

Abstract
The process by which the periderm transitions to stratified epidermis with the establishment of the skin barrier is unknown. Understanding the cellular and molecular processes involved is crucial for the treatment of human pathologies, where abnormal skin development and barrier dysfunction are associated with hypothermia and perinatal dehydration. For the first time, we demonstrate that retinoic acid (RA) levels are important for periderm desquamation, embryonic skin differentiation and barrier formation. Although excess exogenous RA has been known to have teratogenic effects, little is known about the consequences of elevated endogenous retinoids in skin during embryogenesis. Absence of cytochrome P450, family 26, subfamily b, polypeptide 1 (Cyp26b1), a retinoic-acid-degrading enzyme, results in aberrant epidermal differentiation and filaggrin expression, defective cornified envelopes and skin barrier formation, in conjunction with peridermal retention. We show that these alterations are RA dependent because administration of exogenous RA in vivo and to organotypic skin cultures phenocopy Cyp26b1−/− skin abnormalities. Furthermore, utilizing the Flaky tail (Ft/Ft) mice, a mouse model for human ichthyosis, characterized by mutations in the filaggrin gene, we establish that proper differentiation and barrier formation is a prerequisite for periderm sloughing. These results are important in understanding pathologies associated with abnormal embryonic skin development and barrier dysfunction.
Key words: Retinoic acid, Cyp26b1, Periderm, Skin differentiation, Skin barrier formation, Filaggrin
Introduction
Retinoic acid (RA) is the most biologically active retinoid, being also a potent teratogen in rodent and human fetuses, with severe effects in craniofacial and limb development (Lammer et al., 1985; Ross et al., 2000). The endogenous level of RA is determined by the balance of the activity between the enzymes that synthesize RA (retinaldehyde dehydrogenates) and the enzymes that degrade RA (CYP26s). RA mediates its activity through the binding to RA receptors (RARs) (Niederreither and Dollé, 2008; Ross et al., 2000).
Retinoids are well known to influence skin development; however, contrasting results have been reported between in vivo and in vitro results (Imakado et al., 1995; Lee et al., 2009; Saitou et al., 1995; Yuspa et al., 1983; Yuspa and Harris, 1974), and the specific role of RA during embryonic skin differentiation has not been determined. Skin functions as a barrier to the physical environment, preventing dehydration and invasion of pathogenic microorganisms. During murine embryogenesis, the first barrier is a transitional superficial layer, termed periderm, that emerges at approximately embryonic day (E)8.5 (M'Boneko and Merker, 1988). The induction of the keratin-associated proteins (KAP) complex genes is associated with peridermal development (Cui et al., 2007), and coincides with the formation of a functional barrier by the underlying developing stratified epidermis. It has been proposed that when peridermal cells start to slough off at approximately E16.5, the cornified layers of the fully differentiated epidermis replace it as a barrier (Hardman et al., 1999; Segre, 2006). Keratinocytes in the epidermis undergo a differentiation process that culminates with the formation of the cornified layers. This process is associated with expression of differentiation-specific proteins, such as filaggrin (FLG), loricrin (LOR), S100 proteins, late cornified envelope (LCE) proteins and the small proline-rich (SPRR) proteins, encoded by genes clustered in the epidermal differentiation complex (Jackson et al., 2005; Marshall et al., 2001; Mischke et al., 1996). Cornified layers consist of corneocytes, which are terminally differentiated keratinocytes characterized by the cornified envelope (CE) (Segre, 2006).
Utilizing mouse models with embryonic deletion of cytochrome P450, family 26, subfamily b, polypeptide 1 (Cyp26b1), which encodes an RA-degrading enzyme, in vivo administration of RA to pregnant mice and to organotypic skin cultures, we have addressed, for the first time, the effects of RA in developing mammalian skin. We demonstrate that alterations in the desquamation of the peridermal layer are concomitant with RA-dependent abnormal epidermal morphology and differentiation, and decreased filaggrin expression, resulting in impairment of the skin barrier formation. Our findings establish the requirement of a functional barrier for peridermal sloughing, and are validated by the analysis of the Flaky tail (Ft/Ft) mouse model (Fallon et al., 2009; Presland et al., 2000; Scharschmidt et al., 2009). In this model, characterized by filaggrin mutations and consequential barrier defects, we have determined peridermal retention.
Our results establish a close relationship between stratum corneum maturation, skin barrier formation, periderm retention and how alterations in the interplay of these processes might lead to consequential pathologies of the skin.
