

Abstract
Novel 3,5-bis(benzylidene)-1-[3-(2-hydroxyethylthio)propanoyl]piperidin-4-ones (3a–e) display potent cytotoxicity and a preferential lethality toward various neoplasms compared to some normal cells. The corresponding sulfonic acid analogues 5a–e and an isostere 4 demonstrated substantially lower activity. The leads 3d and 3e possess very high activity against colon cancer and leukemia cell lines, caused DNA fragmentation, and activated caspase-3 in HL-60 cells. The enones 3b–e were well tolerated in a short-term toxicity screen in mice.
INTRODUCTION
The principal aim of this laboratory is the designing of novel conjugated arylidene ketones as candidate antineoplastic agents. The arylidene ketones display preferential affinity for thiols rather than amino or hydroxy groups.1,2 Since nucleic acids do not contain sulfhydryl groups, conjugated enones are likely to be devoid of the genotoxic effects produced by many contemporary anticancer drugs.3 In addition, various studies have shown that certain neoplasms are more susceptible to successive chemical insults than normal cells.4,5 In other words, after initial drug interactions take place, greater chemosensitization occurs with malignancies, making them more vulnerable to a second chemical insult than normal tissues. Such considerations led to the development of 3,5-bis(benzylidene)-4-piperidones 1 (Figure 1) as cellular thiol alkylators, and in general they possess promising cytotoxic potencies toward a number of neoplastic and transformed cells.6,7
Figure 1.

Structures of series 1–5.
The cytotoxic properties of 1 are believed to be due principally to the 1,5-diaryl-3-oxo-1,4-pentadienyl pharmacophore which is designed to interact with the primary binding site of the molecular target. The areas surrounding these locations will vary, and it is possible that portions of the ligand molecules will interact at one or more auxiliary binding sites that are close to the primary sites. Thus, by a judicious choice of substituents on the piperidyl nitrogen atom of 1, two positive outcomes may be realized. First, interactions at the primary and auxiliary binding sites may increase cytotoxic potencies compared to only interactions with the 1,5-diaryl-3-oxo-1,4-pentadienyl group. Second, the nature of the primary and auxiliary binding sites may be different in malignant and normal cells, and hence greater toxicity to neoplasms may take place. An initial study to evaluate these hypotheses involved the attachment of various substituents on the piperidyl nitrogen atom which have van der Waals and hydrogen bonding capacities leading to series 2. A comparison between the analogues in series 1 and 2, which have the same substituents in the arylidene aryl rings, reveals that in 48% of the comparisons greater potencies were noted in series 2 while in 35% of the cases, equipotencies are noted.7 Series 2 compounds are highly lipophilic (average log P = 6.7 8) which is likely to contribute to poor oral bioavailability. Hence the objective of the present study is to develop a novel cluster of N-acyl-3,5-bis(benzylidene)-4-piperidones that will display promising cytotoxicity, selective toxicity toward neoplasms compared to normal cells, and suitable pharmacokinetic properties. These considerations led to the decision to develop series 3 as candidate antineoplastic agents by retaining the polar and hydrophobic groups in the N-acyl side chain but to reduce the lipophilicity. In addition, a comparison of the cytotoxic potencies of series 3 against the related analogues in series 5 which contain the very hydrophilic sulfonic acid group and an isostere 4 was undertaken.
RESULTS
Series 3 compounds were synthesized from 4-piperidone hydrochloride by the route outlined in Scheme 1. The synthesis of 4 and 5 has been reported from our laboratory previously.9 The enones 3a–e, 4, and 5a–e were evaluated against human Molt 4/C8 and CEM T-lymphocytes and murine L1210 leukemic cells, and these data are presented in Table 1. The enones 3c–e and 5c,d were examined against a panel of 57 human tumor cell lines, and some of the data generated are summarized in Table 2. All of the compounds in series 3 were evaluated against human HL-60 promyelocytic leukemic cells and human oral squamous cell carcinomas (HSC-2, HSC-3, and HSC-4). In addition, these compounds were assayed against three normal human cell lines: HGF gingival fibroblasts, HPC pulp cells, and HPLF periodontal ligament fibroblasts. A summary of these biodata is in Table 3. Both 3d and 3e caused deoxyribonucleic acid (DNA) fragmentation and activated caspase-3 in HL-60 cells but not in HSC-2 carcinomas. These results are in Figures 2 and 3. Doses of 30, 100, and 300 mg/kg of the enones 3b–d, 4, and 5a–e were injected intraperitoneally to mice, and the animals were observed for mortalities and neurotoxicity after 0.5 and 4 h. A dose of 50 mg/kg of 3b was administered per os to rats that were monitored over a 4 h time frame for any display of fatalities or neurological deficit.
