

Abstract
Background
Mutations in the leucine-rich repeat kinase 2 gene (LRRK2), located at 12q12, are the most common known genetic causes of Parkinson’s disease (PD). Studies of LRRK2 mutation carriers have shown incomplete and age-dependent penetrance and previous studies have suggested that inherited susceptibility factors may modify the penetrance of LRRK2 mutations.
Methods
Genomewide linkage to age of onset of LRRK2-related PD was evaluated in a sample of 113 LRRK2 mutation carriers from 64 families using single nucleotide polymorphism data from the Illumina HumanCNV370 genotyping array. Association between onset age and SNPs located under suggestive linkage peaks was also evaluated.
Results
The top LOD-score for onset age (LOD-score=2.43) was located in the chromosome 1q32.1 region. Moderate linkage to onset was also identified at 16q12.1 (LOD-score=1.58). Examination of single nucleotide polymorphism association to PD onset under the linkage peaks revealed no statistically significant SNP associations.
Conclusions
The two novel genomic regions identified may harbor modifiers of LRRK2-related PD onset age or penetrance and further study of these regions may provide important insight into LRRK2-related PD.
Keywords: Parkinson’s Disease, LRRK2, Linkage
INTRODUCTION
Parkinson’s disease (PD) is a neurodegenerative disorder affecting approximately 1.8% of individuals over the age of 65 1. While some cases of PD have a known genetic or environmental cause, most appear to be due to complex interactions among unidentified genetic and environmental susceptibility factors. Mutations in the leucine-rich repeat kinase 2 gene (LRRK2) are the most common known genetic cause of PD, with the most frequent LRRK2 mutation, G2019S, estimated to be associated with 5% to 6% of familial PD and 1% to 2% of idiopathic cases in populations of European descent 2-5. However, studies of the G2019S mutation have reported a wide range of penetrance estimates. Early studies performed in large families with multiple affected members reported high lifetime penetrance for LRRK2 mutations, ranging from 70% 6 to 100% 7 and further examination of the age-dependent penetrance in families with multiple affected members ranged from 17-33% penetrance at age 50 increasing to 85-100% at ages above 70 8, 9. However, more recent studies examining cohorts of PD patients not ascertained for familial history of the disease have generally reported both lower lifetime penetrance estimates (22-32%) 10-12 and lower age-dependent estimates (2% at age 50 to 33% at age 80) 13.
While the early familial penetrance estimates may have been upwardly biased due to ascertainment strategies, we have recently shown in an unbiased analysis of unascertained parents of PD affected siblings, that the penetrance of LRRK2 mutations in families with multiple affected members may be substantially higher than in randomly ascertained idiopathic PD cases (67% versus 33% at age 85) 13, 14. This suggests the presence of additional genetic susceptibility factors influencing the risk of LRRK2-related PD.
While younger onset of symptoms is commonly observed with some other PD related genes including PARK1 (SNCA) and PARK2 (parkin) 15-17, PD associated with LRRK2 mutations presents an onset distribution very similar to that seen in idiopathic PD 4, 13, 18. The range in onset age and clear age-dependent penetrance of LRRK2 mutations suggest that genetic variants associated with onset age of LRRK2-related PD may represent a primary mechanism by which penetrance is modified. These genetic modifiers are potential targets not only for treatment, but also for prevention of LRRK2-related PD.
We have therefore undertaken the first genomewide linkage and association studies aimed at identifying modifiers of penetrance in LRRK2-related PD.
METHODS
Ethics Statement
These studies were approved by the Institutional Review Boards of Boston University and Indiana University. Appropriate written informed consent was obtained for all participants included in this study.
Sample
A sample of 113 LRRK2 mutation carriers from 64 families was identified from two ongoing studies of familial PD, the GenePD study and the PROGENI study, and was included in a genomewide association study (GWAS) with a large additional sample of familial PD cases that did not carry any known pathogenic LRRK2 mutations and healthy controls. Most of the LRRK2 mutation carriers were ascertained in pedigrees with multiple members (90 carriers from 41 pedigrees), but 23 singletons with familial PD were also studied. Four of the LRRK2 carriers showed no signs of PD, though they had multiple relatives with PD; the remaining 109 carriers were verified PD cases. PD cases underwent a uniform neurological evaluation that employed PD diagnostic criteria based upon a modified version of the United Kingdom PD Society Brain Bank Criteria 19. Onset age of LRRK2-related PD was determined by interview and reflected the age of first PD symptom, which commonly preceded age of physician diagnosis.
LRRK2 mutations were identified through the genotyping of known mutations (G2019S, R1441C, R1441H, Y1699C, I1192V, L1795F, Q1111H, E334K and I2020T) using TaqMan technology implemented on either the ABI PRISM® 7900HT or the ABI PRISM® 7300 Sequence Detection system 4, 5, 18. The majority of the LRRK2 carriers identified were G2019S carriers (99 carriers from 57 families). In addition we identified 7 families with mutations at either I1192V(1 family, 2 carriers), L1795F (1 family, 2 carriers), Q1111H (1 family, 2 carriers), E334K (1 family, 2 carriers) and R1441C (3 families, 6 carriers).