Results
The absence of Cyp26b1 leads to aberrant skin morphology and upregulation of RA-induced genes in epidermis
Cyp26b1 expression begins in the mesenchyme surrounding hair follicles at E14.5 and is strongly expressed in the dermis by E18.5 (Abu-Abed et al., 2002). To evaluate the expression of Cyp26b1 in skin, we performed real-time PCR and demonstrated that it is present in both epidermal and dermal fractions of E17.5 skin (Fig. 1A). The expression of Cyp26b1 in the dermis was five-fold higher than in epidermis. Cyp26a1 and Cyp26c1, other members of the Cyp26 family, were undetectable in skin (data not shown). Keratin 1 and vimentin mRNAs were used to confirm purity of the epidermal and dermal samples, respectively (Fig. 1B).
Fig. 1.

Altered skin morphology and RA-induced genes in Cyp26b1−/− developing epidermis. (A,B) Relative Cyp26b1 (A), and keratin 1 (K1) and vimentin (Vim) expression (B) in E17.5 epidermis and dermis, determined by real-time PCR. Values are means ± s.d., relative to epidermis (set as 1). Keratin 1 (K1) and vimentin (Vim) mRNAs were at undetected level in dermis and epidermis, respectively. (C) Absence of Cyp26b1 leads to significant Rarb upregulation in epidermis and dermis. Values are means ± s.d., relative to WT epidermis (set as 1). (D–I) Histology of developing skin revealed peridermal cells (arrows) in E16.5 WT and Cyp26b1−/− skin, but altered epidermal thickness and lack of detectable cornified layers at E17.5 (G) and E18.5 in Cyp26b1−/− skin. (J,K) Gross appearance of E18.5 skin. Cyp26b1−/− dorsal skin surface has protrusions. (L,M) Keratin 19 (brown) is upregulated in Cyp26b1−/− epidermis. Epi, epidermis; Der, dermis. **P<0.01. Scale bars; 50 μm (D–I,L,M); 0.2 mm (J,K).
RA is a small diffusible molecule, and its concentration is difficult to measure directly in tissues. Because alterations in RA concentration correlate to Rarb expression, quantification of Rarb has been used to determine levels of RA (Johannesson et al., 2009; Noji et al., 1991). We examined Rarb expression and determined that it was six-fold higher in Cyp26b1−/− than in wild-type (WT) epidermis and 14-fold higher in the dermis (Fig. 1C).
Histological analysis of E16.5 Cyp26b1−/− epidermis showed similar morphology to WT epidermis, with nucleated peridermal cells on the surface (Fig. 1D,E). However, by E17.5, morphological alterations were evident in Cyp26b1−/− epidermis (Fig. 1F,G), and by E18.5, cornified layers were remarkably reduced and hair follicle growth arrested at the germ stage (Fig. 1H,I). To determine whether there were differences in proliferation, we performed immunohistochemistry with anti-Ki67, which showed an increase in proliferative cells in E18.5 Cyp26b1−/− basal layer (supplementary material Fig. S1). Immunofluorescent staining with anti-caspase 3 antibody, which was used to detect apoptotic cells, showed no major difference between WT and Cyp26b1−/− skin during embryogenesis (data not shown). Light microscopy of the surface of Cyp26b1−/− skin showed numerous protrusions not observed in WT skin (Fig. 1J,K).
Keratin 19 (K19), a keratin not normally expressed in stratified epithelia, is induced by RA in epithelial cell lines and its expression is dependent on the extracellular RA concentration (Crowe, 1993). The altered endogenous RA levels linked to the absence of Cyp26b1, strongly induced K19 in E18.5 Cyp26b1−/− epidermis (Fig. 1L,M). Glycogen has been detected in the epidermis of RA-treated chick embryonic skin culture using periodic acid–Schiff (PAS) staining (Obinata et al., 1991). We detected strong PAS staining in suprabasal layers of Cyp26b1−/− skin but not in WT skin (supplementary material Fig. S2).
To establish whether the observed phenotype was attributable to increased dermal RA levels, we investigated the consequences of dermal-specific Cyp26b1 deletion. For this purpose, we established Hoxb6Cre;Cyp26b1f/− mice by crosses between mice carrying floxed Cyp26b1 alleles (Cyp26b1f/f) and Hoxb6Cre;Cyp26b1+/−. Cyp26b1f/+ littermates were used as control. Targeting vector strategy for the generation of Cyp26b1f/f mice and the genotyping results for Hoxb6Cre;Cyp26b1f/− mice are shown in Fig. 2A and described in the Materials and Methods. Dermal-specific Hoxb6Cre recombination was confirmed at E14.5, the embryonic stage that coincided with the onset of Cyp26b1 expression, by crosses with R26R reporter mice (Fig. 2B). The recombination was still detectable at E18.5 (Fig. 2B). There was no morphological difference between E18.5 control (Cyp26b1f/+) and Hoxb6Cre;Cyp26b1f/− epidermis (Fig. 2C, top panels) and immunohistochemical analysis of keratin 5 (K5) and filaggrin showed comparable expression patterns (Fig. 2C, bottom panels). Skeletal preparations showed abnormal hindlimb development in Hoxb6Cre;Cyp26b1f/− mice (supplementary material Fig. S3), a phenotype that corroborated previously determined effects of RA excess in limb development (Campbell et al., 2004; Dranse et al., 2011; Yashiro et al., 2004). These results support that dermal deletion of Cyp26b1 is not a sufficient effector for the development of the epidermal phenotype observed in Cyp26b1−/− skin.
