

Abstract
Enterococcus faecalis is a low-GC Gram-positive bacterium that is intrinsically resistant to cephalosporins, antibiotics that target cell wall biosynthesis. To probe the mechanistic basis for intrinsic resistance, a library of transposon mutants was screened to identify E. faecalis strains that are highly susceptible to ceftriaxone, revealing a transposon mutant with a disruption in murAA. murAA is predicted to encode a UDP-N-acetylglucosamine 1-carboxyvinyl transferase that catalyzes the first committed step in peptidoglycan synthesis: phosphoenolpyruvate (PEP)-dependent conversion of UDP-N-acetylglucosamine to UDP-N-acetylglucosamine-enolpyruvate. In-frame deletion of murAA, but not its homolog in the E. faecalis genome (murAB), led to increased susceptibility of E. faecalis to cephalosporins. Furthermore, expression of murAA enhanced cephalosporin resistance in an E. faecalis mutant lacking IreK (formerly PrkC), a key kinase required for cephalosporin resistance. Further genetic analysis revealed that MurAA catalytic activity is necessary but not sufficient for this role. Collectively, our data indicate that MurAA and MurAB have distinct roles in E. faecalis physiology and suggest that MurAA possesses a unique property or activity that enables it to enhance intrinsic resistance of E. faecalis to cephalosporins.
INTRODUCTION
Enterococcus faecalis is a Gram-positive, opportunistic pathogen found ubiquitously in nature and as a commensal in the human gastrointestinal tract (1, 30, 39). Enterococci are among the leading causes of nosocomial infections (17, 37), particularly surgical wound infections, urinary tract infections, bacteremia, and endocarditis. Treatment of enterococcal infections can be complicated by the well-known ability of enterococci to acquire new resistance to multiple antibacterial agents (16, 26) as well as the intrinsic resistance of enterococci to certain classes of antibiotics. For example, enterococci are intrinsically resistant to cephalosporins (30), antibiotics in the β-lactam family, which target bacterial cell wall biosynthesis by inactivating penicillin-binding proteins (PBPs) and preventing the critical cross-linking step required for peptidoglycan integrity (41, 45). Although enterococci have been known to be intrinsically resistant to cephalosporins for decades, the mechanism of cephalosporin resistance remains incompletely understood.
Three determinants for cephalosporin resistance in E. faecalis have been described. The first is Pbp5, defined as the “low-affinity” penicillin-binding protein that is thought to catalyze peptidoglycan cross-linking in the presence of cephalosporins when all other PBPs are inactivated (6, 35, 44). However, Pbp5 is not sufficient for cephalosporin resistance, because mutations in a two-component signal transduction pathway (CroRS) render E. faecalis susceptible to cephalosporins (8, 15). Presumably, the CroRS signaling system regulates the expression of a gene(s) that promotes cephalosporin resistance, although the identity of any such CroRS-dependent genes remains unknown. The third determinant of cephalosporin resistance is IreK (formerly PrkC), a Ser/Thr kinase whose kinase activity is required for cephalosporin resistance (23, 25). Although the substrates of IreK kinase in E. faecalis remain unknown, in other species of bacteria enzymes of the peptidoglycan synthesis pathway are known to be phosphorylated on Thr residues (14, 40) with functional consequences for enzymatic activity (14), suggesting that some aspects of peptidoglycan synthesis could be regulated posttranslationally.
Peptidoglycan synthesis requires the sequential action of over 7 dedicated cytoplasmic enzymes (reviewed in reference 2). The first committed step in the synthesis pathway is catalyzed by a UDP-N-acetylglucosamine 1-carboxyvinyl transferase (EC 2.5.1.7), a family of enzymes that transfers the enolpyruvyl moiety of phosphoenolpyruvate (PEP) to UDP-N-acetylglucosamine (UDP-GlcNAc) with concomitant release of inorganic phosphate (Pi). In Escherichia coli, this reaction is catalyzed by the product of the murA gene, which is essential for viability (5, 27). MurA is the target of the antibiotic fosfomycin, a PEP analog, which irreversibly binds and interferes with MurA function (18, 36).
The genomes of most low-GC Gram-positive bacteria carry two homologs of murA (10), although in most cases the physiological functions of each homolog have not been well defined. Studies conducted in Staphylococcus aureus and Streptococcus pneumoniae demonstrate that either of the respective murA homologs can be genetically deleted, suggesting that they are at least partially functionally redundant (4, 10), whereas in Bacillus subtilis and Bacillus anthracis, one of the homologs (murA1) appears to be essential for viability (19–21). The genome of E. faecalis carries two homologs of murA, annotated as murAA (EF2605) and murAB (EF1169), whose gene products exhibit ∼45% identity to each other (32). To our knowledge, the physiological functions of MurAA and MurAB in E. faecalis have not been studied.
To probe the mechanistic basis for intrinsic cephalosporin resistance in E. faecalis, transposon mutagenesis was used to identify mutants that are highly susceptible to ceftriaxone, an expanded-spectrum cephalosporin. Here we report that mutations in murAA, but not murAB, render E. faecalis susceptible to cephalosporins. Furthermore, expression of murAA enhanced cephalosporin resistance in an E. faecalis mutant lacking IreK, and catalytic activity of MurAA is necessary but not sufficient for this role. Collectively, our data indicate that MurAA and MurAB have distinct roles in E. faecalis physiology and suggest that MurAA possesses a unique property or activity that enables it to enhance intrinsic resistance of E. faecalis to cephalosporins.
MATERIALS AND METHODS
Bacterial strains, media, and oligonucleotides.
The strains used in this study are listed in Table 1. All of the oligonucleotides used in plasmid construction were designed on the basis of the E. faecalis V583 genome sequence available at The J. Craig Venter Institute Comprehensive Microbial Resource. Oligonucleotides were synthesized by Integrated DNA Technologies, Inc. E. coli strains were grown in either LB (Difco) or brain heart infusion (BHI) (Difco), at 37°C, with shaking at 225 rpm. E. faecalis was cultured in Mueller-Hinton broth (MHB) or BHI prepared as described by the manufacturer and grown at 37°C unless otherwise specified, with shaking at 225 rpm. The solid medium used is the same as above with addition of 1.6% (wt/vol) Bacto agar (Difco). When required, antibiotics were added at the following final concentrations: for E. coli, kanamycin (Kn), 50 μg/ml; erythromycin (Em), 100 μg/ml; spectinomycin (Sp), 200 μg/ml; for E. faecalis, spectinomycin, 100 μg/ml or 1,000 μg/ml, and erythromycin, 10 μg/ml. All chemicals and antibiotics were purchased from Sigma unless otherwise indicated.
Table 1.
Strains and plasmids used in this study
| Strain or plasmid | Relevant genotype or description | Source or reference |
|---|---|---|
| Strains | ||
| E. coli | ||
| TOP10 | E. coli cloning host | Invitrogen |
| BL21(DE3) | E. coli protein expression host | Lab stock |
| E. faecalis | ||
| OG1RF | Wild-type reference strain | 11 |
| CK119 | OG1RF ΔireK2 | 25 |
| CK128 | OG1RF croR2::EfaMarTn | This work |
| CK153 | OG1RF murAA3 (C-terminally Strep-tagged murAA allele) | This work |
| CK161 | OG1RF ΔireK2 murAA3 (C-terminally Strep-tagged murAA allele) | This work |
| DV1-4 | OG1RF ΔmurAB2 | This work |
| DV75-2 | OG1RF ΔmurAA2 | This work |
| DV77 | OG1RF ΔmurAA2 (derived independently from DV75-2) | This work |
| 39G11 | OG1RF murAA4::EfaMarTn | This work |
| JL104 | OG1RF Δpbp5-2 | This work |
| E. faecium | ||
| Com12 | Fecal isolate | 28 |
| 1,231,501 | Clinical isolate | 31 |
| Plasmids | ||
| pDL278p23 | E. faecalis expression vector, constitutive P23 promoter (Spr) | 7 |
| pET28b | E. coli expression vector (Knr) | Novagen |
| pCJK47 | E. faecalis allelic-exchange vector with pheS* counterselectable marker (Emr) | 22 |
| pJRG32 | pCJK47 derivative with a synthetic P-pheS* cassette (Emr) | This work |
| pJRG36 | ΔmurAB2 (ΔK22-L417) in pCJK47 | This work |
| pDV14-9 | pDL278p23::murAA C120S | This work |
| pDV15-1 | pDL278p23::murAA C120S with C-terminal Strep tag | This work |
| pDV20-1 | pET28b::murAA | This work |
| pDV26 | ΔmurAA2 (ΔV17-Q410) in pJRG32 | This work |
| pDV27-4 | pDL278p23::murAA with C-terminal Strep tag | This work |
| pDV44-1 | pET28b::murAA C120S | This work |
| pJMK4 | pDL278p23::murA (E. coli murA with C-terminal Strep tag) | This work |
| pJLL21 | Δpbp5-2 (ΔS11-A677) in pJRG32 | This work |
| pCJK163 | murAA3 (C-terminal Strep tag) in pCJK47 | This work |
Transposon mutagenesis screen.