Scheme 1. Synthesis of 3a–ea.

a Reagents: (i) CH2=CH–COCl; (ii) HSCH2CH2OH; (iii) R1-C6H4-CHO.
Table 1.
Evaluations of Series 3–5 against Molt 4/C8, CEM, and L1210 Cells
| compd | Molt 4/C8
|
CEM
|
L1210
|
|||
|---|---|---|---|---|---|---|
| IC50 (μM) | Δ1a | IC50 (μM) | Δ1a | IC50 (μM) | Δ1a | |
| 3a | 0.66 ± 0.18 | 2.5 | 0.57 ± 0.17 | 3.0 | 3.80 ± 0.59 | 2.1 |
| 3b | 1.85 ± 1.20 | 0.9b | 1.10 ± 0.56 | 1.7 | 5.91 ± 2.13 | 1.4 |
| 3c | 1.66 ± 0.54 | 174 | 3.51 ± 1.56 | 47 | 19.60 ± 2.3 | 13 |
| 3d | 0.42 ± 0.32 | 32 | 0.69 ± 0.45 | 13 | 6.47 ± 1.72 | 6.4 |
| 3e | 0.49 ± 0.11 | 17 | 0.48 ± 0.18 | 9.2 | 3.92 ± 0.96 | 8.4 |
| 4 | 2.77 ± 0.92 | 104 | 3.20 ± 1.34 | 51 | 10.90 ± 7.3 | 22 |
| 5a | 143 ± 15 | 0.01 | 125 ± 6 | 0.01 | 234 ± 1 | 0.03 |
| 5b | 79.7 ± 21.8 | 0.02 | 194 ± 1 | 0.01 | 198 ± 1 | 0.04 |
| 5c | 156 ± 22 | 1.9 | 199 ± 18 | 0.8b | 227 ± 1 | 1.1b |
| 5d | 35.8 ± 2.3 | 0.4 | 41.6 ± 1.2 | 0.2 | 100 ± 30 | 0.4 |
| 5e | 6.98 ± 2.34 | 1.2b | 7.44 ± 1.06 | 0.6b | 11.4 ± 1.4 | 2.9 |
| melphalan | 3.24 ± 0.56 | 2.47 ± 0.21 | 2.13 ± 0.02 | |||
The Δ1 values are the quotients of the IC50 of the compound in series 1 and the analogue in series 3–5, which have the same aryl substituents.
The Δ1 value does not represent a statistically significant difference between the IC50 of the compound and the analogue in series 1 having the same aryl groups.
Table 2.
Evaluation of 3c–e and 5c,d against a Panel of Human Tumor Cell Lines
| compd | all cell lines
|
colon cancers, IC50 (μM)
|
leukemic cell lines, IC50 (μM)
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| av GI50a (μM) | COLO-205 | HCC-2998 | HCT-116 | HCT-15 | HT-29 | KM-12 | SW-620 | av IC50 | CCRF-CEM | HL-60 (TB) | K562 | RPMI-8226 | av IC50 | |
| 3c | 4.07 | 2.51 | 2.00 | 1.51 | 2.19 | 2.40 | 1.98 | 2.14 | 2.10 | 0.47 | 1.70 | 2.88 | 0.33 | 1.35 |
| 3d | 0.62 | 0.46 | 1.10 | 0.17 | 0.26 | 0.26 | 0.19 | 0.17 | 0.37 | 0.21 | 0.32 | 0.10 | 0.08 | 0.18 |
| 3e | 0.28 | 0.06 | 0.28 | 0.11 | 0.21 | 0.10 | 0.17 | 0.07 | 0.14 | 0.04 | 0.10 | 0.06 | 0.02 | 0.06 |
| 5c | >72.5 | >100 | 30.9 | 33.1 | >100 | 41.7 | 30.2 | 38.0 | >53.4 | >100 | >100 | 55.0 | 24.5 | >69.9 |
| 5d | >32.4 | 24.0 | 22.4 | 28.2 | 30.2 | 25.1 | 25.1 | 29.5 | 26.4 | 9.33 | 25.7 | 28.2 | 18.2 | 20.4 |
| 5-FU | >56.2 | 4.17 | 13.5 | 8.51 | 9.71 | 10.2 | 8.32 | >100 | >22.1 | 33.1 | >100 | 2.40 | >45.2 | |
| melphalan | 26.9 | 66.1 | 41.7 | 30.2 | 36.3 | 46.8 | 43.7 | 38.9 | 43.4 | 6.17 | 2.04 | 43.7 | 66.1 | 29.5 |
The average GI50 is the average IC50 against 57 tumor cell lines. The term GI50 rather than IC50 is utilized in some cases. For example, in the cases of 5c, d and 5-fluorouracil where 50% inhibition of growth was not achieved at the highest concentration of 100 μM, the value 100 μM was used in determining the average inhibition concentration.