Microarray Genotyping and Quality Assessment
Genotyping was performed by the Center for Inherited Disease Research (CIDR) using the Illumina HumanCNV370 version1_C BeadChips (Illumina, San Diego, CA, USA) and the Illumina Infinium II assay protocol 20 as part of a larger study which also included 895 control and 935 non-LRRK2 PD cases 21, 22. SNP quality control, cryptic relatedness and population stratification were evaluated in the entire sample of genotyped cases and controls as describe in previous manuscripts 21, 22. One LRRK2-related PD case was removed due to low call rate, leaving 108 LRRK2-related PD cases for study. All samples were self-reported as white, non-hispanic, however, seven LRRK2-related PD cases, from four families, were flagged for non-Caucasian ancestry during population stratification analyses and were removed from subsequent association analyses, but not linkage analysis.
Statistical Methods
In order to identify modifiers of age-dependent penetrance of LRRK2 mutations, linkage and association analyses of PD onset age were conducted. For linkage analyses, 23 singletons, two sib-pairs discordant on LRRK2 carrier status (case-phenocopy pairs) and one pedigree with only parent offspring pairs were excluded, leaving 85 individuals from 38 pedigrees for analysis (Table 1).
Table 1.
Description of linkage and association study samples
| Genotyped Sample | Individuals | Families | PD affected carriers | Non-penetrant | |
|---|---|---|---|---|---|
| total | 113 | 64 | 109 | 4 | |
| In Families | 92 | 41 | 86 | 4 | |
| Singletons | 23 | 23 | 23 | 0 | |
| Analysis Subsets | Individuals | Families | average age (SD) of PD onset or unaffected enrollment | %Male | |
| Linkage | Families | 85 | 38 | - | - |
| PD affected carriers | 81 | 61 (10.15) | 53% | ||
| Non-penetrant carriers | 4 | 61 (9.42) | 0% | ||
| Association | PD affected carriers | 99 | 59 | 62.6 (9.8) | 54.5% |
Distinct exclusion criteria, appropriate for association analysis, were applied in order to generate the final sample used in the association analyses. The seven LRRK2-related PD cases (from four families) who were flagged during the initial population stratification analyses as having Hispanic or Asian ancestry were excluded from the association analyses (described previously)21. In addition, two cases were removed due to a lack of known age of onset, leaving 99 cases from 59 families.
Principal Component Analysis
As described above, extreme population outliers representing individuals of likely Hispanic, Asian, or African descent were identified in population stratification analyses during our initial sample QC 21, 22. To identify any additional population stratification related to onset age in LRRK2 cases, principal components were generated from the final set of 99 LRRK2-related PD cases only. The region surrounding the LRRK2 locus on chromosome 12 was excluded from the calculations of the principal components. All principal components were generated using Eigenstrat 23 and only one member per family was included in the analysis. Eigenstrat was then used to apply principal components to the remaining family members who were not originally used to define the components. Principal components identified to be associated with age of onset with a p-value less than 0.10 were included as covariates in the regression analyses where appropriate.
Linkage Analysis
A subset of the 328,189 SNPs passing all quality control measures was selected for the linkage analyses to avoid inflation of LOD scores caused by linkage disequilibrium between SNPs 24. Plink 25was used to select markers with MAF>0.20, and pairwise correlation < 0.04, identifying 10,129 informative and independent markers for use in the linkage analyses. Information content across the chromosomes was calculated using Merlin.
Estimated heritability of onset age for LRRK2-related PD was obtained for this sample using SOLAR26. Linkage analysis to onset age was performed using two methods: (1) multipoint non-parametric quantitative trait linkage (QTL) implemented in Merlin 27 and (2) a robust score statistic based on variance components (RSS) 28 implemented in R.
Association Analysis
Two principal components were identified to be associated with onset age (p <0.10) in the association sample, and thus were included in subsequent analyses. Association to onset age was tested under an additive mode of inheritance for SNPs with a MAF greater than 0.2 and a dominant model for SNPs with a MAF less than or equal to 0.2. A linear mixed effects model using the kinship coefficient matrix (implemented using the kinship package in R) was used to account for the familial relationships. These analyses were also repeated in the subset of cases with G2019S mutations only.
To test whether there were significant associations in the linkage regions, the p-values for SNPs in each region were adjusted for multiple comparisons using a Bonferroni correction for the number of SNPs under the peak. The boundaries of the linkage peaks were defined as the positions where the LOD score dropped to one-half of the peak LOD score in that region. To test for significant SNP associations genomewide, the commonly accepted criterion for genomewide significance of p< 5×10-8 29, based on recent estimates of independent genomewide sequence variation to maintain 5% genomewide type I error rate 30, 31 was used.
RESULTS
For the linkage analyses, sib-pairs concordant for LRRK2 mutation status were included providing 85 individuals from 38 pedigrees for analysis (Table 1). Four non-penetrant siblings were observed, all of whom were female, and had an average age of exam similar to the affected group (actual ages 52, 54, 67, 71).
Principal Components Analysis
For the association study, extreme population outliers identified in population stratification analyses were excluded. A second set of principal components recalculated using only the LRRK2 case sample was tested for association to onset age. PC 2 and 10 were significantly associated with onset age of PD and were included in the onset-age association analyses as covariates (p= 0.002, p= 0.06 respectively).
Linkage Analysis
The estimated heritability of onset age in these families was 60.6% and was found to be significantly different than zero (p=0.02; 95% CI 8%-100%), supporting the hypothesis that genetic modifiers play an important role in onset and penetrance for LRRK2 carriers in PD.