Approximately 8,000 transposon insertion mutants derived from a previously described library (24) were screened to identify mutants exhibiting reduced resistance toward the expanded-spectrum cephalosporin ceftriaxone. Mutants were inoculated from frozen stock cultures into 100 μl of tryptic soy broth in 96-well plates and incubated overnight at 37°C. The resulting cell suspensions were inoculated onto the surface of MM9YEG (22) agar plates supplemented or not with ceftriaxone using a 96-prong device. Mutants that did not grow on the ceftriaxone-supplemented plates after 24 h at 37°C were chosen for further analysis.
Plasmid construction.
Plasmids for expression of proteins in E. faecalis were generated in the following manner. The open reading frame (ORF) of E. coli murA was amplified from E. coli DH5α genomic DNA and introduced into pDL278p23 using primer-specified restriction sites, giving rise to pJMK4; the open reading frame of E. faecalis murAA was amplified from OG1RF genomic DNA and cloned into pDL278p23 using primer-specified restriction sites, giving rise to pDV27-4. For epitope-tagged proteins, streptomycin (Strep) tag epitope tags (WSHPQFEK) were encoded in the primers as well. To construct the murA C120S allele, we used a previously described BsaI-based cloning strategy (22) to seamlessly fuse two PCR amplicons carrying the desired mutation. The resulting fragment was cloned into pDL278p23 using primer-specified restriction sites, giving rise to pDV14-9. This plasmid was subsequently used as a template for PCR to generate a Strep-tagged version of the murA C120S allele that was cloned into pDL278p23, leading to pDV15-1.
Plasmids for protein overexpression in E. coli were constructed as follows. E. faecalis murAA was amplified from OG1RF genomic DNA, and murAA C120S was amplified from pDV14-9. Primer-specified NcoI/XhoI restriction sites were used to clone the alleles into pET28b to create C-terminally His-tagged recombinants: pDV20-1 for MurAA-His6 and pDV44-1 for MurAA C120S-His6 expression.
Modification of chromosomal murAA, murAB, and pbp5 loci.
Modification of murAA, murAB, and pbp5 in the E. faecalis chromosome was performed via markerless genetic exchange as previously described (22) using either the original markerless allelic exchange plasmid (pCJK47) or a derivative of pCJK47 (pJRG32) in which the pheS* counterselectable marker was replaced by a synthetic pheS* allele (custom synthesized at GenScript) containing synonymous substitutions at the third position of most codons (to prevent recombination with pheS carried in the genome of E. faecalis). A brief summary of specific plasmids follows. A derivative of plasmid pCJK47 carrying an in-frame deletion allele of murAB (pJRG36) was constructed using a BsaI-based cloning strategy to seamlessly fuse two PCR amplicons flanking murAB to form the in-frame deletion. The deletion allele was designed such that the first 21 and last 13 codons remained (92% of the open reading frame [ORF] was deleted), in an effort to avoid perturbing expression of neighboring genes, and was transferred to the OG1RF chromosome via conjugation-based allelic exchange. For manipulation of murAA, a derivative of pJRG32 carrying an in-frame deletion of murAA (pDV26) was constructed and transferred to the OG1RF chromosome as described above. The deletion was designed such that the first 16 and last 23 codons of murAA remained (91% of the ORF was deleted). Deletion mutants were isolated by plating on counterselection plates (22) at 30°C for 2 to 3 days. To introduce a Strep tag epitope tag at the C terminus of chromosomally encoded MurAA, a derivative of pCJK47 was constructed (pCJK163) carrying murAA in which the 8-codon Strep tag was appended to the end of the annotated gene. The allele was transferred to the murAA locus of OG1RF as described above, precisely replacing the wild-type (WT) copy of murAA. For deletion of pbp5, a derivative of pJRG32 carrying an in-frame deletion of pbp5 (pJLL21) was constructed and transferred to the OG1RF chromosome as described above. The deletion was designed such that the first 10 and last 3 codons of pbp5 remained (98% of the ORF was deleted).
Growth rates and antibiotic susceptibility.
Growth rates were determined from exponentially growing cultures in the wells of a 100-well honeycomb plate at 37°C using a Bioscreen C plate reader (Oy Growth Curves Ab, Ltd.). The reported mean generation times are a result of three independent experiments and were calculated using data derived exclusively from the exponential phase of growth. MICs for antibiotics were determined in aerobic liquid cultures using a microtiter plate serial dilution method in the Bioscreen C plate reader. Twofold dilutions of antibiotics in MHB were prepared in the wells of a 100-well honeycomb microtiter plate. Bacteria from stationary-phase cultures in MHB were normalized to equivalent optical densities at 600 nm (OD600) for a given experiment and subjected to a 20,000-fold dilution upon inoculation into the multiwell plates containing antibiotic dilutions. The final bacterial density of the inocula was therefore consistent for each experiment but varied between experiments in a range from ∼5 × 103 to ∼5 × 104 CFU/ml. This variability in the size of the inocula likely accounts for the slight differences in observed MICs for the wild-type strain between experiments (see Tables 3 and 4), consistent with the known phenomenon of the “inoculum effect” (33). As a control for this, each susceptibility experiment included a wild-type control strain examined in parallel to facilitate relative comparisons. Plates were incubated at 37°C for 24 h with brief shaking and measurement of OD600 at 15-min intervals. The lowest concentration of antibiotic that prevented growth was recorded as the MIC. Plasmid-bearing strains were cultured in the presence of spectinomycin (100 μg/ml) for plasmid maintenance. All experiments were performed a minimum of three times.
Table 3.
Complementation analysis of the ΔmurAA mutant
| Straina | MIC of ceftriaxone (μg/ml)b | MGT (min)c |
|---|---|---|
| WT (vector) | 4 | 128 ± 3 |
| WT (P-murAA) | 16 | 133 ± 5 |
| ΔmurAA2 (vector) | 0.5 | 170 ± 3 |
| ΔmurAA2 (P-murAA) class I | 16 | 129 ± 1 |
| ΔmurAA2 (P-murAA) class II | >128 | 126 ± 2 |
Strains were WT (OG1RF) and ΔmurAA2 (DV75-2) mutant. Plasmids were vector (pDL278p23) and P-murAA (pDV27-4).
Determined in MHB supplemented with Sp (100 μg/ml) after 24 h of incubation at 37°C from a minimum of 3 independent experiments.
MGT, mean generation time, determined in MHB supplemented with Sp (100 μg/ml) incubated at 37°C, from a minimum of two experiments.
Table 4.
Median MICs
| Straina | MIC (μg/ml) of ceftriaxoneb |
|---|---|
| WT (vector) | 8 |
| ΔireK (vector) | 2 |
| ΔireK (P-murAA) | 16 |
| ΔireK (P-murAA C120S) | 4 |
| ΔireK (P-EcmurA) | 4 |
| croR::Tn (vector) | 4 |
| croR::Tn (P-murAA) | 4 |
| Δpbp5 (vector) | 1 |
| Δpbp5 (P-murAA) | 1 |
Strains were WT (OG1RF), ΔireK mutant (CK119), croR::Tn strain (CK128), and Δpbp5 mutant (JL104). Plasmids were vector, pDL278p23, P-murAA C120S (pDV15-1), P-EcmurA (pJMK4), and P-murAA (pDV27-4).
MIC determined in MHB supplemented with Sp (100 μg/ml) after 24 h of incubation at 37°C from a minimum of three independent experiments.
Checkerboard susceptibility assays to assess combinations of antimicrobials were performed in MHB at 37°C as previously described (12, 33), except in the wells of a 100-well honeycomb plate. The fractional inhibitory concentration (FIC) indices were calculated according to the following formula: FIC index = FICA + FICB, where FICA = [MIC of drug A in combination]/[MIC of drug A alone] and FICB = [MIC of drug B in combination]/[MIC of drug B alone]. Conservative interpretation of the FIC index has traditionally defined synergism as an FIC index of ≤0.5.