Table 3.
Evaluation of 3a–e against Human Tumor and Normal Cell Lines
| compd | human tumor cell lines
|
human normal cell lines, CC50(μM)
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HL-60
|
HSC-2
|
HSC-3
|
HSC-4
|
|||||||||
| CC50 (μM) | SIa | CC50 (μM) | SIa | CC50 (μM) | SIa | CC50 (μM) | SIa | HGF | HPC | HPLF | avb | |
| 3a | 1.7 ± 0.0 | 8.7 | 1.60 ± 0.20 | 9.2 | 2.8 ± 0.69 | 5.3 | 1.3 ± 0.54 | 11.3 | 17 ± 2.1 | 14 ± 4.2 | 13 ± 3.6 | 14.7 |
| 3b | 2.1 ± 0.40 | 4.0 | 0.89 ± 0.04 | 9.4 | 1.7 ± 0.31 | 4.9 | 1.2 ± 0.47 | 6.9 | 9.1 ± 0.49 | 7.5 ± 1.0 | 8.4 ± 1.8 | 8.33 |
| 3c | 3.2 ± 0.35 | 4.9 | 2.41 ± 0.40 | 6.5 | 3.3 ± 0.70 | 4.8 | 2.4 ± 0.81 | 6.5 | 17 ± 2.10 | 14 ± 1.4 | 16 ± 2.6 | 15.7 |
| 3d | 1.0 ± 0.10 | 8.0 | 0.47 ± 0.04 | 17.1 | 0.97 ± 0.23 | 8.3 | 0.51 ± 0.18 | 15.8 | 8.3 ± 0.73 | 7.2 ± 0.26 | 8.6 ± 2.3 | 8.03 |
| 3e | 0.44 ± 0.01 | 13.3 | 0.34 ± 0.05 | 17.3 | 0.69 ± 0.27 | 8.5 | 0.38 ± 0.19 | 15.5 | 5.4 ± 1.3 | 5.8 ± 1.9 | 6.4 ± 2.8 | 5.87 |
| melphalan | 1.4 ± 1.20 | 150 | 8.7 ± 4.20 | 24.1 | 25 ± 7.70 | 8.4 | 32 ± 8.8 | 6.6 | 161 ± 27 | 269 ± 153 | 199 ± 60 | 210 |
The SI (selective index) values are obtained by dividing the average CC50 of the compound toward the three normal cell lines by the CC50 obtained using a specific cancer cell line.
These values are the average CC50 in μM of each compound toward HGF, HPC, and HPLF cells.
Figure 2.

Effect of 3d and 3e on DNA fragmentation in HL-60 and HSC-2 cells. Cells were incubated for 6 h with different concentrations of 3d and 3e or exposed to UV radiation for 1 min. The treated and control cells were incubated for 3 h in the regular culture media, after which time the DNA was prepared and then applied to 2% agarose gel electrophoresis.
Figure 3.