The maximum LOD score for onset age observed using the QTL method was 2.43 and the maximum score using the RSS method was 1.94, both located in the chromosome 1q32.1 region at 199 cM (Figure 1A). Moderate linkage peaks were also identified at 16q12.1 using both methods (QTL LOD=1.58 at 63 cM, RSS LOD=1.10 at 60 cM) (Figure 1B). No linkage to onset age was observed in the area of the LRRK2 locus on Chromosome 12.
Figure 1.
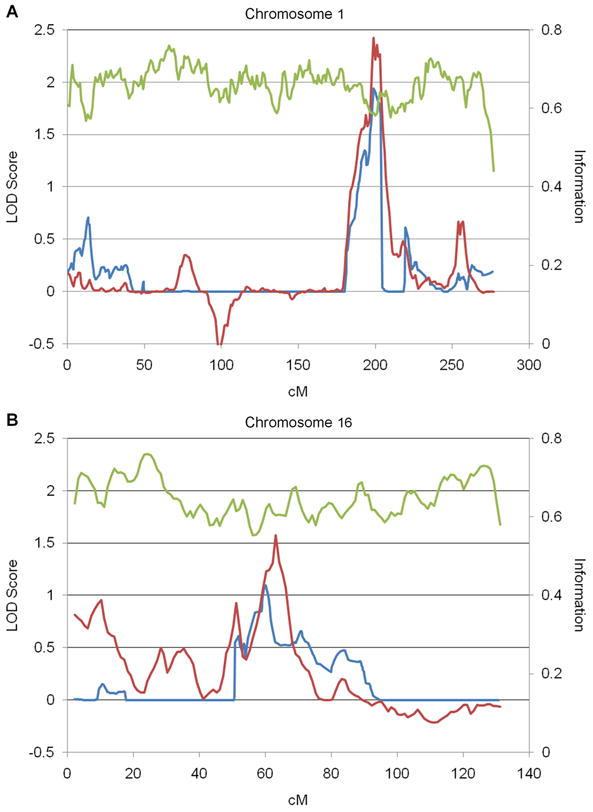
Linkage to LRRK2-related PD onset age. Quantitative trait linkage (red) and Robust score statistic (blue) LOD scores for onset age and information content (green) are shown for A. chromosome 1 and B. chromosome 16.
Association Analysis
Association to onset age was examined in the regions showing evidence of linkage. After Bonferroni correction for multiple comparisons under the peaks (onset: comparisons=3101, α=1.6 × 10-5), no statistically significant SNP associations were observed.
All associations with onset age yielding a p-value less than 0.005, in either the full sample or in the subset of G2019S carriers, under the chromosome 1 and chromosome 16 peaks are shown in Table 2A and 2B, respectively.
Table 2.
A: SNP Association to onset age under linkage peaks on Chromosomes 1.
| SNP | ref allele | chr | BP position | cM position | gene symbol | Control MAF | LRRK2 MAF | All effect estimate | All p-value | G2019S only effect estimate | G2019S only p-value |
|---|---|---|---|---|---|---|---|---|---|---|---|
| rs2186024 | C | 1 | 190,696,708 | 190.66 | RGS1 | 0.45 | 0.43 | -4.60 | 0.00068 | -3.13 | 0.03575 |
| rs2494354 | G | 1 | 192,721,321 | 193.26 | 0.10 | 0.06 | -8.31 | 0.0047 | -6.30 | 0.0759 | |
| rs2494312 | C | 1 | 192,763,213 | 193.28 | 0.11 | 0.07 | -7.85 | 0.0043 | -6.40 | 0.0521 | |
| rs927724 | C | 1 | 194,441,413 | 193.87 | KCNT2 | 0.12 | 0.11 | 7.13 | 0.0030 | 7.06 | 0.0059 |
| rs599779 | A | 1 | 197,604,771 | 195.46 | 0.40 | 0.39 | 4.52 | 0.0023 | 4.16 | 0.0109 | |
| rs1898240 | G | 1 | 197,613,703 | 195.47 | 0.20 | 0.15 | 5.85 | 0.0046 | 5.24 | 0.0148 | |
| rs487359 | A | 1 | 197,648,234 | 195.49 | 0.37 | 0.31 | 4.58 | 0.0015 | 4.65 | 0.0032 | |
| rs1890133 | T | 1 | 197,721,501 | 195.54 | 0.24 | 0.23 | -4.40 | 0.0048 | -2.49 | 0.1374 | |
| rs10919967 | T | 1 | 198,769,294 | 196.41 | 0.19 | 0.15 | 1.82 | 0.4443 | 7.93 | 0.0022 | |
| rs1400875 | G | 1 | 200,088,066 | 199.67 | IPO9 | 0.32 | 0.29 | -4.40 | 0.0043 | -2.65 | 0.1394 |
| rs2820312 | T | 1 | 200,135,880 | 199.86 | LMOD1 | 0.32 | 0.29 | -4.40 | 0.0043 | -2.65 | 0.1394 |
| rs3087949 | C | 1 | 201,314,244 | 202.52 | PPFIA4 | 0.29 | 0.32 | -2.99 | 0.0395 | -4.57 | 0.0024 |
| rs2050935 | C | 1 | 201,598,170 | 203.24 | FMOD | 0.20 | 0.18 | -5.80 | 0.0037 | -5.13 | 0.0244 |
| rs4950978 | T | 1 | 203,291,196 | 205.91 | CNTN2 | 0.23 | 0.23 | -4.63 | 0.0028 | -3.74 | 0.0211 |
| rs1470637 | T | 1 | 203,299,906 | 205.92 | CNTN2 | 0.34 | 0.36 | -3.73 | 0.0045 | -2.85 | 0.0367 |
| B: SNP Association to onset age under linkage peaks on Chromosomes 16. | |||||||||||
| rs7200879 | G | 16 | 30,855,073 | 56.80 | FBXL19 | 0.23 | 0.23 | 3.44 | 0.04620 | 5.29 | 0.00485 |
| rs1344528 | G | 16 | 48,099,429 | 59.05 | ZNF423 | 0.45 | 0.43 | -3.69 | 0.0080 | -4.58 | 0.0025 |