Antibiotic susceptibility on agar plates was evaluated in the following manner. For plasmid-free strains, overnight cultures grown in MHB were subjected to serial 10-fold dilutions in fresh MHB, and 4 μl of each dilution was inoculated onto Mueller-Hinton (MH) agar plates supplemented or not with ceftriaxone (1 μg/ml) or ceftazidime (7.5 μg/ml) and incubated at 37°C overnight. For plasmid-bearing strains, overnight cultures were grown in BHI (or MHB) supplemented with spectinomycin (for plasmid maintenance), diluted, and plated as above onto plates amended with spectinomycin. Plates were incubated at 37°C, overnight.
Overexpression and purification of E. faecalis MurAA and MurAA C120S.
Overnight cultures of E. coli BL21(DE3)(pDV20-1) and BL21(DE3)(pDV44-1) grown at 37°C in LB supplemented with 50 μg/ml kanamycin were diluted 50-fold into 200 ml of the same medium and incubated for 3 h at 37°C with shaking (250 rpm). Protein expression was induced with 0.5 mM IPTG (isopropyl-β-d-thiogalactopyranoside; Gold Biotechnology) for 1 h at 30°C. Bacteria were harvested by centrifugation (10,816 × g, 8 min, 4°C), and cells were suspended in 10 ml binding buffer (50 mM Tris, pH 8, 300 mM NaCl, 5 mM imidazole, pH 8). Cell suspensions were treated with lysozyme (1 mg/ml) in lysozyme buffer (10 mM Tris, pH 8, 50 mM NaCl, 10 mM EDTA, pH 8) for 20 min at 37°C. Cells were disrupted by sonication. Debris was collected by centrifugation (26,892 × g, 15 min at 4°C). The supernatant was filtered through an 0.22-μm filter and subsequently loaded onto a Ni column (Profinity IMAC Ni charged resin; Bio-Rad) equilibrated with the binding buffer. Columns were treated with 5 column volumes of binding buffer and then washed with 5 column volumes of wash buffer (50 mM Tris, pH 8, 300 mM NaCl, 20 mM imidazole, pH 8) and eluted in elution buffer (50 mM Tris, pH 8, 300 mM NaCl, 500 mM imidazole, pH 8). The eluted fractions were analyzed on a 10% SDS-PAGE gel, and fractions containing the protein were pooled. Samples were dialyzed against dialysis buffer (50 mM Tris, pH 8, 150 mM NaCl, 10% glycerol). Protein was mixed 1:1 with 80% glycerol and either used immediately or stored at −80°C.
Preparation of E. faecalis lysates for enzymatic assays.
Cultures growing exponentially in MHB were collected by centrifugation. Cells were suspended in TD buffer (50 mM Tris, pH 7.5, 2 mM dithiothreitol [DTT] [9, 29]) and disrupted by bead beating with 0.1-mm zirconia-silica beads (Biospec Products, Inc.). Cell debris and beads were removed by centrifugation (16,100 × g, 3 min, 4°C). Cell lysates were desalted using Micro Bio-Spin6 chromatography columns (Bio-Rad) equilibrated in TD buffer. Total protein concentration was determined using the Coomassie Plus protein assay reagent (Pierce) with bovine serum albumin (Pierce) as a standard. Lysates were used immediately in the enzyme assay.
Determination of UDP-N-acetylglucosamine 1-carboxyvinyl transferase activity.
UDP-N-acetylglucosamine 1-carboxyvinyl transferase activity was determined by measuring inorganic phosphate (Pi) released during the reaction, as previously described (9, 27, 29). Reaction mixtures (100 μl) contained 20 mM UDP-GlcNAc, 2 mM PEP, TD buffer, and either purified recombinant protein or processed E. faecalis cell lysates as a source of enzyme. In the case of lysates, 50 μg of total protein was used per reaction; for pure proteins, either 1 μg or 5 μg of the pure proteins was used per reaction. UDP-GlcNAc, protein, and TD buffer were preincubated at 37°C for 15 min, after which PEP was added to start the reaction. At desired intervals, aliquots were removed and assayed for Pi content using the Ser/Thr phosphatase assay system (Promega). After addition of molybdate dye mix, color development proceeded for 12 min at room temperature and absorbance was measured at 600 nm using a SpectraMax M5 spectrophotometer (Molecular Devices). Values were corrected for the background reading in the absence of UDP-GlcNAc. Results are expressed as Pi concentration determined using a standard curve generated per the manufacturer's instructions. All of the experiments were performed a minimum of two times.
Assay for cell wall integrity.
Susceptibility of exponentially growing cells to SDS-mediated lysis was monitored as a measure of cell wall integrity (38). Overnight cultures of plasmid-bearing strains were diluted to an OD600 of 0.01 in MHB supplemented with spectinomycin (100 μg/ml) and cultured until OD600 reached 0.2 to 0.3. Bacteria were harvested by centrifugation (10,816 × g, 8 min, 4°C). Cells were resuspended in 50 μl of TE buffer (20 mM Tris, 10 mM EDTA, pH 8) and normalized to equivalent OD600 (control experiments established that total viable CFU were equivalent among the strains at identical OD600 under these conditions). Cell suspensions were split into two equal aliquots, one of which was subsequently treated with lysozyme (5 mg/ml). All samples were incubated for 30 min at 37°C. Laemmli SDS buffer was added, and samples were subjected to SDS-PAGE followed by staining for total protein using GelCode blue stain reagent (Pierce) according to the manufacturer's instructions.
Immunoblot analysis.
Overnight cultures of plasmid-bearing strains were diluted to an OD600 of 0.01 in BHI supplemented with spectinomycin (100 μg/ml) and cultured until OD600 reached 0.5 to 0.6. Bacteria were harvested by centrifugation (10,816 × g, 8 min, 4°C). Cells were resuspended in TE buffer (20 mM Tris, 10 mM EDTA, pH 8), normalized to equivalent OD600, treated with lysozyme (5 mg/ml) for 30 min at 37°C, mixed with Laemmli SDS sample buffer, and subjected to SDS-PAGE. After electrophoresis, the proteins were transferred onto a polyvinylidene difluoride (PVDF) membrane (Bio-Rad), blocked with 5% milk in Tris-buffered saline (TBS) for 1 h at room temperature, and probed with rabbit anti-Strep tag antibody (catalog no. A00626; Genscript), rabbit anti-kinase antibody (dilution 1/10,000 in TBS-0.1% Tween 20) (23), or anti-sigma-70 monoclonal antibody 2G10 (catalog no. AB12088; Abcam). Detection was performed with goat anti-rabbit or anti-mouse secondary antibodies conjugated to horseradish peroxidase (Invitrogen). When a membrane was probed sequentially with multiple antibodies, membranes were stripped in between using Restore PLUS Western blot stripping buffer (Pierce) for 30 min at room temperature and blocked with 5% milk in TBS.
RESULTS
To probe the genetic basis for intrinsic cephalosporin resistance in E. faecalis, we screened ∼8,000 transposon mutants (24) in wild-type E. faecalis OG1RF to identify isolates exhibiting reduced resistance toward the expanded-spectrum cephalosporin ceftriaxone. This initial screen yielded a surprisingly small number of authentic ceftriaxone-susceptible mutants (6 total). The reason for this low yield is unknown, but we suspect the particular environmental conditions used for the screen were not ideal. In any case, genomic DNA was isolated from the ceftriaxone-susceptible mutants and subjected to DNA sequencing using a transposon-specific primer, revealing transposon insertions in 3 genetic loci. We obtained multiple insertions in croR and pbp5, which was expected given their known roles in cephalosporin resistance. The third locus to carry a transposon insertion was EF2605 (strain 39G11). EF2605 is a putative UDP-N-acetylglucosamine 1-carboxyvinyl transferase (annotated MurAA in the E. faecalis V583 genome), predicted to catalyze the first committed step in peptidoglycan synthesis. As with other low-GC Gram-positive bacteria, the genome of E. faecalis carries two homologs of murA, annotated as murAA (EF2605) and murAB (EF1169), whose gene products exhibit ∼45% identity to each other. The results of our transposon screen prompted us to examine the role of E. faecalis murAA, and its homolog murAB, in more detail.
The ΔmurAA2 mutant is susceptible to cephalosporins.