Fig. 2.

Dermal-specific Cyp26b1 deletion is not sufficient for development of skin phenotype. (A) Schematic representation of targeting vector and generation of Cyp26b1f/f mice. H, HincII; N, NsiI; RV, EcoRV; S, SpeI. (Right) Genotyping of E18.5 Hoxb6Cre;Cyp26b1f/− mice by PCR determined Cyp26b1 deletion by Cre recombinase in dermis and hindlimb but not in epidermis and brain. (B) X-gal staining in Hoxb6Cre;R26R revealed dermal-specific Cre recombination in the skin. Dotted line indicates the border between the epidermis and the dermis. (C) H&E histological and immunohistological analysis showing no phenotypic difference between E18.5 WT and Hoxb6Cre;Cyp26b1f/− epidermis. Fila, filaggrin (green); K5, keratin 5 (red); DAPI (blue). Scale bars: 50 μm.
Loss of Cyp26b1 in developing skin leads to decreased filaggrin expression and defective skin barrier formation
To determine the molecular effectors and mechanisms involved in the epidermal phenotype in Cyp26b1−/− skin, we performed microarray analysis comparing E18.5 WT and Cyp26b1−/− skin. We found upregulation of genes involved in CE assembly, particularly genes of the Lce and Sprr families (Fig. 3A). Lce3a, Lce3b, Lce3c and Lce3f were prominently upregulated, and Lce3c has been recently shown to be upregulated in response to skin barrier disruption (de Cid et al., 2009). The retinoic-acid-binding proteins cellular retinoic acid binding protein II (CrabpII) and Rarβ were upregulated 13.6- and 7.54-fold, respectively. Interestingly, when CRABPII was increased by RA treatment of human keratinocytes it was associated with absence of terminal differentiation (Siegenthaler et al., 1992). Lipid metabolism is known to be an integral part of barrier formation (Elias, 2005). LipK, LipM and Alox12b, which encode enzymes with essential function in lipid metabolism, were upregulated in Cyp26b1−/− skin (Fig. 3A). The most downregulated genes were keratins associated with hair follicle morphogenesis, which is consistent with our finding that hair follicle development arrested at germ stage in Cyp26b1−/− skin (supplementary material Table S1).
Fig. 3.

Cyp26b1−/− skin is defective in CE and skin barrier formation. (A) Microarray results of altered genes with possible relevance to barrier function in E18.5 Cyp26b1−/− skin compared with WT skin. (B) Dye exclusion assay revealed absence of functional barrier in Cyp26b1−/− fetuses. Transepidermal water loss (TEWL) measurements showed increased water loss in E18.5 Cyp26b1−/− dorsal skin (**P<0.01). Values are means ± s.d., relative to control (set as 1). (C) Transmission electron micrographs of E18.5 dorsal skin revealed lack of keratohyalin granules in Cyp26b1−/− skin. The number and thickness of cornified layers (C1–8) was reduced in Cyp26b1−/− skin. (D) CEs isolated from E18.5 WT skin were rigid and polygonal, whereas Cyp26b1−/− CEs were irregular in shape and many retained nuclei (arrow, and inset at a higher magnification). Scale bars: 200 nm (upper panels in C); 150 nm (lower panels in C).
Because expression of many genes involved in CE formation was altered, we performed skin dye permeability assays and transepidermal water loss (TEWL) measurements to determine whether Cyp26b1−/− fetuses developed a functional skin barrier. Normally, the skin barrier begins to form in the dorsal side of the embryo at approximately E16, and is identified by absence of dye penetration (Hardman et al., 1998). Dye penetration occurred over the whole body in Cyp26b1−/− fetuses from E16.25 to E19, indicating an impairment of skin barrier formation (Fig. 3B). TEWL was significantly higher (P<0.01) in Cyp26b1−/− fetuses at E18.5, demonstrating that the skin barrier was impaired (Fig. 3B).
Transmission electron microscopy (TEM) of E18.5 mutant epidermis showed that keratohyalin granules, generally seen in the granular layer in WT skin (Fig. 3C, white arrows) were not present in the Cyp26b1−/− granular layer. Cornified layers that were hard to distinguish under the light microscope in the mutant epidermis were observed by TEM. However, the thickness and number of Cyp26b1−/− cornified layers was notably reduced, and intact nuclei were seen underneath the stratum corneum (Fig. 3C, bottom panels).