| rs7204293 | A | 16 | 48,585,606 | 59.77 | TMEM188 | 0.08 | 0.09 | -8.27 | 0.0025 | -7.28 | 0.0188 |
| rs1861662 | T | 16 | 48,664,884 | 59.84 | HEATR3 | 0.08 | 0.08 | -7.52 | 0.0045 | -6.55 | 0.0319 |
| rs4785239 | T | 16 | 49,758,032 | 61.67 | SALL1 | 0.15 | 0.14 | 6.57 | 0.0013 | 6.70 | 0.0034 |
| rs11645369 | C | 16 | 51,507,565 | 65.31 | 0.25 | 0.20 | -6.12 | 0.0017 | -5.12 | 0.0216 | |
| rs3095635 | G | 16 | 52,098,599 | 66.77 | AKTIP | 0.18 | 0.10 | -6.69 | 0.0031 | -6.52 | 0.0128 |
| rs1075440 | C | 16 | 52,348,407 | 67.17 | FTO | 0.32 | 0.24 | -4.53 | 0.0017 | -3.03 | 0.0848 |
| rs17224310 | A | 16 | 52,548,454 | 67.49 | FTO | 0.17 | 0.20 | 4.52 | 0.0107 | 5.22 | 0.0043 |
| rs7206456 | A | 16 | 52,562,990 | 67.51 | FTO | 0.29 | 0.33 | -3.77 | 0.0048 | -3.96 | 0.0047 |
| rs4784384 | T | 16 | 53,029,562 | 68.25 | 0.32 | 0.35 | 4.34 | 0.0022 | 4.51 | 0.0033 | |
| rs893265 | G | 16 | 53,864,590 | 70.30 | IRX6 | 0.37 | 0.38 | -3.72 | 0.0025 | -4.11 | 0.0026 |
| rs2397442 | T | 16 | 53,866,761 | 70.31 | IRX6 | 0.50 | 0.44 | 4.43 | 0.0004 | 4.06 | 0.0038 |
| rs30935 | C | 16 | 53,878,002 | 70.37 | IRX6 | 0.43 | 0.42 | 5.11 | 0.0001 | 4.67 | 0.0010 |
| rs733140 | C | 16 | 53,881,595 | 70.39 | IRX6 | 0.23 | 0.20 | -3.73 | 0.0105 | -4.63 | 0.0028 |
| rs11860394 | G | 16 | 53,884,305 | 70.40 | IRX6 | 0.35 | 0.35 | -3.87 | 0.0037 | -3.04 | 0.0420 |
| rs10521323 | A | 16 | 54,038,722 | 70.70 | MMP2 | 0.25 | 0.28 | 4.58 | 0.0005 | 4.15 | 0.0033 |
| rs1420228 | C | 16 | 54,042,674 | 70.70 | MMP2 | 0.11 | 0.18 | -5.83 | 0.0022 | -5.23 | 0.0109 |
| rs243842 | C | 16 | 54,084,923 | 70.70 | MMP2 | 0.39 | 0.37 | 4.52 | 0.0010 | 4.34 | 0.0029 |
| rs899228 | G | 16 | 54,944,789 | 71.69 | GNAO1 | 0.22 | 0.20 | 4.84 | 0.0041 | 4.69 | 0.0078 |
Nearest genes within 100kb of SNP
In the genomewide association analyses for onset age, no SNPs reached a genomewide level of significance (p< 5×10-8). Supplementary Table S1 lists all SNP associations to onset age with p<0.005. We reviewed the results for SNPs located within 500kb of the LRRK2 gene to see if there were any trans- allele influence on age of onset of LRRK2 related PD, however no SNPs within this region demonstrated association to PD at even a nominal level (p≤0.05).
DISCUSSION
This study represents the first genomewide linkage scan and association study for modifiers of LRRK2-related PD. We have demonstrated significant heritability of onset age of LRRK2-related PD and identified two chromosomal regions with suggestive evidence of linkage to LRRK2 onset age. Association analysis of SNPs under these peaks showed no statistically significant associations after correction for multiple comparisons. We also observed no evidence that additional variability in the LRRK2 locus influences age of onset of LRRK2-related PD.
The methods used to define the linkage peaks allows the peaks to encompass a broad range, with 121 and 185 annotated genes in the linked regions on chromosomes 1 and 16, respectively. Using an inclusive definition to define the linkage region results in a larger interval and may detract from our power to detect association in these regions. However, this approach was appropriate due to the imprecision of linkage mapping.