To probe the contribution of MurAA and MurAB to cephalosporin resistance, markerless, in-frame deletions of murAA and murAB were constructed in an otherwise wild-type E. faecalis strain (OG1RF). We assessed the susceptibilities of the original transposon mutant and the in-frame deletion mutants to two expanded-spectrum cephalosporins (ceftriaxone and ceftazidime) on MH agar plates (see Fig. S1A in the supplemental material). Deletion of the murAA gene led to an increased susceptibility of the mutant to both ceftriaxone and ceftazidime, similar to that observed with the original transposon mutant, whereas the ΔmurAB2 mutant was indistinguishable from the wild type. Measurement of MICs for the mutants confirmed the agar plate susceptibility results (Table 2), revealing that whereas the ΔmurAB2 mutant was just as resistant as the wild type, the ΔmurAA2 mutant was 16-fold more susceptible than the wild type to both ceftriaxone and ceftazidime. Complementation analysis was performed by introducing a plasmid-borne allele of murAA into the ΔmurAA2 mutant. After electroporation of the expression vector, we obtained 2 classes of plasmid-bearing strains: class I exhibited levels of cephalosporin resistance equivalent to those of the wild type carrying the expression vector, while class II exhibited a cephalosporin hyperresistance phenotype (Table 3). This phenomenon was observed only upon introduction of the murAA expression vector into the ΔmurAA2 mutant (i.e., we did not observe 2 classes of transformants upon introduction of the expression plasmid into the wild type), and we suspect that the electroporation or outgrowth process may have inadvertently led to the selection of spontaneous mutants in this genetic background that resulted in the aberrantly high level of cephalosporin resistance observed in class II. The class II mutants were not investigated further. In any case, while detailed analysis of such mutants is beyond the scope of this work, these results confirm that the cephalosporin resistance defect observed for the ΔmurAA2 mutant is indeed due to the absence of the murAA gene. Overall, the susceptibility results indicate that MurAA, but not MurAB, plays an important role in cephalosporin resistance of E. faecalis.
Table 2.
Median MICs for ΔmurAA and ΔmurAB mutants
| Antibiotic | MIC (μg/ml)a |
||
|---|---|---|---|
| WT (OG1RF) | ΔmurAA2 (DV75-2) | ΔmurAB2 (DV1-4) | |
| Cell wall targets | |||
| Ceftriaxone | 16 | 1 | 16 |
| Ceftazidime | 64 | 4 | 64 |
| d-Cycloserine | 128 | 128 | 128 |
| Bacitracin | 64 | 32 | 64 |
| Vancomycin | 2 | 2 | 2 |
| Ampicillin | 1 | 0.5 | 1 |
| Fosfomycin | 64 | 8 | 64 |
| Other targets | |||
| Norfloxacin | 4 | 4 | 4 |
| Tetracycline | 0.25 | 0.25 | 0.25 |
| Erythromycin | 0.5 | 0.5 | 0.5 |
| Chloramphenicol | 4 | 4 | 4 |
| Kanamycin | 128 | 128 | 128 |
Determined in MHB after 24 h of incubation at 37°C from a minimum of three independent experiments.
Enhanced susceptibility of the ΔmurAA2 mutant is specific to a subset of cell wall-active antibiotics.
Analyses of growth kinetics of the mutants (see Fig. S2 in the supplemental material) revealed that two independently derived ΔmurAA2 mutants exhibit a growth defect relative to the otherwise isogenic wild type. Complementation of the ΔmurAA2 mutation restored the growth rate to that of the wild type (Table 3). Given the growth defect of the ΔmurAA2 mutant, we sought to determine if its enhanced cephalosporin susceptibility was a general trait or was specific to cephalosporins. To do so, we measured MICs for antibiotics with a variety of distinct cellular targets (Table 2). The ΔmurAB2 mutant exhibited no differences in susceptibility from the wild type toward any antibiotic tested. Similarly, no differences in susceptibility were observed for the ΔmurAA2 mutant toward antibiotics targeting protein synthesis or DNA gyrase. In contrast, the ΔmurAA2 mutant exhibited altered susceptibility to some cell wall-active antibiotics, but not others (Table 2). In particular, the ΔmurAA2 mutant retained essentially wild-type resistance to d-cycloserine, vancomycin, bacitracin, and ampicillin but was substantially more susceptible to fosfomycin and cephalosporins. The enhanced susceptibility of the E. faecalis ΔmurAA2 mutant toward fosfomycin was not surprising, as MurA is a known target for fosfomycin. In both S. aureus and B. anthracis, inactivation (or depletion) of murA homologs results in enhanced susceptibility to fosfomycin (4, 19). Collectively, these results indicate that the ΔmurAA2 mutant is not sensitized to antibiotics in general—or even to all antibiotics that inhibit cell wall biosynthesis—but that MurAA has a specific role in promoting resistance to cephalosporins.
To test this hypothesis pharmacologically, we adopted a chemical genetics approach to probe the involvement of MurAA using a small-molecule inhibitor of MurA activity, fosfomycin, which we hypothesized would enhance cephalosporin activity against E. faecalis (i.e., act in a synergistic manner with cephalosporins). Checkerboard susceptibility assays were performed to probe for evidence of synergism between fosfomycin and ceftriaxone, and fractional inhibitory concentration (FIC) indices were determined as previously described (12, 33), where an FIC index of ≤0.5 is traditionally interpreted as strong evidence of synergism between two compounds. Consistent with the results from our genetic studies, the combination of ceftriaxone and fosfomycin exhibited synergism against wild-type E. faecalis (FIC = 0.375), indicating that fosfomycin does indeed enhance the activity of a cephalosporin. No synergism was observed with the combination of ceftriaxone and chloramphenicol (FIC = 0.75), an antibiotic that targets ribosomal function (an FIC index between 0.5 and 2 is traditionally considered to represent an additive or indifferent effect). Furthermore, the synergistic effect of fosfomycin with ceftriaxone was eliminated in the ΔmurAA2 mutant (FIC = 0.75), a strain lacking the presumed primary cellular target for fosfomycin. As a further test for synergy of fosfomycin and ceftriaxone, we performed combination time-kill studies to probe survival of E. faecalis after treatment with antibiotics (Fig. 1). The results revealed that neither fosfomycin nor ceftriaxone alone inhibited growth, but the combination led to a rapid and substantial decline in viability. Collectively, these results indicate that pharmacological inhibition of MurAA activity in wild-type E. faecalis with a small-molecule probe compromises cephalosporin resistance, just as deletion of the gene encoding MurAA does, supporting the hypothesis that MurAA has a specific role in promoting resistance to cephalosporins.
Fig 1.
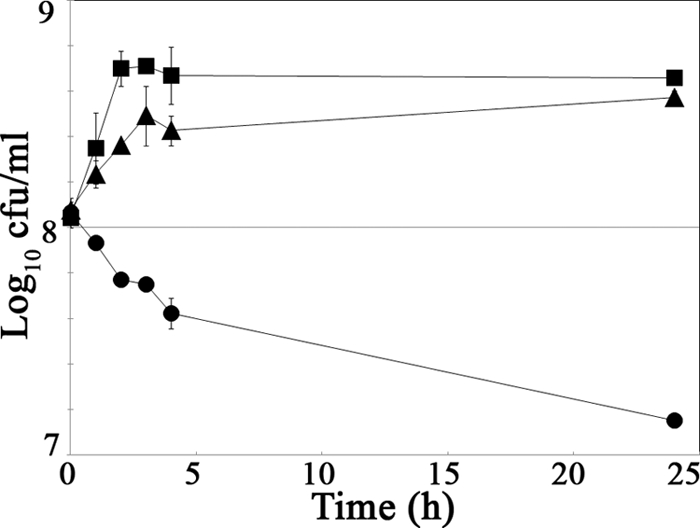
Time-kill analysis reveals that fosfomycin and ceftriaxone act synergistically. An overnight culture of WT (OG1RF) was diluted to an OD600 of 0.004 and grown in MHB at 37°C with shaking until OD600 reached 0.13 to 0.14. Three aliquots were removed and treated with a corresponding antibiotic. Samples were removed at intervals, and viable bacteria were enumerated on MH agar. Antibiotics and concentrations used: 32 μg/ml fosfomycin (squares); 512 μg/ml ceftriaxone (triangles); combination of 32 μg/ml fosfomycin and 512 μg/ml ceftriaxone (circles). Data represent the geometric means ± standard errors from independent experiments.
