptase clones in plasma encode functional reverse transcriptase enzymes. Antivir. Ther. 6(Suppl. 1):42 [Google Scholar]
- 13. Grivel JC, Margolis L. 2009. Use of human tissue explants to study human infectious agents. Nat. Protoc. 4:256–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ioannidis JP, et al. 1998. Clinical efficacy of high-dose acyclovir in patients with human immunodeficiency virus infection: a meta-analysis of randomized individual patient data. J. Infect. Dis. 178:349–359 [DOI] [PubMed] [Google Scholar]
- 15. Jen JF, Glue P, Gupta S, Zambas D, Hajian G. 2000. Population pharmacokinetic and pharmacodynamic analysis of ribavirin in patients with chronic hepatitis C. Ther. Drug Monit. 22:555–565 [DOI] [PubMed] [Google Scholar]
- 16. Johnston E, et al. 2005. Panel of prototypical infectious molecular HIV-1 clones containing multiple nucleoside reverse transcriptase inhibitor resistance mutations. AIDS 19:731–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Katzenstein DA, et al. 1996. The relation of virologic and immunologic markers to clinical outcomes after nucleoside therapy in HIV-infected adults with 200 to 500 CD4 cells per cubic millimeter. AIDS Clinical Trials Group Study 175 Virology Study Team. N. Engl. J. Med. 335:1091–1098 [DOI] [PubMed] [Google Scholar]
- 18. Larder BA, Kemp SD. 1989. Multiple mutations in HIV-1 reverse transcriptase confer high-level resistance to zidovudine (AZT). Science 246:1155–1158 [DOI] [PubMed] [Google Scholar]
- 19. LeGoff J, et al. 2010. No selection of nucleoside reverse transcriptase inhibitor resistance associated mutations by acyclovir suppressive therapy in herpes simplex virus-2/HIV-1 dually infected persons. AIDS 24:2595–2596 [DOI] [PubMed] [Google Scholar]
- 20. Lingappa JR, et al. 2010. Daily acyclovir for HIV-1 disease progression in people dually infected with HIV-1 and herpes simplex virus type 2: a randomised placebo-controlled trial. Lancet 375:824–833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lisco A, et al. 2008. Acyclovir is activated into a HIV-1 reverse transcriptase inhibitor in herpesvirus-infected human tissues. Cell Host Microbe 4:260–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ludeman C, Cole SR, Poole C, Chu H, Eron JJ. 2011. Meta-analysis of randomized trials on the association of prophylactic acyclovir and HIV-1 viral load in individuals coinfected with herpes simplex virus-2. AIDS 25:1265–1269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lycke J, Malmestrom C, Stahle L. 2003. Acyclovir levels in serum and cerebrospinal fluid after oral administration of valacyclovir. Antimicrob. Agents Chemother. 47:2438–2441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McMahon MA, Parsons TL, Shen L, Siliciano JD, Siliciano RF. 2011. Consistent inhibition of HIV-1 replication in CD4+ T cells by acyclovir without detection of human herpesviruses. J. Virol. 85:4618–4622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McMahon MA, et al. 2008. The antiherpetic drug acyclovir inhibits HIV replication and selects the V75I reverse transcriptase multidrug resistance mutation. J. Biol. Chem. 283:31289–31293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mugwanya K, et al. 2011. High-dose valacyclovir HSV-2 suppression results in greater reduction in plasma HIV-1 levels compared with standard dose acyclovir among HIV-1/HSV-2 coinfected persons: a randomized, crossover trial. 12:1912–1917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nagot N, et al. 2007. Reduction of HIV-1 RNA levels with therapy to suppress herpes simplex virus. N. Engl. J. Med. 356:790–799 [DOI] [PubMed] [Google Scholar]