An earlier study of onset age in 44 LRRK2-related PD cases 32 that focused on the previously identified PD related genes SNCA and MAPT, identified a SNP located in MAPT that showed a significant increase in age of onset in LRRK2 positive subjects who carried the minor allele. This SNP (rs2435207) was not genotyped in our GWAS platform. A nearby SNP in strong LD with it (rs2435211, R2=0.97 from HapMap CEU 33) was genotyped, but was not significantly associated with onset age in our study (p=0.10) and the estimate was not in the same direction of effect as the prior study. No MAPT or SNCA SNPs genotyped in this study showed significant association to onset age.
In the genomewide SNP association analyses, no SNPs reached the commonly accepted level of genomewide significance of 5×10-8. The 99 LRRK2-related PD cases (Table 1) included were the only cases available for study; therefore, the genomewide association studies were underpowered. Nevertheless, genomewide SNP association results in this well characterized sample may provide a valuable resource for researchers who have other LRRK2 cohorts to evaluate whether these same regions are implicated in their samples. These results may also provide important prioritization information for use in other gene characterization experiments. Therefore, an extended list of the top association study results (p<0.005) are provided in the supplementary tables S1.
While the association analyses in this study were unable to provide additional support for more localized regions underneath either of the linkage peaks, recent reports from two large GWAS of idiopathic PD implicate a region on chromosome 1q32, overlapping the onset age linkage peak observed in this study 34, 35. The observation of association to PD risk in the same region as the linkage to age of onset in LRRK2-related PD supports the hypothesis that genetic modifiers of penetrance of known disese-causing genes may also be involved in the disease process of idiopathic PD. Furthermore, this convergence suggests that continued study of these genes in the less heterogeneous LRRK2-related PD cohort may provide a powerful method for the functional characterization of this region.
In conclusion, we have identified two genomic regions that may harbor modifiers of LRRK2-related PD onset age or penetrance. Further study of these regions may provide important insight into the function and etiology of LRRK2-related PD, as well as potential therapeutic targets for the treatment and possible prevention of this form of PD. Such insights would undoubtedly affect the understanding of idiopathic PD as well.
Supplementary Material
Acknowledgments
This study used samples from the NINDS Human Genetics Resource Center DNA and Cell Line Repository (http://ccr.coriell.org/ninds), as well as clinical data.
DNA samples contributed by the Parkinson Institute - Istituti Clinici di Perfezionamento, Milan, Italy were from the “Human genetic bank of patients affected by PD and parkinsonisms”, supported by Italian Telethon grant n. GTB07001 and by the “Fondazione Grigioni per il Morbo di Parkinson”.
A portion of this research was conducted using the Linux Cluster for Genetic Analysis (LinGA-II) funded by the Robert Dawson Evans Endowment of the Department of Medicine at Boston University School of Medicine and Boston Medical Center.
Genotyping services were provided by the Center for Inherited Disease Research (CIDR). CIDR is fully funded through a federal contract from the National Institutes of Health to The Johns Hopkins University, contract number HHSN268200782096C. We particularly thank Drs. Kimberly Doheny and Elizabeth Pugh from CIDR and Mr. Justin Paschall from NCBI for their assistance. The data used in this study is available at http://www.ncbi.nlm.nih.gov/sites/entrez?db=gap through dbGaP accession number: phs000126.v1.p1.
The following are members of the PROGENI Steering Committee. University of Tennessee Health Science Center: R. F. Pfeiffer; University of Rochester: F. Marshall, D. Oakes, A. Rudolph, A. Shinaman; Columbia University Medical Center: K. Marder; Indiana University School of Medicine: P.M. Conneally, T. Foroud, C. Halter; University of Kansas Medical Center: K. Lyons; Eli Lilly & Company: E. Siemers; Medical College of Ohio: L. Elmers; University of California, Irvine: N. Hermanowicz.
The following are members of the GenePD Steering Committee. University of Virginia Health System: G.F. Wooten; UMDNJ-Robert Wood Johnson Medical School: L. Golbe; Center for Human Genetic Research, Massachusetts General Hospital, Harvard Medical School: J.F. Gusella; Boston University School of Medicine: R.H. Myers.
We thank the subjects for their participation in this research study.