To test if this role for MurAA is unique to E. faecalis, we performed checkerboard susceptibility assays on two strains of Enterococcus faecium, another species of enterococci that exhibits intrinsic cephalosporin resistance. The combination of ceftriaxone and fosfomycin exhibited synergism on both strains of E. faecium (FIC = 0.375 for E. faecium Com12; FIC = 0.188 for E. faecium 1,231,501), suggesting that MurAA of E. faecium promotes cephalosporin resistance in this species as well.
Integrity of the ΔmurAA2 mutant cell wall is compromised.
Because MurAA was predicted to play a key role in peptidoglycan synthesis and the ΔmurAA2 mutant exhibited a growth defect, we hypothesized that loss of MurAA might result in a defect in cell wall integrity. As a simple test for this, we assessed susceptibility of intact bacteria to lysis by SDS (38). Exponentially growing bacteria were harvested and either left intact or subjected to treatment with lysozyme (to digest peptidoglycan) prior to exposure to 2% SDS in Laemmli SDS loading buffer. Samples were boiled and subjected to SDS-PAGE to assess protein release as a measure of the efficiency of lysis (Fig. 2). As expected, all lysozyme-treated samples were efficiently lysed due to lysozyme-mediated digestion of peptidoglycan. Very little, if any, lysis was detected with wild-type or ΔmurAB2 cells that were not treated with lysozyme, indicating that the cell wall of these strains was predominantly intact. The ΔmurAA2 mutant underwent substantial lysis without lysozyme pretreatment, suggesting that the integrity of its cell wall is compromised.
Fig 2.
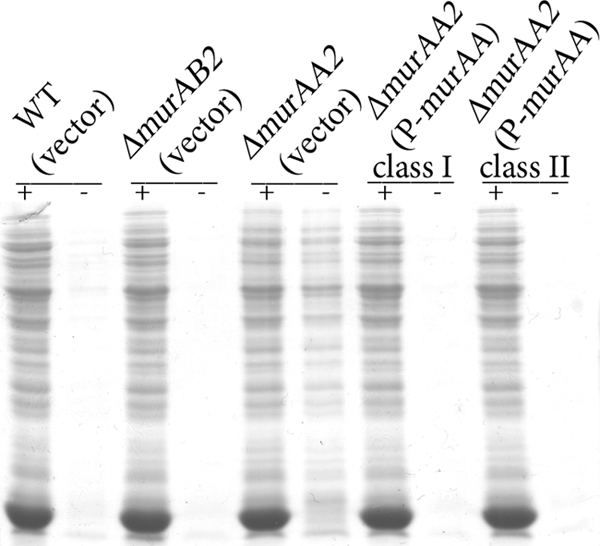
Cell wall integrity is compromised in the ΔmurAA2 mutant. Bacteria from exponentially growing cultures of plasmid-bearing WT (OG1RF), ΔmurAA2 (DV75-2), and ΔmurAB2 (DV1-4) E. faecalis were collected and split. Aliquots were pretreated with or without lysozyme, as indicated, prior to addition of Laemmli buffer containing 2% SDS. Samples were subjected to SDS-PAGE, and total protein stain was used to assess the extent of lysis. Vector, pDL278p23; P-murAA, pDV27-4.
Expression of murAA enhances cephalosporin resistance in the E. faecalis ΔireK kinase mutant.
The susceptibility results above suggest that MurAA acts to promote cephalosporin resistance in E. faecalis. To probe the relationship of MurAA to other known determinants that are critical for cephalosporin resistance, we introduced a murAA expression plasmid encoding an epitope-tagged MurAA into several cephalosporin-susceptible mutants to determine if murAA expression could alter their susceptibility. The susceptible mutants chosen for analysis each contained a single lesion in one of the 3 determinants previously established to be critical for cephalosporin resistance: ireK, pbp5, or croR. Immunoblot analysis confirmed that MurAA was expressed at comparable levels in all mutant backgrounds (see Fig. S3b in the supplemental material). Antibiotic susceptibility tests revealed that MurAA production substantially enhanced ceftriaxone resistance of the ΔireK mutant but had essentially no effect on that of the croR or Δpbp5 mutant (Table 4; see also Fig. S3a). Thus, MurAA provides a function that is capable of overcoming the cephalosporin susceptibility defect of a strain specifically lacking the IreK kinase, suggesting that MurAA may act downstream of IreK in a pathway leading to cephalosporin resistance.
One hypothesis to explain MurAA-mediated enhancement of resistance in the ΔireK mutant is that the ΔireK mutant does not synthesize MurAA (from its normal chromosomal locus) at wild-type levels. To test this, a murAA allele encoding a C-terminal Strep tag was introduced into the chromosome of both wild-type E. faecalis and the ΔireK mutant, using markerless allelic exchange to replace the wild-type murAA allele such that the epitope-tagged allele could be expressed using its natural regulatory elements. Expression of Strep-tagged MurAA was assessed via immunoblotting, revealing that the wild type (WT) and the ΔireK mutant express MurAA at comparable levels (Fig. 3). Thus, cephalosporin susceptibility of the ΔireK mutant is not due to the absence of MurAA.
Fig 3.
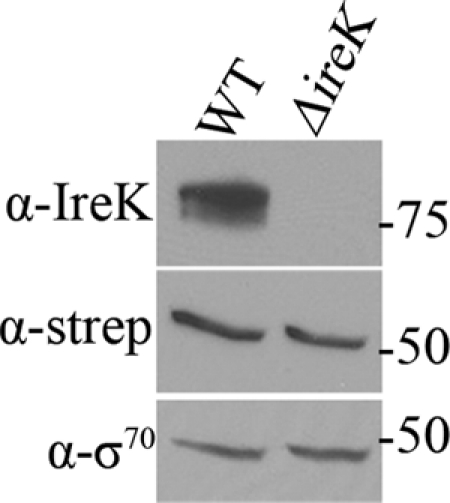
MurAA is present in the ΔireK mutant. Cultures of WT (CK153) and ΔireK (CK161) mutant with chromosomally borne Strep-tagged murAA alleles were grown in BHI. Whole-cell lysates were prepared and subjected to immunoblot analysis. Membranes were probed with IreK (α-IreK), Strep tag (α-strep), and sigma-70 (α-σ70) antibodies.
Catalytic activity of MurAA is necessary but not sufficient to enhance cephalosporin resistance in the E. faecalis ΔireK kinase mutant.
Based on sequence homology, E. faecalis MurAA is predicted to exhibit UDP-N-acetylglucosamine 1-carboxyvinyl transferase activity. To test this experimentally, in vitro enzyme assays were performed using recombinant E. faecalis MurAA-His6 purified from E. coli. PEP-dependent conversion of UDP-GlcNAc to UDP-GlcNAc-EP was followed by measuring the inorganic phosphate released using a malachite green-based method (Materials and Methods), confirming that MurAA is indeed a UDP-N-acetylglucosamine 1-carboxyvinyl transferase (Fig. 4). To ensure that the observed activity was due to MurAA and not an undetected copurifying protein, we used an identical approach to purify and test a mutant MurAA-His6 bearing a C120S substitution. The MurAA C120S mutant was predicted to be catalytically inactive based on sequence comparison with the Enterobacter cloacae MurA, for which it was previously established that a Cys-to-Ser alteration at the equivalent site (Cys115 in E. cloacae MurA) abolishes activity (43). No activity was observed with purified E. faecalis MurAA C120S-His6, as expected.
Fig 4.
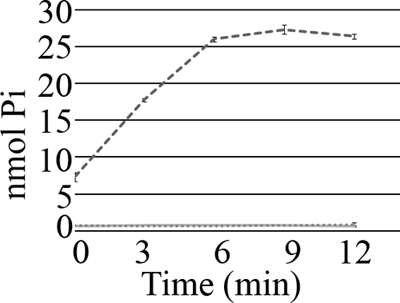
Purified E. faecalis MurAA, but not MurAA C120S, is catalytically active. His6-tagged E. faecalis MurAA and MurAA C120S were purified from E. coli BL21(DE3) cells and assayed for UDP-N-acetylglucosamine 1-carboxyvinyl transferase activity. One microgram of freshly purified MurAA (dashed line) was used in the assay. No activity was observed with either 1 μg (full line) or 5 μg (spotted line) of freshly purified MurAA C120S.