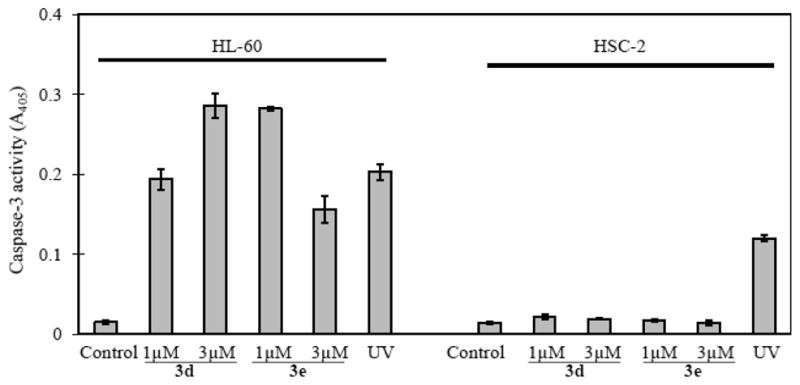
Effect of 3d and 3e on the activation of caspase-3. The HL-60 and HSC-2 cells were incubated with the substrate for 2 h, and 100 μg of protein was used in the assay. The results are the mean ± SD of three experiments.
DISCUSSION
The aryl substitutions in 3a–e and 5a–e were chosen from all four quadrants of the Craig scatter diagram,10 which ensured substantial variations in the electronic and hydrophobic properties of these groups. Series 3–5 were assessed against Molt 4/C8 and CEM cells to glean some idea of their cytotoxicity toward human transformed cells. The murine L1210 bioassay was chosen, since a number of anticancer drugs show efficacy against this cell line;11 therefore, this screen may predict useful anti-neoplastic agents. The following observations were made pertaining to the biodata in Table 1. First, series 3 compounds display potent cytotoxicity toward Molt 4/C8 and CEM T-lymphocytes. All the IC50 values are below 4 μM, and 60% are submicromolar. The enones 3a–e are less cytotoxic toward L1210 cells, although the IC50 values are below 20 μM. Second, the bioisosteres 3c and 4 are equipotent in all three bioassays while, with the exception of 5e, the sulfonic acids 5 display substantially reduced cytotoxic potencies compared to series 3 and 4. Third, melphalan is an alkylating agent used in cancer chemotherapy, and a comparison of its potency with 3–5 was made. The following 4-piperidones are more potent than melphalan (fold increase in potency in parentheses): 3a (4.9), 3c (2.0), 3d (7.6), 3e (6.7) in the Molt 4/C8 screen and 3a (4.3), 3b (2.3), 3d (3.6), and 3e (5.1) in the CEM test. The thiol 4 is equipotent with melphalan in the Molt 4/C8 and CEM assays. One may conclude that 3 is a promising series of novel cytotoxins. Fourth, the marked reduction in the potencies of series 5 may be due to the strongly acidic sulfonic acid group which could impede the penetration of the molecules via the cell membranes.
To ascertain the effect of N-acylation of 1a–e to produce 3–5, Δ1 values are presented in Table 1. These data are obtained by dividing the IC50 of series 1 by the IC50 of analogues of 3–5 which have the same aryl substituent. With the exception of 1b and 3b being equipotent in the Molt 4/C8 screen, all of the members of 3 and 4 are more potent than the analogues 1a–e, i.e., in 94% of the comparisons made. This observation is in contrast to the comparison between 1a–e and 5a–e. In this case, 60% of the Δ1 values reveal that greater potency resides in series 1, while 27% of the Δ1 values reveal equipotency and only 5c (Molt 4/C8 screen) and 5e (L1210 assay), or 13% of the comparisons, denote greater potencies than their counterparts in series 1. Overall, conversion of 1a–e into the N-acyl analogues 3–5 led to lower IC50 or equipotency in 58% and 15%, respectively, of the comparisons made (a 48% increase in potency of series 2 over the analogues in series 1, vide supra). One may note that the average log P of 3–5 is 2.91 (ref 9 and Table 4 in Supporting Information) in contrast to 6.7 for series 2.8
The 4-piperidones 3c,d and the analogues in series 5 having the same aryl substituents, namely, 5c,d and 3e, were evaluated against a panel of 57 human tumor cell lines from nine different neoplastic conditions: colon, non-small-cell lung, central nervous system, ovarian, renal, prostate, and breast cancers and leukemic and melanotic cells.12 The results are presented in Table 2. The average GI50 results against all cell lines reveal the marked potencies of 3d and 3e. In fact, the percentages of IC50 that are submicromolar are 54 and 87 for 3d and 3e, respectively. On the other hand, the biodata for 5c and 5d are congruent with the results portrayed in Table 1, namely, that 5d has lower IC50 than 5c and both compounds are substantially less potent than the analogues 3c and 3d. The average potencies of 3c–e toward all cell lines are 6.6, 43, and 96 times greater than that of melphalan, respectively. An examination of the mean graphs13 revealed that in general colon cancers and leukemic cells are especially sensitive to 3c–e and 5c,d. The IC50 values of 3d and 3e toward the colon cancers are all submicromolar with the exception of 3d in the HCC-2998 bioassay. Of particular note is the observation that the IC50 values of 3e toward COLO 205 and SW-620 tumors are in the double digit nanomolar range. The average IC50 values of 3c–e are 11, 60, and 158 times lower than the average IC50 for 5-fluorouracil, which is used clinically in the treatment of colon cancer.14 In the case of the leukemic cell lines, the IC50 values of 3a–e are generally submicromolar. Melphalan is used to treat certain types of leukemia,15 and the average IC50 values of 3c–e are 22, 164, and 492 times lower than the average IC50 of melphalan. One may conclude that 3c–e demonstrate high potencies toward a wide range of human tumor cell lines, especially colon cancers and leukemias.