The atypical cornified layer and disrupted barrier led us to isolate CEs from E18.5 Cyp26b1−/− skin. There were considerably fewer CEs and they had a fragile and irregular morphology, and many contained nuclei (Fig. 3D; arrow, inset shown at higher magnification). By contrast, CEs from WT skin were mostly rigid and had polygonal morphology (Fig. 3D).
To analyze alterations in the epidermal differentiation process, we examined the expression of differentiation proteins (Fig. 4A–H). Immunohistochemistry of keratin 5 (K5), keratin 10 (K10), involucrin and loricrin showed comparable strata expression pattern in Cyp26b1−/− and WT epidermis (Fig. 4A–F). However, filaggrin was expressed at lower levels with considerably fewer and smaller filaggrin granules in the Cyp26b1−/− epidermis (Fig. 4G,H). Western blot analysis revealed that profilaggrin was processed to filaggrin similarly in E18.5 and postnatal 3 days WT skin, and conversely, no filaggrin was detected in Cyp26b1−/− skin (Fig. 4I). By contrast, loricrin was more abundant in Cyp26b1−/− skin than in E18.5 and postnatal 3 days WT skin (Fig. 4I). The downregulation of filaggrin expression was corroborated at the RNA level by real-time PCR (Fig. 4J).
Fig. 4.

Altered skin differentiation with decreased filaggrin expression in Cyp26b1−/− epidermis. (A–F) Keratin 5 (K5; red), keratin 10 (K10; green), involucrin (Inv; green) and loricrin (Lori; green) were detected by immunohistochemistry in E18.5 WT and Cyp26b1−/− skin. Pan-Krt; pan-keratin (red). (G,H) Filaggrin (Fila; green) was reduced and distribution was irregular on Cyp26b1−/− skin, as observed by immunohistochemistry. DAPI staining is blue. (I) Western blot analysis showed that profilaggrin is processed to filaggrin in E18.5 WT and in postnatal 3 days (P3) WT skin, but no filaggrin is detected in Cyp26b1−/− skin. Increased loricrin (Lori) was detected in E18.5 Cyp26b1−/− skin. (J) Significant filaggrin downregulation was detected by real-time PCR on Cyp26b1−/− E16.5, E17.5 and E18.5 skin samples. Values are means ± s.d., relative to E16.5 WT skin (set as 1); **P<0.01. Scale bars: 50 μm.
Altogether, our results demonstrate that the absence of Cyp26b1 during embryogenesis is associated with altered filaggrin expression accompanied by abnormal CE formation and skin barrier defects.
Increased RA in epidermis affects peridermal development
We identified that the onset of the epidermal phenotype of Cyp26b1−/− fetuses correlated with the timing of peridermal desquamation. Histological analysis revealed that, whereas cornified layers were formed in E17.5 WT skin, peridermal cells were still present in Cyp26b1−/− skin, with hardly detectable cornified layers at this stage (Fig. 5A,B). Keratin 6 (K6), a characterized peridermal marker (Mazzalupo and Coulombe, 2001), was observed on the surface of both WT and Cyp26b1−/− E16.5 epidermis and was still robustly detected in E17.5 Cyp26b1−/− epidermis (Fig. 5C–F).
Fig. 5.

Increased RA levels affect peridermal desquamation. (A,B) Histological analysis revealed peridermal cells in E17.5 Cyp26b1−/− skin (B, black arrows). (C–F) Keratin 6 (K6; brown), was detected in the uppermost layer of E17.5 Cyp26b1−/− skin. Counterstaining was performed with Hematoxylin (blue). (G) Top panel; microarray results of expression of peridermal-associated genes in E18.5 Cyp26b1−/− skin. Bottom panel: KAP cluster location on mouse chromosome 16. Highly upregulated KAP genes are highlighted in red. (H–K) In situ hybridization (red grains) analysis showed expression of Krtap13 in WT and Cyp26b1−/− peridermal layer at E16.5 (H,I), with persistent expression in Cyp26b1−/− E17.5 (K) epidermis. Scale bars: 50 μm. (L) Real-time PCR showed marked increase in relative expression of Krtap13, 2310034C09Rik and 2310057N15Rik in E17.5 Cyp26b1−/− skin. Values are means ± s.d.; the expression level of each gene is given relative to that in E16.5 WT skin (set as 1); **P<0.01.