PSG-PROGENI Investigators and Coordinators: Albany Medical College: S Factor, D Higgins, S Evans; Barrow Neurological Institute: H Shill, M Stacy, J Danielson, L Marlor, K Williamson; Baylor College of Medicine: J Jankovic, C Hunter; Beth Israel Deaconess Medical Center: D Simon, P Ryan, L Scollins; Beth Israel Medical Center: R Saunders-Pullman, K Boyar, C Costan-Toth, E Ohmann; Brigham & Women’s Hospital: L Sudarsky, C Joubert; Brown University (Memorial Hospital of RI): J Friedman, K Chou, H Fernandez, M Lannon; Cleveland Clinic Florida-Weston: N Galvez-Jimenez, A Podichetty, K Thompson; Clinical Neuroscience Center: P Lewitt, M DeAngelis; Colorado Neurological Institute: C O’Brien, L Seeberger, C Dingmann, D Judd; Columbia University Medical Center: K Marder, J Fraser, J Harris; Creighton University: J Bertoni, C Peterson; Evanston Northwestern Healthcare: M Rezak, G Medalle; Hotel-Dieu Hospital-Chum: S Chouinard, M Panisset, J Hall, H Poiffaut; Hunter Homes McGuire Veterans Medical Center: V Calabrese, P Roberge; Indiana University School of Medicine: J Wojcieszek, J Belden; Institute For Neurodegenerative Disorders: D Jennings, K Marek, S Mendick; Johns Hopkins University: S Reich, B Dunlop; London Health Sciences Centre: M Jog, C Horn; Mayo Clinic Jacksonville: R Uitti, M Turk; McFarland Neurosciences: T Ajax, J Mannetter; Medical College of Georgia: K Sethi, J Carpenter, B Dill, L Hatch, K Ligon, S Narayan; Medical College of Wisconsin: K Blindauer, K Abou-Samra, J Petit; Medical University of Ohio: L Elmer, E Aiken, K Davis, C Schell, S Wilson; Mount Sinai School of Medicine: M Velickovic, W Koller (deceased), S Phipps; North Shore-LIJ Health System: A Feigin, M Gordon, J Hamann, E Licari, M Marotta-Kollarus, B Shannon, R Winnick; Northwestern University: T Simuni, A Videnovic, A Kaczmarek, K Williams, M Wolff; Ochsner Clinic Foundation: J Rao, M Cook; Ohio State University: M Fernandez, S Kostyk, J Hubble, A Campbell, C Reider, A Seward; Oregon Health & Science University: R Camicioli, J Carter, J Nutt, P Andrews, S Morehouse, C Stone; Ottawa Hospital Civic Site: T Mendis, D Grimes, C Alcorn-Costa, P Gray, K Haas, J Vendette; Pacific Neuroscience Medical Group: J Sutton, B Hutchinson, J Young; Saskatoon Dist Health Board Royal Univ Hosp: A Rajput, A Rajput, L Klassen, T Shirley; Scott & White Hospital/Texas A&M University: B Manyam, P Simpson, J Whetteckey, B Wulbrecht; The Parkinson’s & Movement Disorder Institute: D Truong, M Pathak, K Frei, N Luong, T Tra, A Tran, J Vo; Toronto Western Hospital, University Health: A Lang, G Kleiner-Fisman, A Nieves, L Johnston, J So; UMDNJ-School of Osteopathic Medicine: G Podskalny, L Giffin; University of Alabama at Birmingham: P Atchison, C Allen; University of Alberta: W Martin, M Wieler; University of Calgary: O Suchowersky, M Klimek; University of California Irvine: N Hermanowicz, S Niswonger; University of California San Diego: C Shults (deceased), D Fontaine; University of California San Francisco: M Aminoff, C Christine, M Diminno, J Hevezi; University of Chicago: A Dalvi, U Kang, J Richman, S Uy, J Young; University of Cincinnati: A Dalvi, A Sahay, M Gartner, D Schwieterman; University of Colorado Health Sciences Center: D Hall, M Leehey, S Culver, T Derian; University of Connecticut: T Demarcaida, S Thurlow; University of Iowa: R Rodnitzky, J Dobson; University of Kansas Medical Center: K Lyons, R Pahwa, T Gales, S Thomas; University of Maryland School of Medicine: L Shulman, S Reich, W Weiner, K Dustin; University of Miami: K Lyons, C Singer, W Koller (deceased), W Weiner, L Zelaya; University of Minnesota: P Tuite, V Hagen, S Rolandelli, R Schacherer, J Kosowicz; University of New Mexico: P Gordon, J Werner; University of Puerto Rico School of Medicine: C Serrano, S Roque; University of Rochester: R Kurlan, D Berry, I Gardiner; University of South Florida: R Hauser, J Sanchez-Ramos, T Zesiewicz, H Delgado, K Price, P Rodriguez, S Wolfrath; University of Tennessee Health Science Center: R Pfeiffer, L Davis, B Pfeiffer; University of Texas Southwestern Medical Center: R Dewey, B Hayward, A Johnson, M Meacham, B Estes; Wake Forest University School of Medicine: F Walker, V Hunt, C O’Neill; Washington University: B Racette, L Good, M Rundle
PROGENI Molecular Genetic Laboratory: Division of Human Genetics, Cincinnati Children’s Hospital Medical Center: William C. Nichols, Michael W. Pauciulo, Diane K. Marek, Veronika E. Elsaesser
GenePD Investigators and Coordinators: University Southern California School of Medicine: M. Lew; University of Calgary: O. Suchowersky, S. Furtado; University of Lübeck, Germany: C. Klein; UMDNJ-Robert Wood Johnson Medical School: L. Golbe, M.H. Mark; Massachusetts General Hospital, Harvard Medical School: J. Growdon, N. Huggins; University of Virginia Health System: G.F. Wooten; University of Alabama at Birmingham : R. Watts; University of Toronto: M. Guttman; Washington University School of Medicine: B. Racette, J. Perlmutter; Barrow Neurological Institute: L. Marlor, Sun Health Research Institute: H. Shill; University of Miami: C. Singer; Parkinson Institute, Istituti Clinici di Perfezionamento, Milano, Italy: S. Goldwurm, G. Pezzoli; Boston University School of Medicine: M.H. Saint-Hilaire, T. Massood; Cleveland Clinic Foundation: K. Baker, I. Itin, A. Ahmed; University of Louisville School of Medicine: I. Litvan; University of Sydney ANZAC Research Institute, Concord Hospital, Sydney, Australia: G. Nicholson, A. Corbett; Struthers Parkinson’s Center, Minneapolis: M. Nance; Port City Neurology, Scarborough, ME: E. Drasby; Parkinson’s Disease and Movement Disorder Center of Boca Raton: S. Isaacson; Newcastle University, Newcastle upon Tyne, UK: D. Burn, P. Chinnery; General Regional Hospital Bolzano, Bolzano, Italy: P. Pramstaller; University of Arkansas for Medical Sciences: J. Al-hinti; Aarhus University Hospital, Aarhus, Denmark: A. Moller, K. Ostergaard; University of Arizona: S. Sherman; Auckland City Hospital, Auckland, New Zealand: R. Roxburgh, B. Snow; University of Kentucky College of Medicine: J. Slevin, F. Cambi.