To ascertain if series 3 exert a greater toxicity to neoplasms than normal cells, the enones were evaluated against human HL-60, HSC-2, HSC-3, and HSC-4 tumor cell lines and human HGF, HPLF, and HPC nonmalignant cells. Under clinical conditions, a neoplasm will be surrounded by different types of normal cells. Hence, to probe for selective toxicity to cancers compared to normal tissues, a comparison was made between the CC50 of a compound toward a specific malignant cell line and the average CC50 against the three normal cells. In this manner, selectivity index (SI) values were generated, and the data indicate that selectivity is demonstrated by all of the compounds in series 3. The order of selectivity (average SI values in parentheses) is 3e (13.7) > 3d (12.3) > 3a (8.6) > 3b (6.3) > 3c (5.7). One may also note that all the CC50 values toward the cancer cell lines are less than 4 μM and a little over half of these figures are in the submicromolar range. The relative potencies (average CC50 values in μM of each compound toward HL-60, HSC-2, HSC-3, and HSC-4 cells in parentheses) are 3e (0.46) > 3d (0.74) > 3b (1.47) > 3a (1.85) > 3c (2.83). From the SI and potency data, 3d and 3e emerge as lead molecules for further studies.
Statistical analyses were undertaken to ascertain whether correlations (p < 0.05) or trends to significance (p < 0.1) are found between certain physicochemical descriptors such as Hammett σ, Hansch π, and molecular refractivity (MR) constants of the aryl substituents in series 3 and the CC50 toward the malignant cell lines. Negative correlations between the σ values of the aryl substituents in 3a–e and the CC50 in the HL-60, HSC-3, and HSC-4 cells and a negative trend to significance in the case of the HSC-2 cell line are noted. These observations reveal that potency increases as the electron-attracting capacity of the aryl substituent rises. Positive correlations were observed between the σ values and the SI generated in the HL-60, HSC-2, and HSC-3 bioassays, while a positive trend toward a correlation was noted using HSC-4 cells. Thus, greater toxicity toward the malignant cells than normal cells is associated with increases in the electron-attracting properties of the aryl substituents. The fact that lipophilicity does not appear to govern cytotoxic potencies or the SI is reinforced by there being no correlation or trend to significance noted between the log P of 3a–e (presented in Table 4) and the CC50 or SI. The correlations noted are consistent with the probability that thiol alkylation is an important way in which bioactivity is mediated. In other words, a rise in the electron-withdrawing properties of the aryl groups leading to an increased fractional positive charge on the olefinic carbon atoms should enhance the rate of reaction with cellular thiols.
The next phase of the study investigated the effect of the promising leads 3d and 3e on DNA and caspase-3 activity. Caspase-3 is an effector caspase that plays an important role in apoptotic cell death. Both compounds induced DNA fragmentation (Figure 2) and activated caspase-3 (Figure 3) in HL-60 cells, suggesting that apoptosis is taking place via the mitochondrial pathway. On the other hand, as indicated in Figures 2 and 3, neither 3d nor 3e caused any DNA fragmentation or activation of caspase-3 in the HSC-2 carcinoma. Thus, 3d and 3e appear to cause cell death in HL-60 and HSC-2 cells by different mechanisms. This property of chemoselectivity may allow greater toxicity to be displayed toward neoplasms than nonmalignant cells which is likely the reason for the encouraging SI presented in Table 3.