Microarray analysis determined that the most highly upregulated genes in mutant skin, Krtap13, 2310034C09Rik and 2310057N15Rik (406-fold, 345-fold and 126-fold, respectively), were members of the KAP (keratin-associated proteins) complex (Fig. 5G). KAP genes are clustered on mouse chromosome 16 and have been shown to be markers of periderm development (Fig. 5G) (Cui et al., 2007). During normal embryogenesis, expression of Krtap13 is highest at E16.5 and markedly decreases by E17.5, timings that coincide with peridermal desquamation (Takaishi et al., 1998). In situ hybridization analysis showed that Krtap13 was restricted to the periderm in E16.5 Cyp26b1−/− skin, as in the WT, but that substantial Krtap13 expression was still detected in the upper layers of E17.5 Cyp26b1−/− epidermis (Fig. 5H–K). Real-time PCR determined that Krtap13, 2310034C09Rik and 2310057N15Rik were expressed and maintained at high levels in Cyp26b1−/− skin from E16.5 onwards, whereas expression for those genes was markedly decreased in E17.5 WT skin (Fig. 5L).
Peridermal-specific Krtap13 expression is directly linked to increased RA levels
Next, we addressed the effect of increased in vivo RA levels on the epidermal barrier formation and peridermal development, by feeding RA through gavage to WT pregnant mice with embryos at E15.4, E16.5 and E18.4 of gestation (Fig. 6A). Defective barrier formation in RA-treated fetuses became evident by dye penetration assay, and skin from E18.5 RA-treated fetuses had fewer CEs that presented the characteristic fragile morphology of immature CEs with retention of nuclei seen in Cyp26b1−/− fetuses (Fig. 6B). Histological analysis showed that there were substantial differences between E18.5 vehicle- and RA-treated groups: the RA-treated skin phenocopied the Cyp26b1−/− skin, with fewer cornified layers and protrusions on the skin surface (Fig. 6C–F). There was also downregulation of filaggrin expression in RA-treated E18.5 skin (Fig. 6G,H). This finding was corroborated by real-time PCR analysis, which revealed significant (P<0.01) downregulation of filaggrin expression in RA-treated skin compared with vehicle (oil) alone-treated skin (Fig. 6I). In addition, RA-treated skin showed expression of K6 (Fig. 6J,K) and upregulated Krtap13 expression, demonstrating that Krtap13 responds to increased RA levels (Fig. 6L). Krtap13 RA-dependent induction decreased at E17.5, correlating with the timing of RA administration (no RA administration between E16.5 and E18.4).
Fig. 6.

RA excess in WT embryos phenocopies the epidermal defects of Cyp26b1−/− mouse embryos. (A) Timetable for RA administration to WT pregnant mice (red arrows) and for skin sample isolation (black arrowheads). (B) Dye exclusion assay revealed lack of barrier function in RA-treated fetuses collected at E16.8 and E18.5 (ii and iv). CEs isolated at E18.5 (iv) from RA-treated fetuses were irregular in shape and retained nuclei (arrow and inset at a higher magnification). (C–F) RA-treated fetal skin phenocopied Cyp26b1−/− skin at E18.5 (iv) both at the histological level and in gross appearance. (G,H) Immunohistochemical analysis showed that filaggrin (Fila; green) expression was downregulated in RA-treated skin (bottom panel). K5, keratin 5 (red). (I) Downregulation of filaggrin expression observed in E16.8, E17.5 and E18.5 Cyp26b1−/− skin and in RA-treated embryos was also corroborated by real-time PCR. Values are means ± s.d. relative to that in E16.8 WT skin (set as 1). (J–K) K6 (brown) was positive in RA-treated skin. Counterstaining was performed with Hematoxylin (blue). (L) Real-time PCR showed persistent expression of Krtap13 at E17.5 and significant Krtap13 upregulation 2 hours after RA administration at E18.4; **P<0.01. Scale bars: 50 μm (C,D,G,H). 0.2 mm (E,F).
Using another approach to address the effects of RA on skin development, we performed studies on skin organotypic cultures with back skin of E15.5 embryos. A functional skin barrier was established 48 hours after initiation of the culture in the control group (O'Shaughnessy et al., 2007), revealed by the dye Lucifer Yellow, in the upper layers of the stratum corneum (Fig. 7A). However, dye diffused through all epidermal layers in RA-treated organotypic cultures and was present in the dermis and hypodermis (Fig. 7B). Filaggrin expression was detected in control skin but not in RA-treated skin (Fig. 7C,D). Real-time PCR revealed that Krtap13 was upregulated 15.7-fold in RA-treated skin explants compared with control skin explants, with Rarb expression also upregulated (35.5-fold; Fig. 7E). By contrast, Krtap13 was not induced in RA-treated primary mouse keratinocytes overexpressing Rarb with RA treatment (data not shown). These results support our findings in Cyp26b1−/− mice and in WT mice administered RA in vivo, establishing a link between elevated levels of RA, altered filaggrin expression, disruption of barrier formation and periderm retention, evidenced by upregulated Krtap13 and sustained expression of K6. These results also establish a direct association between Krtap13 expression and the concomitant downregulation of filaggrin expression.
Fig. 7.