To test if catalytic activity of MurAA was required for its ability to enhance cephalosporin resistance of the ΔireK kinase mutant, we constructed the C120S mutation in our murAA expression plasmid and introduced the resulting vector into the ΔireK kinase mutant. Although MurAA C120S was produced at levels comparable to wild-type MurAA (see Fig. S4b in the supplemental material), susceptibility tests revealed that MurAA C120S essentially could not enhance cephalosporin resistance (Table 4; see also Fig. S4a), suggesting that MurAA catalytic activity is indeed required for this property. To determine if the UDP-N-acetylglucosamine 1-carboxyvinyl transferase activity of MurAA is sufficient to enhance cephalosporin resistance of the ΔireK kinase mutant, we engineered a plasmid to express E. coli murA in E. faecalis cells. Our rationale was that while E. coli MurA exhibits UDP-N-acetylglucosamine 1-carboxyvinyl transferase activity, it is likely sufficiently divergent from E. faecalis MurAA (∼47% identical) that any additional (unknown) MurAA-specific functions or IreK-dependent regulation would be absent (especially because E. coli lacks a homolog of IreK). Although E. coli MurA was expressed at levels comparable to those of E. faecalis MurAA from our expression plasmid (see Fig. S4b), susceptibility tests revealed that E. coli MurA was also unable to enhance cephalosporin resistance (Table 4; see also Fig. S4a).
To confirm that E. coli MurA was catalytically active in E. faecalis, enzymatic assays were performed on lysates of plasmid-containing strains to measure UDP-N-acetylglucosamine 1-carboxyvinyl transferase activity. Note that we were unable to detect activity from the chromosomally expressed E. faecalis MurAA under our assay conditions. We found that E. coli MurA exhibited substantial activity in E. faecalis lysates (more than E. faecalis MurAA, in fact) (Fig. 5) but was nevertheless unable to enhance cephalosporin resistance of the ΔireK mutant. Collectively, these results indicate that UDP-N-acetylglucosamine 1-carboxyvinyl transferase activity of MurAA is required but not sufficient for its role in cephalosporin resistance, suggesting that a unique property or activity of MurAA contributes to its role in cephalosporin resistance.
Fig 5.
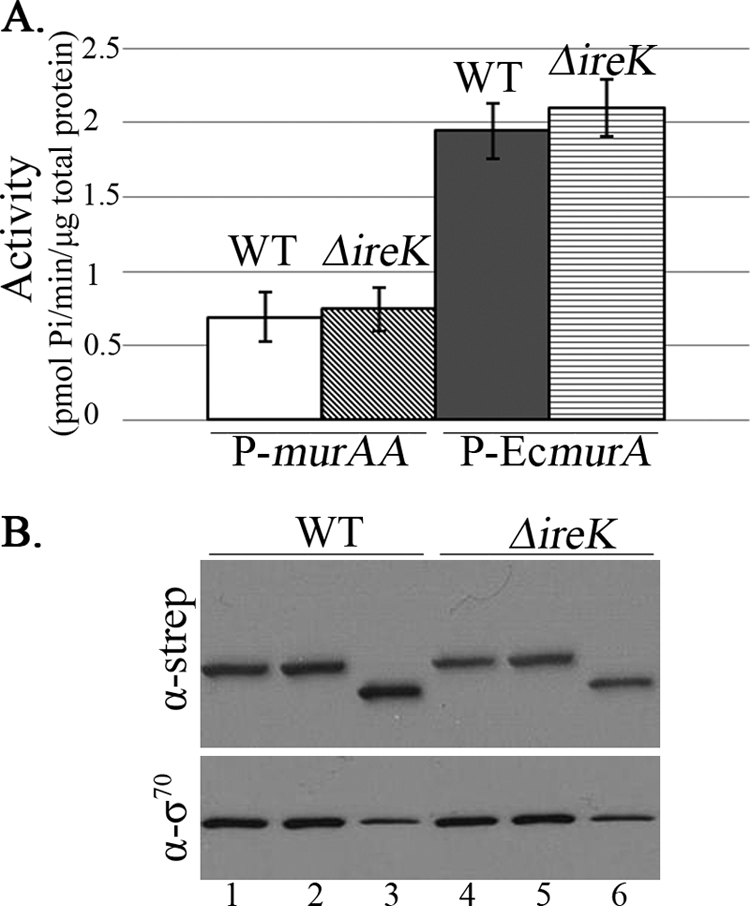
Both E. faecalis MurAA and E. coli MurA are catalytically active in E. faecalis lysates. (A) Assay for UDP-N-acetylglucosamine 1-carboxyvinyl transferase activity. Cultures of plasmid-bearing WT (OG1RF) and ΔireK (CK119) E. faecalis strains were grown in MHB supplemented with spectinomycin (100 μg/ml). Whole-cell lysates were assayed for UDP-N-acetylglucosamine 1-carboxyvinyl transferase activity. Data represent the means ± standard errors. Activity was not detected in lysates expressing murAA C120S. (B) Immunoblot analysis. Aliquots were removed from each enzyme reaction and subjected to immunoblot analysis with anti-Strep tag (α-strep; for epitope-tagged MurAA or EcMurA) and anti-sigma-70 (α-σ70; loading control) antibodies. Lanes 1 and 4, strains carrying plasmid that expresses E. faecalis MurAA (pDV27-4); lanes 2 and 5, strains carrying plasmid that expresses MurAA C120S (pDV15-1); lanes 3 and 6, strains carrying plasmid that expresses the E. coli MurA (pJMK4).
DISCUSSION
The goal of this work was to gain a better understanding of the underlying mechanisms of intrinsic cephalosporin resistance in E. faecalis. Experiments here demonstrate three key findings. The first is that the two murA homologs carried in the E. faecalis genome are not entirely functionally redundant. We were able to construct deletion mutants individually lacking either murAA or murAB, suggesting that each is capable of providing some UDP-N-acetylglucosamine 1-carboxyvinyl transferase activity to support peptidoglycan synthesis; however, numerous other phenotypic differences between the mutants argue that the 2 murA homologs possess divergent functions. In particular, the ΔmurAA mutant exhibited a pronounced growth defect, a reduction in cell wall integrity, and enhanced susceptibility toward a subset of antibiotics that the ΔmurAB mutant did not share. Indeed, we were unable to identify a phenotypic defect of the ΔmurAB mutant under any condition that we evaluated. These results argue that MurAA plays a key role in the underlying physiology of E. faecalis that MurAB cannot provide. Despite several attempts, we were unable to construct a double mutant lacking both murAA and murAB, suggesting that MurAB is expressed and can indeed provide some UDP-N-acetylglucosamine 1-carboxyvinyl transferase activity in the absence of MurAA, albeit at an apparently significantly reduced level compared to MurAA (inferred from the growth defect of the ΔmurAA mutant). Thus, although both MurAA and MurAB can provide sufficient UDP-N-acetylglucosamine 1-carboxyvinyl transferase activity to support viability, they have clearly evolved to serve different physiological roles in E. faecalis. In both B. anthracis and S. aureus, the two MurA homologs encoded in the genome also appear to possess distinct physiological roles, as in both organisms mutation (or depletion) of one homolog (murA in S. aureus and murA1 in B. anthracis) leads to defects in growth while deletion of the second homolog has less of an effect (4, 19). In the absence of any detectable phenotype of the E. faecalis ΔmurAB mutant, the physiological role of MurAB in E. faecalis remains a mystery.
The second major finding of this work is that murAA is a specific determinant of cephalosporin resistance in E. faecalis. Deletion of murAA in E. faecalis led to a 16-fold reduction in MIC for both cephalosporins tested. Our data argue that the enhanced cephalosporin susceptibility of the ΔmurAA mutant is not simply the result of a general defect in growth or tolerance to stress, as no differences in susceptibility were observed toward antibiotics that have non-cell wall targets (i.e., DNA gyrase or the ribosome). Similarly, although the integrity of the cell wall of the ΔmurAA mutant appears to be impaired, this does not result in a general enhancement of susceptibility to antibiotics that target the cell wall, as the ΔmurAA mutant retains essentially wild-type resistance to several antibiotics with cell wall targets (d-cycloserine, bacitracin, and vancomycin), including another antibiotic in the β-lactam family (ampicillin). Instead, the susceptibility data collectively argue that murAA is specifically required for intrinsic resistance of E. faecalis toward cephalosporins. Chemical genetics experiments demonstrating MurAA-dependent synergy between ceftriaxone and the MurAA-inactivating small molecule fosfomycin are consistent with this model. In addition, our synergism studies suggest that this role for MurAA is not restricted to E. faecalis but that MurAA of E. faecium possesses a similar property. Of note, synergism between fosfomycin and a β-lactam antibiotic has been described for methicillin-resistant S. aureus (34, 42), and genetic depletion of murA1 renders B. anthracis hypersusceptible to β-lactams as well (19). Whether these phenomena are related in a mechanistic way to MurAA-enhanced cephalosporin resistance in E. faecalis is unknown, particularly in light of the observation that deletion of E. faecalis murAA does not dramatically affect susceptibility to another β-lactam (ampicillin). It is worth noting that a recent study of clinical isolates of E. faecalis reported a synergistic effect of fosfomycin with ceftriaxone (13), consistent with our results. Further studies are required to elucidate the mechanism(s) underlying this phenomenon.