GenePD Molecular Genetics Laboratories: Center for Human Genetic Research, Massachusetts General Hospital, Harvard Medical School: J.F. Gusella, M.E. McDonald, M. Sun, L. Mysore, M.A, Anderson, D. Lucente; Neurogenetics Laboratory, Boston University School of Medicine: S. Williamson, M.W. Nagle, R.H. Myers.
AUTHOR ROLES Jeanne C. Latourelle: Conception, organization and execution of the research project; design, execution, and review and critique of the statistical analyses, writing the first draft of the manuscript. Audrey E. Hendricks: Conception and execution of the research project; design, execution, and review and critique of the statistical analyses, review and critique of the manuscript. Nathan Pankratz: Conception, organization, and execution of the research project; design, and review and critique of the statistical analyses; review and critique of the manuscript. Jemma B. Wilk: Conception, organization, and execution of the research project; design, and review and critique of the statistical analyses; review and critique of the manuscript. Cheryl Halter: Execution and organization of the research project; review and critique of the manuscript. William C. Nichols: Conception, organization, and execution of the research project; design, and review and critique of the statistical analyses; review and critique of the manuscript. James F. Gusella: Conception, organization, and execution of the research project; design, and review and critique of the statistical analyses; review and critique of the manuscript. Anita L. Destefano: Conception, organization, and execution of the research project; design, and review and critique of the statistical analyses; review and critique of the manuscript. Richard H. Myers: Conception, organization, and execution of the research project; design, and review and critique of the statistical analyses; review and critique of the manuscript. Tatiana Foroud: Conception, organization, and execution of the research project; design, and review and critique of the statistical analyses; review and critique of the manuscript. The Psg –Progeni And Genepd Investigators, Coordinators And Molecular Genetic Laboratories: Conception, organization, and execution of the research project; review and critique of the statistical analyses; review and critique of the manuscript.
Funding sources: This project was supported by R01 NS37167, R01 NS036711, the Robert P. & Judith N. Goldberg Foundation, the Bumpus Foundation and the Harvard NeuroDiscovery Center.
Footnotes
Financial Disclosure /Conflict of Interest: The authors report no financial disclosure or conflicts of interest.
References
- 1.Mayeux R. Epidemiology of neurodegeneration. Annual review of neuroscience. 2003;26:81–104. doi: 10.1146/annurev.neuro.26.043002.094919. [DOI] [PubMed] [Google Scholar]
- 2.Tan EK, Skipper LM. Pathogenic mutations in Parkinson disease. Human mutation. 2007;28(7):641–653. doi: 10.1002/humu.20507. [DOI] [PubMed] [Google Scholar]
- 3.Singleton AB. Altered alpha-synuclein homeostasis causing Parkinson’s disease: the potential roles of dardarin. Trends in neurosciences. 2005;28(8):416–421. doi: 10.1016/j.tins.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 4.Latourelle JC, Sun M, Lew MF, et al. The Gly2019Ser mutation in LRRK2 is not fully penetrant in familial Parkinson’s Disease: the GenePD study. BMC Med. 2008;6(1):32. doi: 10.1186/1741-7015-6-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pankratz N, Pauciulo MW, Elsaesser VE, et al. Mutations in LRRK2 other than G2019S are rare in a north American-based sample of familial Parkinson’s disease. Mov Disord. 2006;21(12):2257–2260. doi: 10.1002/mds.21162. [DOI] [PubMed] [Google Scholar]
- 6.Funayama M, Hasegawa K, Kowa H, Saito M, Tsuji S, Obata F. A new locus for Parkinson’s disease (PARK8) maps to chromosome 12p11.2-q13.1. Annals of neurology. 2002;51(3):296–301. doi: 10.1002/ana.10113. [DOI] [PubMed] [Google Scholar]
- 7.Paisan-Ruiz C, Jain S, Evans EW, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron. 2004;44(4):595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- 8.Kachergus J, Mata IF, Hulihan M, et al. Identification of a novel LRRK2 mutation linked to autosomal dominant parkinsonism: evidence of a common founder across European populations. American journal of human genetics. 2005;76(4):672–680. doi: 10.1086/429256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lesage S, Ibanez P, Lohmann E, et al. G2019S LRRK2 mutation in French and North African families with Parkinson’s disease. Annals of neurology. 2005;58(5):784–787. doi: 10.1002/ana.20636. [DOI] [PubMed] [Google Scholar]
- 10.Ferreira JJ, Guedes LC, Rosa MM, et al. High prevalence of LRRK2 mutations in familial and sporadic Parkinson’s disease in Portugal. Mov Disord. 2007;22(8):1194–1201. doi: 10.1002/mds.21525. [DOI] [PubMed] [Google Scholar]