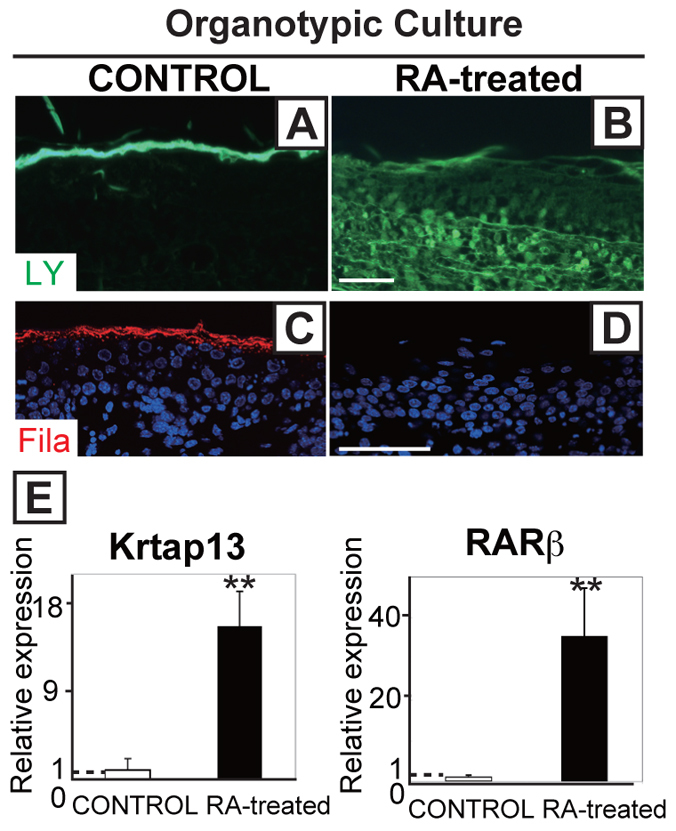
RA excess in organotypic skin cultures phenocopies the epidermal defects of Cyp26b1−/− mouse embryos. (A,B) Lucifer Yellow dye (LY; green) assays applied onto skin explants showed penetration of the dye into the hypodermis in RA-treated skin. (C,D) Filaggrin (red) was detected in 48-hour control cultures but not in RA-treated skin explants. (E) Krtap13 and Rarb were significantly upregulated in RA-treated skin. Values are means ± s.d., relative to control skin (set as 1). **P<0.01. Scale bars: 50 μm.
Finally, we examined Ft/Ft fetuses, which are homozygous for a frameshift mutation in the filaggrin gene (Fallon et al., 2009) (Fig. 8A), to address the link between decreased filaggrin, barrier defects and periderm desquamation. Ft/Ft mice skin barrier defects after birth have been reported (Moniaga et al., 2010; Scharschmidt et al., 2009), but we demonstrated barrier dysfunction during Ft/Ft fetal development with a dye exclusion assay (Fig. 8B). C57B6 (B6) mice were used as control because Ft/Ft mice were backcrossed onto the B6 background (Moniaga et al., 2010; Scharschmidt et al., 2009) (Fig. 8B). We corroborated that filaggrin was not detectable in Ft/Ft skin, although it was present in suprabasal layer in B6 skin (Fig. 8C). Based on the results of our Cyp26b1−/− model, we hypothesized that the barrier defects in Ft/Ft mice would be accompanied by alterations in peridermal development. We addressed this by assaying the expression of K6 and Krtap13 in Ft/Ft mice, and determined that both of these markers were upregulated at E17.75 (Fig. 8C,D). Altogether, these results show a link between impaired barrier function and periderm retention.
Fig. 8.