Further investigation was made to evaluate the tolerance of most of the compounds in series 3–5 in mice. A number of anticancer drugs display significant toxicity to rodents, e.g., the LD50 values of melphalan, mechlorethamine, and cisplatin are 21.7, 3.6, and 26.8 mg/kg, respectively.16 Hence a short-term toxicity evaluation of 3b–e, 4, and 5a–e was undertaken by injecting doses of 30, 100, and 300 mg/kg into mice and observing the animals for survival and neurotoxicity after 0.5 and 4 h. None of the compounds caused mortalities at the doses employed, and no neurotoxic symptoms were noted with 3b–e or 5d,e. On the other hand, neurological deficit was observed with 4 and 5a–c after 0.5 h and also after 4 h in the case of 5c. Thus, the evidence points to series 3 having more favorable mammalian tolerability than the analogues of 4 and 5. In addition, administration of 50 mg/kg 3b to rats did not evoke fatalities or neurotoxicity when the animals were observed intermittently over a 4 h period.
Finally assessment of druglike properties of 3a–e was made in light of the guidelines developed by Lipinski et al.17 and Veber et al.18 The physicochemical data suggest that 3a–d achieve all of the criteria for druglike properties except 3e (data in Table 4 in the Supporting Information). Of these four compounds, 3d demonstrated excellent cytotoxic potencies and greater lethality for neoplasms than normal cells, thereby confirming it to be an excellent lead molecule for preclinical evaluations.
CONCLUSIONS
1-[3-(2-Hydroxyethylthio)propanoyl]-3,5-bis(benzylidene) piperidin-4-ones 3, in particular 3d and 3e, are a novel cluster of tumor-specific cytotoxins. 3d and 3e display excellent growth-inhibiting properties against a number of human cancer cell lines, are especially effective against colon cancers and leukemic cells, and are more potent cytotoxins than 5-fluorouracil and melphalan. The increased toxicity of 3a–e toward certain neoplasms compared with normal cells was revealed; in particular 3d and 3e demonstrate greater toxicity to the tumors. The observation that 3d and 3e exert their lethal effects in different ways depending on the cell line under consideration probably contributes significantly to them being tumor-selective cytotoxins. Doses up to and including 300 mg/kg 3b–e were administered to mice without causing fatalities or neurotoxicity, which further enhances their potential for development.
Supplementary Material
Acknowledgments
The following agencies are thanked for providing support for this study: Canadian Institutes of Health Research for an operating grant to J.R.D., Ministry of Education, Science, Sports and Culture for a Grant-in-Aid (No. 19592156) to H.S., the Geconcerteerde Onderzoeksacties (GOA 05/19), which provided funds to J.B., U.S. National Cancer Institute, which generated the data in Table 2, and J. P. Stables and the National Institute of Neurological Disorders and Stroke, which undertook the toxicity evaluations in rodents. L. Van Berckelaer of Rega Institute, Belgium, kindly carried out the Molt 4/C8, CEM and L1210 bioassays. The authors thank Beryl McCullough and Gwen Korte who typed several drafts of the manuscript.