- 11.Clark LN, Wang Y, Karlins E, et al. Frequency of LRRK2 mutations in early- and late-onset Parkinson disease. Neurology. 2006 doi: 10.1212/01.wnl.0000244345.49809.36. [DOI] [PubMed] [Google Scholar]
- 12.Ozelius LJ, Senthil G, Saunders-Pullman R, et al. LRRK2 G2019S as a cause of Parkinson’s disease in Ashkenazi Jews. The New England journal of medicine. 2006;354(4):424–425. doi: 10.1056/NEJMc055509. [DOI] [PubMed] [Google Scholar]
- 13.Goldwurm S, Zini M, Mariani L, et al. Evaluation of LRRK2 G2019S penetrance: relevance for genetic counseling in Parkinson disease. Neurology. 2007;68(14):1141–1143. doi: 10.1212/01.wnl.0000254483.19854.ef. [DOI] [PubMed] [Google Scholar]
- 14.Djousse L, Knowlton B, Hayden MR, et al. Evidence for a modifier of onset age in Huntington disease linked to the HD gene in 4p16. Neurogenetics. 2004;5(2):109–114. doi: 10.1007/s10048-004-0175-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Polymeropoulos MH, Lavedan C, Leroy E, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276(5321):2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 16.Sun M, Latourelle JC, Wooten GF, et al. Influence of heterozygosity for parkin mutation on onset age in familial Parkinson disease: the GenePD study. Archives of neurology. 2006;63(6):826–832. doi: 10.1001/archneur.63.6.826. [DOI] [PubMed] [Google Scholar]
- 17.Foroud T, Uniacke SK, Liu L, et al. Heterozygosity for a mutation in the parkin gene leads to later onset Parkinson disease. Neurology. 2003;60(5):796–801. doi: 10.1212/01.wnl.0000049470.00180.07. [DOI] [PubMed] [Google Scholar]
- 18.Nichols WC, Pankratz N, Hernandez D, et al. Genetic screening for a single common LRRK2 mutation in familial Parkinson’s disease. Lancet. 2005;365(9457):410–412. doi: 10.1016/S0140-6736(05)17828-3. [DOI] [PubMed] [Google Scholar]
- 19.Gibb WR, Lees AJ. The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson’s disease. Journal of neurology, neurosurgery, and psychiatry. 1988;51(6):745–752. doi: 10.1136/jnnp.51.6.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gunderson KL, Steemers FJ, Ren H, et al. Whole-genome genotyping. Methods in enzymology. 2006;410:359–376. doi: 10.1016/S0076-6879(06)10017-8. [DOI] [PubMed] [Google Scholar]
- 21.Pankratz N, Wilk JB, Latourelle JC, et al. Genomewide association study for susceptibility genes contributing to familial Parkinson disease. Human genetics. 2009;124(6):593–605. doi: 10.1007/s00439-008-0582-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Latourelle JC, Pankratz N, Dumitriu A, et al. Genomewide association study for onset age in Parkinson disease. BMC Med Genet. 2009;10:98. doi: 10.1186/1471-2350-10-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38(8):904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 24.Abecasis GR, Wigginton JE. Handling marker-marker linkage disequilibrium: pedigree analysis with clustered markers. American journal of human genetics. 2005;77(5):754–767. doi: 10.1086/497345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. American journal of human genetics. 2007;81(3):559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Almasy L, Blangero J. Multipoint quantitative-trait linkage analysis in general pedigrees. American journal of human genetics. 1998;62(5):1198–1211. doi: 10.1086/301844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin--rapid analysis of dense genetic maps using sparse gene flow trees. Nature genetics. 2002;30(1):97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 28.Dupuis J, Shi J, Manning AK, et al. Mapping quantitative traits in unselected families: algorithms and examples. Genet Epidemiol. 2009 doi: 10.1002/gepi.20413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Risch N, Merikangas K. The future of genetic studies of complex human diseases. Science. 1996;273(5281):1516–1517. doi: 10.1126/science.273.5281.1516. [DOI] [PubMed] [Google Scholar]
- 30.Pe’er I, Yelensky R, Altshuler D, Daly MJ. Estimation of the multiple testing burden for genomewide association studies of nearly all common variants. Genetic epidemiology. 2008;32(4):381–385. doi: 10.1002/gepi.20303. [DOI] [PubMed] [Google Scholar]
- 31.Dudbridge F, Gusnanto A. Estimation of significance thresholds for genomewide association scans. Genetic epidemiology. 2008;32(3):227–234. doi: 10.1002/gepi.20297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Golub Y, Berg D, Calne DB, et al. Genetic factors influencing age at onset in LRRK2-linked Parkinson disease. Parkinsonism Relat Disord. 2009;15(7):539–541. doi: 10.1016/j.parkreldis.2008.10.008. [DOI] [PubMed] [Google Scholar]
- 33.The International HapMap Project. Nature. 2003;426(6968):789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 34.Satake W, Nakabayashi Y, Mizuta I, et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet. 2009 doi: 10.1038/ng.485. [DOI] [PubMed] [Google Scholar]
- 35.Simon-Sanchez J, Schulte C, Bras JM, et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet. 2009 doi: 10.1038/ng.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.