The third key finding is that the catalytic activity of MurAA is necessary but not sufficient for enhancement of cephalosporin resistance. We determined that, as expected, E. faecalis MurAA possesses UDP-N-acetylglucosamine 1-carboxyvinyl transferase activity and further that a MurAA mutant bearing a C120S substitution was catalytically inactive (Fig. 4). Overexpression of wild-type murAA enhanced cephalosporin resistance of the ΔireK mutant, but overexpression of the catalytically inactive murAA was unable to do so, arguing that MurAA catalytic activity is required for this effect. However, the inability of E. coli murA to enhance cephalosporin resistance in the ΔireK mutant—despite providing high levels of UDP-N-acetylglucosamine 1-carboxyvinyl transferase activity—supports the hypothesis that E. faecalis MurAA has additional properties, in addition to its basic catalytic ability, that enable it to promote resistance to cephalosporins. We note that immunoblot analyses established that expression levels of the various MurAA constructs were comparable, so differences in protein abundance do not account for the substantial differences in cephalosporin resistance in any obvious way. The nature of the proposed additional MurAA activity is unknown, but conceivably it could be a regulatory function that modulates other aspects of the signaling network to control cephalosporin resistance in E. faecalis. The fact that MurAA was able to enhance resistance of the ΔireK mutant, but not that of the croR or pbp5 mutants, suggests that MurAA provides a function that is specifically lacking in the ΔireK mutant and may therefore be downstream of IreK in a pathway leading to intrinsic cephalosporin resistance.
These findings raise an unresolved question: if MurAA can enhance cephalosporin resistance of the ΔireK mutant, why doesn't expression of endogenous murAA in the ΔireK mutant (Fig. 3) confer resistance? We suggest three speculative models that could explain why plasmid-based overproduction of MurAA enhances cephalosporin resistance of the ΔireK mutant. All three models require that MurAA possess an additional (as-yet-unknown) activity that is regulated by IreK and is distinct from its UDP-N-acetylglucosamine 1-carboxyvinyl transferase activity, in order to account for the observation that expression of E. coli murA (and the high level of UDP-N-acetylglucosamine 1-carboxyvinyl transferase activity that it provides) cannot promote cephalosporin resistance in the ΔireK mutant. In the discussion below, “activity” refers to this hypothetical new function, not the known UDP-N-acetylglucosamine 1-carboxyvinyl transferase activity.
The first possibility is that MurAA itself requires an IreK-dependent modification (for example, phosphorylation) to control the proposed new activity. Two other enzymes of the peptidoglycan biosynthesis pathway are known to be phosphorylated on Thr residues (MurC of Corynebacterium glutamicum [14] and MurD of Mycobacterium smegmatis [40]), and phosphorylation of C. glutamicum MurC affects its catalytic activity (14). In this scenario, E. faecalis MurAA might only be partially “active” in the absence of IreK and unable to promote resistance. Upon MurAA overexpression in the ΔireK mutant, although the specific activity of MurAA would remain low due to the absence of IreK-dependent modification, there may be a sufficient amount of partially active MurAA in the cell to meet some minimum requirement for cephalosporin resistance. We attempted to determine if IreK could phosphorylate MurAA using in vitro kinase reactions with purified components but did not observe any evidence for phosphorylation under our conditions.
A second possibility is that enhancement of resistance upon overexpression of MurAA could be explained by the presence of an IreK-regulated inhibitor of MurAA activity. In this scenario, the inhibitory factor might interfere with MurAA function (perhaps by forming a protein-protein complex with MurAA) in the absence of IreK, preventing MurAA from promoting resistance. Upon overexpression of MurAA, excess MurAA could titrate the inhibitory factor, freeing up some MurAA molecules to exert their function and promote cephalosporin resistance. Phage-encoded protein inhibitors of E. coli MurA have been identified (3), suggesting that the strategy of using protein inhibitors of MurAA would be a reasonable means of controlling its biological functions.
A third possibility is that MurAA may participate in protein-protein interactions to form multiprotein complexes or to enable appropriate subcellular localization, possibly with other members of the peptidoglycan biosynthetic pathway, and that proper complex formation and/or localization is required to promote cephalosporin resistance. If IreK-dependent modification (of MurAA or other complex members) is normally required for proper complex formation and/or localization, then overexpression of MurAA may promote complex formation (and hence cephalosporin resistance) despite the lack of IreK regulation. Additional studies are necessary to test these models and unravel the role of MurAA in the pathway leading to cephalosporin resistance of E. faecalis.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by program development funds from the MCW Department of Microbiology and Molecular Genetics, by the Advancing a Healthier Wisconsin program, and by grant AI081692 from NIH (to C.J.K.).
We are grateful to Vy Nguyen for assistance with screening of the transposon library and to Gary Dunny for logistical support, helpful advice, and critical review of the manuscript. We thank Jamie Genthe and Joe Kulinski for plasmid construction, Jaime Little for construction of E. faecalis strain JL104, and anonymous reviewers for helpful comments on the manuscript.
Footnotes
Published ahead of print 30 January 2012
Supplemental material for this article may be found at http://aac.asm.org/.
REFERENCES
- 1. Aarestrup FM, Butaye P, Witte W. 2002. Nonhuman reservoirs of enterococci, p 55–99 In Gilmore MS, et al. (ed), The enterococci: pathogenesis, molecular biology, and antibiotic resistance. ASM Press, Washington, DC [Google Scholar]
- 2. Barreteau H, et al. 2008. Cytoplasmic steps of peptidoglycan biosynthesis. FEMS Microbiol. Rev. 32:168–207 [DOI] [PubMed] [Google Scholar]
- 3. Bernhardt TG, Wang IN, Struck DK, Young R. 2001. A protein antibiotic in the phage Qbeta virion: diversity in lysis targets. Science 292:2326–2329 [DOI] [PubMed] [Google Scholar]
- 4. Blake KL, et al. 2009. The nature of Staphylococcus aureus MurA and MurZ and approaches for detection of peptidoglycan biosynthesis inhibitors. Mol. Microbiol. 72:335–343 [DOI] [PubMed] [Google Scholar]
- 5. Brown ED, Vivas EI, Walsh CT, Kolter R. 1995. MurA (MurZ), the enzyme that catalyzes the first committed step in peptidoglycan biosynthesis, is essential in Escherichia coli. J. Bacteriol. 177:4194–4197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Canepari P, Lleo MM, Cornaglia G, Fontana R, Satta G. 1986. In Streptococcus faecium penicillin-binding protein 5 alone is sufficient for growth at sub-maximal but not at maximal rate. J. Gen. Microbiol. 132:625–631 [DOI] [PubMed] [Google Scholar]
- 7. Chen Y, Staddon JH, Dunny GM. 2007. Specificity determinants of conjugative DNA processing in the Enterococcus faecalis plasmid pCF10 and the Lactococcus lactis plasmid pRS01. Mol. Microbiol. 63:1549–1564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Comenge Y, et al. 2003. The CroRS two-component regulatory system is required for intrinsic beta-lactam resistance in Enterococcus faecalis. J. Bacteriol. 185:7184–7192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. De Smet KA, Kempsell KE, Gallagher A, Duncan K, Young DB. 1999. Alteration of a single amino acid residue reverses fosfomycin resistance of recombinant MurA from Mycobacterium tuberculosis. Microbiology 145:3177–3184 [DOI] [PubMed] [Google Scholar]