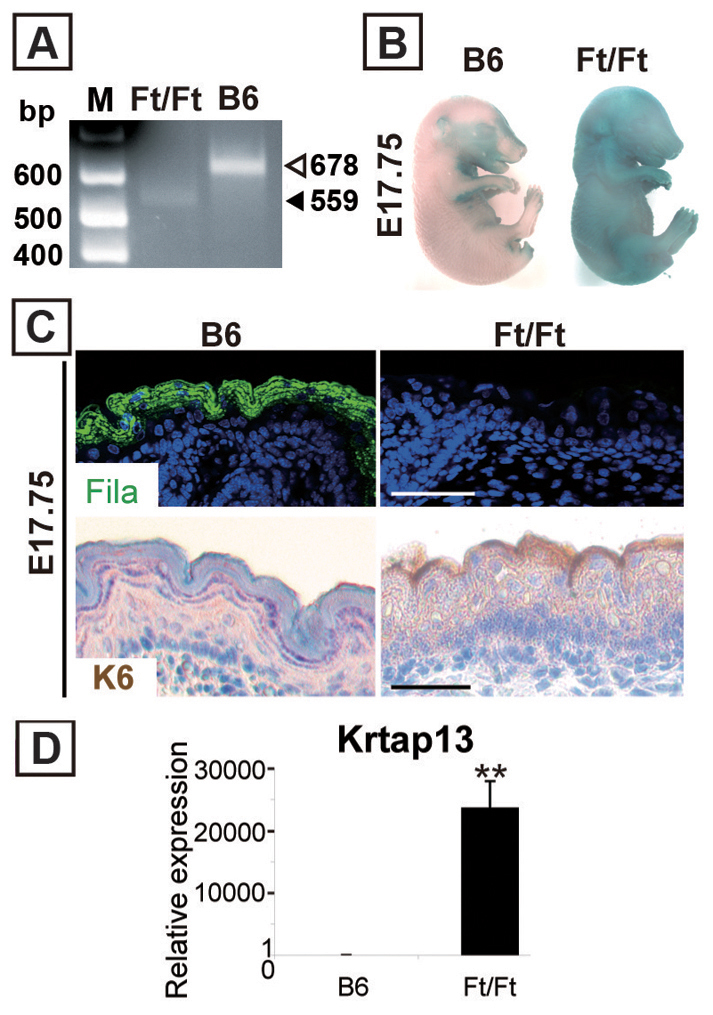
Loss of filaggrin causes skin barrier defects accompanied by peridermal retention. (A) Genotyping as described by Fallon et al. (Fallon et al., 2009) showed a 678 bp band for C57BL/6 (B6) and a 559 bp band for Ft/Ft mice. M, molecular mass marker; bp, base pair. (B) Dye assay revealed a skin barrier defect in Ft/Ft fetuses. (C) In Ft/Ft E17.75 fetal skin, filaggrin protein was not detected (top panels). However, the peridermal marker K6 was detected in the superficial layer (bottom panels). (D) Krtap13 was significantly upregulated in E17.75 Ft/Ft fetal skin. Values are means ± s.d. relative to that in B6 skin (set as 1); **P<0.01.
Discussion
We performed a comprehensive analysis of the effects of high endogenous RA concentration during embryonic skin development that leads to aberrant CE formation, with lack of skin barrier function linked to altered periderm development (Fig. 9), and we demonstrate that they are interrelated processes.
Fig. 9.

Schematic summary of the findings of this study.
The absence of Cyp26b1 in developing skin leads to an aberrant epidermal phenotype
Here we show that in developing Cyp26b1−/− skin barrier formation is defective and cornified layers are markedly decreased.
The normal skin development requires controlled epidermal stratification and CE formation. Abnormal CE assembly linked to defective barrier acquisition is seen in mouse models with targeted ablation of Arnt and loricrin, triple targeted deletion of involucrin, periplakin and envoplakin, and transgenic Ets1 overexpression in the suprabasal layer (Geng et al., 2006; Koch et al., 2000; Nagarajan et al., 2010; Sevilla et al., 2007). However, the phenotype of the CEs in these knockout mouse models is less severe than in Cyp26b1−/− mice, suggesting that RA potentially regulates an encompassing group of effectors and structural proteins required for CE assembly. Other genes, which are part of the epidermal differentiation complex, that are also involved in CE assembly are Lce and Sprr. These genes, in particular Lce, have been recently shown to be upregulated in response to skin barrier disruption (de Cid et al., 2009). Furthermore, Bergboer and collaborators showed that normal skin barrier function correlates with LCE groups 1, 2, 5 and 6, whereas expression of LCE3 genes is linked to barrier repair after injury or inflammation (Bergboer et al., 2011). These findings, in conjunction with the specific and prominent upregulation of expression of the Lce3 group in the Cyp26b1−/− skin, suggest a mechanistic link between barrier disruption and inflammation in this mouse model that correlate with the systematic effects of increased RA levels. In particular, in dominant-negative Rar mice, an RA-deficiency model, there is a loss of epidermal barrier function attributed to disruption of the lipid layer (Imakado et al., 1995). Altogether, the published results and our present findings establish that specific temporal and spatial RA levels are required for proper skin differentiation during development.
We also show that absence of Cyp26b1 and exogenous RA administration correlate with decreased filaggrin expression. Filaggrin is produced during late epidermal differentiation as a large polypeptide precursor that is cleaved, as keratinocytes terminally differentiate, to produce corneocytes in the stratum corneum. RA is known to regulate the conversion of profilaggrin to filaggrin in human keratinocytes (Asselineau et al., 1990), and the human filaggrin promoter has retinoic acid response elements (RAREs), which function to suppress the promoter activity in the presence of RA (Presland et al., 2001; Sandilands et al., 2009). These results and our findings showing decreased filaggrin expression in Cyp26b1−/− skin and RA-treated fetuses, strengthen the connection between excess RA inhibiting profilaggrin, and therefore filaggrin expression, which is also supported by our results on RA-treated organotypic cultures. This phenomenon is specific to filaggrin because loricrin, another established late differentiation marker, was upregulated in Cyp26b1−/− skin. Increased loricrin is also observed in Ft/Ft mice, which have analogous mutations to human filaggrin mutations (Fallon et al., 2009; Presland et al., 2000), and which have been reported to have skin barrier defects (Moniaga et al., 2010; Scharschmidt et al., 2009). Loss-of-function mutations in the filaggrin gene have been identified as a cause for ichthyosis vulgaris and atopic dermatitis (Palmer et al., 2006; Sandilands et al., 2009). These published results and the results presented here, validate the effect of RA on the linked processes of filaggrin expression and development of a functional skin barrier.