Footnotes
ASSOCIATED CONTENT
Supporting Information. Synthetic procedures, biological methods, evaluation of druglike properties of 3a–e. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Mutus B, Wagner JD, Talpas CJ, Dimmock JR, Phillips OA, Reid RS. 1-p-Chlorophenyl-4,4-dimethyl-5-diethylamino-1-penten-3-one hydrobromide, a sulfhydryl-specific compound which reacts irreversibly with protein thiols but reversibly with small molecular weight thiols. Anal Biochem. 1989;177:237–243. doi: 10.1016/0003-2697(89)90045-6. [DOI] [PubMed] [Google Scholar]
- 2.Dimmock JR, Raghavan SK, Logan BM, Bigam GE. Antileukemic evaluation of some Mannich bases dervived from 2-ar-ylidene-1,3-diketones. Eur J Med Chem. 1983;18:248–254. [Google Scholar]
- 3.Benvenuto JA, Connor TA, Monteith DK, Laidlaw JL, Adams SC, Matney TS, Theiss JC. Degradation and inactivation of antitumor drugs. J Pharm Sci. 1993;82:988–991. [PubMed] [Google Scholar]
- 4.Chen G, Waxman DJ. Role of cellular glutathione and glutathione S-transferase in the expression of alkylating agent cytotoxicity in human breast cancer cells. Biochem Pharmacol. 1994;47:1079–1087. doi: 10.1016/0006-2952(94)90420-0. [DOI] [PubMed] [Google Scholar]
- 5.Tsutsui K, Komuro C, Ono K, Nishidia T, Shibamoto Y, Takahashi M, Abe M. Chemosensitization by buthionine sulfoximine in vivo. Int J Radiat Oncol, Biol Phys. 1986;12:1183–1186. doi: 10.1016/0360-3016(86)90254-3. [DOI] [PubMed] [Google Scholar]
- 6.Dimmock JR, Padmanilayam MP, Puthucode RN, Nazarali AJ, Motaganahalli NL, Zello GA, Quail JW, Oloo EO, Kraatz HB, Prisciak JS, Allen TM, Santos CL, Balzarini J, De Clercq E, Manavathu EK. A conformational and quantitative structure–activity relationship study of cytotoxic 3,5-bis-arylidene-4-piperidones and related N-acryloxyl analogs. J Med Chem. 2001;44:586–593. doi: 10.1021/jm0002580. [DOI] [PubMed] [Google Scholar]
- 7.Das U, Alcorn J, Shrivastav A, Sharma RK, De Clercq E, Balzarini J, Dimmock JR. Design, synthesis and cytotoxic properties of novel 1-[4-(2-alkylaminoethoxy)phenylcarbonyl]-3,5-bis(arylidene)-4-piperidones and related compounds. Eur J Med Chem. 2007;42:71–80. doi: 10.1016/j.ejmech.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 8.Das U, Das S, Bandy B, Stables JP, Dimmock JR. N-Aroyl-3,5-bis(benzylidene)-4-piperidones: a novel class of antimycobacterial agents. Bioorg Med Chem. 2008;16:3602–3607. doi: 10.1016/j.bmc.2008.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pati HN, Das U, Quail JW, Kawase M, Sakagami H, Dimmock JR. Cytotoxic 3,5-bis(benzylidene)piperidin-4-ones and N-acyl analogs displaying selective toxicity for malignant cells. Eur J Med Chem. 2008;43:1–7. doi: 10.1016/j.ejmech.2007.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Craig PN. Interdependence between physical parameters and selection of substituent groups for correlation studies. J Med Chem. 1971;14:680–684. doi: 10.1021/jm00290a004. [DOI] [PubMed] [Google Scholar]
- 11.Suffness M, Douros J. Drugs of Plant Origin. In: De Vita VT Jr, Busch H, editors. Methods in Cancer Research. Part A. Vol. 16. Academic Press; New York: 1979. p. 84. [Google Scholar]
- 12.Boyd MR, Paull KD. Some practical considerations and applications of the National Cancer Institute in vitro anticancer drug discovery screen. Drug Dev Res. 1995;34:91–109. [Google Scholar]
- 13.Grever MR, Schepartz SA, Chabner BA. The National Cancer Institute: cancer drug discovery and development program. Semin Oncol. 1992;19:622–638. [PubMed] [Google Scholar]
- 14.Remers WA. Antineoplastic Agents. In: Delgado JN, Remers WA, editors. Wilson and Gisvold’s Textbook of Organic Medicinal and Pharmaceutical Chemistry. 10. Lippincott-Raven; Philadelphia, PA: 1998. p. 367. [Google Scholar]
- 15.Greenspan EM, Bruckner HW. Aspects of Clinical Pharmacology. In: Greenspan EM, editor. Clinical Cancer Chemotherapy. Raven Press; New York: 1975. p. 37. [Google Scholar]
- 16.Quinn FR, Milne GWA. Toxicities derived from anti-tumor screening data. Fundam Appl Toxicol. 1986;6:270–277. doi: 10.1016/0272-0590(86)90240-x. [DOI] [PubMed] [Google Scholar]
- 17.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Delivery Rev. 1997;23:3–25. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 18.Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45:2615–2623. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.