- 28. Neyts J, Andrei G, De Clercq E. 1998. The novel immunosuppressive agent mycophenolate mofetil markedly potentiates the antiherpesvirus activities of acyclovir, ganciclovir, and penciclovir in vitro and in vivo. Antimicrob. Agents Chemother. 42:216–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Patel P, et al. 2012. Prevalence and risk factors associated with herpes simplex virus-2 infection in a contemporary cohort of HIV-infected persons in the United States. Sex. Transm. Dis. 39:154–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Reynolds S, et al. 2011. Abstr. 6th IAS Conference on HIV Pathogenesis, Treatment and Prevention, Rome, Italy, 17 to 20 July 2011, abstr. TUAB0104 [Google Scholar]
- 31. Richman D, et al. 1991. Human immunodeficiency virus type 1 mutants resistant to nonnucleoside inhibitors of reverse transcriptase arise in tissue culture. Proc. Natl. Acad. Sci. U. S. A. 88:11241–11245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rimsky LT, Shugars DC, Matthews TJ. 1998. Determinants of human immunodeficiency virus type 1 resistance to gp41-derived inhibitory peptides. J. Virol. 72:986–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shafer RW, Schapiro JM. 2008. HIV-1 drug resistance mutations: an updated framework for the second decade of HAART. AIDS Rev. 10:67–84 [PMC free article] [PubMed] [Google Scholar]
- 34. Soul-Lawton J, et al. 1995. Absolute bioavailability and metabolic disposition of valaciclovir, the l-valyl ester of acyclovir, following oral administration to humans. Antimicrob. Agents Chemother. 39:2759–2764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Streeter DG, et al. 1973. Mechanism of action of 1-β-d-ribofuranosyl-1,2,4-triazole-3-carboxamide (Virazole), a new broad-spectrum antiviral agent. Proc. Natl. Acad. Sci. U. S. A. 70:1174–1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sullivan PS, et al. 2000. Human immunodeficiency virus (HIV) subtype surveillance of African-born persons at risk for group O and group N HIV infections in the United States. J. Infect. Dis. 181:463–469 [DOI] [PubMed] [Google Scholar]
- 37. Taiwo B, Murphy RL, Katlama C. 2010. Novel antiretroviral combinations in treatment-experienced patients with HIV infection: rationale and results. Drugs 70:1629–1642 [DOI] [PubMed] [Google Scholar]
- 38. Tan DH, et al. 2010. Can herpes simplex virus type 2 suppression slow HIV disease progression: a study protocol for the VALacyclovir In Delaying Antiretroviral Treatment Entry (VALIDATE) trial. Trials 11:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tchesnokov EP, et al. 2009. Mechanisms associated with HIV-1 resistance to acyclovir by the V75I mutation in reverse transcriptase. J. Biol. Chem. 284:21496–21504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Thomas DL. 2008. The challenge of hepatitis C in the HIV-infected person. Annu. Rev. Med. 59:473–485 [DOI] [PubMed] [Google Scholar]
- 41. Van de Perre P, et al. 2008. Herpes simplex virus and HIV-1: deciphering viral synergy. Lancet Infect. Dis. 8:490–497 [DOI] [PubMed] [Google Scholar]
- 42. Vanpouille C, et al. 2010. A new class of dual-targeted antivirals: monophosphorylated acyclovir prodrug derivatives suppress both human immunodeficiency virus type 1 and herpes simplex virus type 2. J. Infect. Dis. 201:635–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vanpouille C, Lisco A, Margolis L. 2009. Acyclovir: a new use for an old drug. Curr. Opin. Infect. Dis. 22:583–587 [DOI] [PubMed] [Google Scholar]
- 44. Ying C, De Clercq E, Neyts J. 2000. Ribavirin and mycophenolic acid potentiate the activity of guanine- and diaminopurine-based nucleoside analogues against hepatitis B virus. Antiviral Res. 48:117–124 [DOI] [PubMed] [Google Scholar]
- 45. Zuckerman RA, et al. 2009. HSV suppression reduces seminal HIV-1 levels in HIV-1/HSV-2 co-infected men who have sex with men. AIDS 23:479–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zuckerman RA, et al. 2007. Herpes simplex virus (HSV) suppression with valacyclovir reduces rectal and blood plasma HIV-1 levels in HIV-1/HSV-2-seropositive men: a randomized, double-blind, placebo-controlled crossover trial. J. Infect. Dis. 196:1500–1508 [DOI] [PubMed] [Google Scholar]