- 10. Du W, et al. 2000. Two active forms of UDP-N-acetylglucosamine enolpyruvyl transferase in gram-positive bacteria. J. Bacteriol. 182:4146–4152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dunny GM, Brown BL, Clewell DB. 1978. Induced cell aggregation and mating in Streptococcus faecalis: evidence for a bacterial sex pheromone. Proc. Natl. Acad. Sci. U. S. A. 75:3479–3483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Elion GB, Singer S, Hitchings GH. 1954. Antagonists of nucleic acid derivatives. VIII. Synergism in combinations of biochemically related antimetabolites. J. Biol. Chem. 208:477–488 [PubMed] [Google Scholar]
- 13. Farina C, et al. 2011. In vitro activity effects of twelve antibiotics alone and in association against twenty-seven Enterococcus faecalis strains isolated from Italian patients with infective endocarditis: high in vitro synergistic effect of the association ceftriaxone-fosfomycin. Chemotherapy 57:426–433 [DOI] [PubMed] [Google Scholar]
- 14. Fiuza M, et al. 2008. The MurC ligase essential for peptidoglycan biosynthesis is regulated by the serine/threonine protein kinase PknA in Corynebacterium glutamicum. J. Biol. Chem. 283:36553–36563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hancock LE, Perego M. 2004. Systematic inactivation and phenotypic characterization of two-component signal transduction systems of Enterococcus faecalis V583. J. Bacteriol. 186:7951–7958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hegstad K, Mikalsen T, Coque TM, Werner G, Sundsfjord A. 2010. Mobile genetic elements and their contribution to the emergence of antimicrobial resistant Enterococcus faecalis and Enterococcus faecium. Clin. Microbiol. Infect. 16:541–554 [DOI] [PubMed] [Google Scholar]
- 17. Hidron AI, et al. 2008. NHSN annual update: antimicrobial-resistant pathogens associated with healthcare-associated infections: annual summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2006–2007. Infect. Control Hosp. Epidemiol. 29:996–1011 [DOI] [PubMed] [Google Scholar]
- 18. Kahan FM, Kahan JS, Cassidy PJ, Kropp H. 1974. The mechanism of action of fosfomycin (phosphonomycin). Ann. N. Y. Acad. Sci. 235:364–386 [DOI] [PubMed] [Google Scholar]
- 19. Kedar GC, et al. 2008. Comparison of the essential cellular functions of the two murA genes of Bacillus anthracis. Antimicrob. Agents Chemother. 52:2009–2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kobayashi K, et al. 2003. Essential Bacillus subtilis genes. Proc. Natl. Acad. Sci. U. S. A. 100:4678–4683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kock H, Gerth U, Hecker M. 2004. MurAA, catalysing the first committed step in peptidoglycan biosynthesis, is a target of Clp-dependent proteolysis in Bacillus subtilis. Mol. Microbiol. 51:1087–1102 [DOI] [PubMed] [Google Scholar]
- 22. Kristich CJ, Chandler JR, Dunny GM. 2007. Development of a host-genotype-independent counterselectable marker and a high-frequency conjugative delivery system and their use in genetic analysis of Enterococcus faecalis. Plasmid 57:131–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kristich CJ, Little JL, Hall CL, Hoff JS. 2011. Reciprocal regulation of cephalosporin resistance in Enterococcus faecalis. mBio 2(6):e00199–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kristich CJ, et al. 2008. Development and use of an efficient system for random mariner transposon mutagenesis to identify novel genetic determinants of biofilm formation in the core Enterococcus faecalis genome. Appl. Environ. Microbiol. 74:3377–3386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kristich CJ, Wells CL, Dunny GM. 2007. A eukaryotic-type Ser/Thr kinase in Enterococcus faecalis mediates antimicrobial resistance and intestinal persistence. Proc. Natl. Acad. Sci. U. S. A. 104:3508–3513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Manson JM, Hancock LE, Gilmore MS. 2010. Mechanism of chromosomal transfer of Enterococcus faecalis pathogenicity island, capsule, antimicrobial resistance, and other traits. Proc. Natl. Acad. Sci. U. S. A. 107:12269–12274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Marquardt JL, Siegele DA, Kolter R, Walsh CT. 1992. Cloning and sequencing of Escherichia coli murZ and purification of its product, a UDP-N-acetylglucosamine enolpyruvyl transferase. J. Bacteriol. 174:5748–5752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. McBride SM, Fischetti VA, Leblanc DJ, Moellering RC, Jr, Gilmore MS. 2007. Genetic diversity among Enterococcus faecalis. PLoS One 2:e582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. McCoy AJ, Sandlin RC, Maurelli AT. 2003. In vitro and in vivo functional activity of Chlamydia MurA, a UDP-N-acetylglucosamine enolpyruvyl transferase involved in peptidoglycan synthesis and fosfomycin resistance. J. Bacteriol. 185:1218–1228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Murray BE. 1990. The life and times of the enterococcus. Clin. Microbiol. Rev. 3:46–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Palmer KL, et al. 2010. High-quality draft genome sequences of 28 Enterococcus sp. isolates. J. Bacteriol. 192:2469–2470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Paulsen IT, et al. 2003. Role of mobile DNA in the evolution of vancomycin-resistant Enterococcus faecalis. Science 299:2071–2074 [DOI] [PubMed] [Google Scholar]
- 33. Pillai SK, Moellering RC, Jr, Eliopoulos GM. 2005. Antimicrobial combinations, p 365–440 In Lorian V. (ed), Antibiotics in laboratory medicine. Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 34. Sieradzki K, Tomasz A. 1997. Suppression of beta-lactam antibiotic resistance in a methicillin-resistant Staphylococcus aureus through synergic action of early cell wall inhibitors and some other antibiotics. J. Antimicrob. Chemother. 39(Suppl. A):47–51 [DOI] [PubMed] [Google Scholar]
- 35. Signoretto C, Boaretti M, Canepari P. 1994. Cloning, sequencing and expression in Escherichia coli of the low-affinity penicillin binding protein of Enterococcus faecalis. FEMS Microbiol. Lett. 123:99–106 [DOI] [PubMed] [Google Scholar]
- 36. Skarzynski T, et al. 1996. Structure of UDP-N-acetylglucosamine enolpyruvyl transferase, an enzyme essential for the synthesis of bacterial peptidoglycan, complexed with substrate UDP-N-acetylglucosamine and the drug fosfomycin. Structure 4:1465–1474 [DOI] [PubMed] [Google Scholar]
- 37. Sood S, Malhotra M, Das BK, Kapil A. 2008. Enterococcal infections & antimicrobial resistance. Indian J. Med. Res. 128:111–121 [PubMed] [Google Scholar]
- 38. Szurmant H, Nelson K, Kim EJ, Perego M, Hoch JA. 2005. YycH regulates the activity of the essential YycFG two-component system in Bacillus subtilis. J. Bacteriol. 187:5419–5426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tannock GW, Cook G. 2002. Enterococci as members of the intestinal microflora of humans, p 101–132 In Gilmore MS, et al. (ed), The enterococci: pathogenesis, molecular biology, and antibiotic resistance. ASM Press, Washington, DC [Google Scholar]
- 40. Thakur M, Chakraborti PK. 2008. Ability of PknA, a mycobacterial eukaryotic-type serine/threonine kinase, to transphosphorylate MurD, a ligase involved in the process of peptidoglycan biosynthesis. Biochem. J. 415:27–33 [DOI] [PubMed] [Google Scholar]
- 41. Tomasz A. 1979. The mechanism of the irreversible antimicrobial effects of penicillins: how the beta-lactam antibiotics kill and lyse bacteria. Annu. Rev. Microbiol. 33:113–137 [DOI] [PubMed] [Google Scholar]
- 42. Utsui Y, Ohya S, Magaribuchi T, Tajima M, Yokota T. 1986. Antibacterial activity of cefmetazole alone and in combination with fosfomycin against methicillin- and cephem-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 30:917–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wanke C, Amrhein N. 1993. Evidence that the reaction of the UDP-N-acetylglucosamine 1-carboxyvinyltransferase proceeds through the O-phosphothioketal of pyruvic acid bound to Cys115 of the enzyme. Eur. J. Biochem. 218:861–870 [DOI] [PubMed] [Google Scholar]
- 44. Williamson R, le Bouguenec C, Gutmann L, Horaud T. 1985. One or two low affinity penicillin-binding proteins may be responsible for the range of susceptibility of Enterococcus faecium to benzylpenicillin. J. Gen. Microbiol. 131:1933–1940 [DOI] [PubMed] [Google Scholar]
- 45. Zapun A, Contreras-Martel C, Vernet T. 2008. Penicillin-binding proteins and beta-lactam resistance. FEMS Microbiol. Rev. 32:361–385 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.