The role of RA in peridermal development and skin barrier formation
The mechanism(s) through which periderm transitions to the stratified epidermis or the pathways and effectors that determine the timing of periderm sloughing have not been established. However, recent reports have demonstrated the correlation between peridermal phenotype and the expression of genes clustered in the KAP complex in mice (Cui et al., 2007). The genes in this complex have been very well characterized with respect to genomic arrangement and expression patterns in human hair (Rogers et al., 2002). Of these clustered genes Krtap13, 2310034C09Rik and 2310057N15Rik, which are specifically expressed in mouse periderm, were the most upregulated genes in Cyp26b1−/− skin.
In lymphotoxin-β-deficient mice, there is premature desquamation of periderm by E15.5, and Krtap13, as well as other genes in the KAP complex, are not detected at E16.5 (Cui et al., 2007). Therefore, the temporal and spatial expression of Krtap13 at E16.5 appears to play a key role in peridermal development, and peridermal retention and/or sloughing are influenced by the stage of the epidermal differentiation process.
Although, using in vivo and in vitro models, we demonstrate that KAP genes respond to RA, the molecular mechanism connecting the expression of KAP-complex genes and RA signaling is at present unknown. We searched a 90 kb genomic region harboring the KAP complex and found several RARE-like and half-RARE sequences, and the mechanistic significance of these sites will be the focus of future research. Although RARE consensus sequences are regarded as direct repeats (DR) of 5′-RGKTCA-3′ separated by one, two or five nucleotides (Umesono et al., 1991), chromatin immunoprecipitation analysis on mouse embryonic fibroblasts recently determined that the majority of RA target genes contain anomalously spaced DRs rather than consensus DRs, suggesting more complexity in RA signaling (Delacroix et al., 2010). Taking these facts into consideration, it remains a challenge to determine the functionality of these regulatory elements in the KAP complex. That both exogenous RA and elevated endogenous RA, caused by the absence of Cyp26b1, induced KAP complex genes supports the idea that upregulated expression of these genes is a direct response to RA and that regulation of the KAP complex is intrinsic to periderm development.
It has been proposed that the development of differentiated stratified epidermis occurs concomitantly with the desquamation of the overlying peridermal cells (Segre, 2006). However, there is no evidence to support that periderm is required for epidermal stratification or alternatively that proper barrier formation by the underlying stratifying epidermis is necessary for timely desquamation of the periderm.
We demonstrate that in Ft/Ft fetuses, which have a homozygous frameshift filaggrin mutation (Fallon et al., 2009) and well-characterized skin barrier defects after birth (Scharschmidt et al., 2009), have periderm retention, as assessed by K6 and Krtap13 upregulation. These results, in conjunction with our RA-dependent findings, strongly suggest that formation of a functional barrier in the developing epidermis is a requisite for a proper temporal peridermal sloughing during embryogenesis. Further studies will be designed to analyze the relationship between periderm desquamation, barrier dysfunction and pathologies of the skin such as ichthyosis and ichthyosis-like conditions and whether RA metabolism during fetal development can contribute to the pathology.
Materials and Methods
Mice
Cyp26b1+/− mice were described previously (Yashiro et al., 2004). Mice carrying the floxed Cyp26b1 allele (Cyp26b1f/f) were generated as described in Fig. 2 and below. The Hoxb6Cre mice were kindly provided by Susan Mackem (Lowe et al., 2000). R26R reporter mice were purchased from Jackson Laboratories (Soriano, 1999). Flaky tail (Ft) mice were kindly provided by John Sundberg (Presland et al., 2000). Embryonic skin samples were photographed using an Olympus SZX9 microscope with a SPOT Pursuit USB Camera (SPOT Imaging Solutions).
Targeting vector map and the strategy of generating Cyp26b1flox/flox mice
The targeting vector consisted of a subcloned 22 kb SpeI fragment with one loxP site inserted at the NsiI site of the intron 4 and an FRT-neo-FRT-loxP cassette inserted at the HincII site in the 3′ untranslated region of Cyp26b1 in a pMCI-DTpA plasmid. All the DNA fragments for generating the vector were obtained as previously described (Uehara et al., 2009; Yashiro et al., 2004). The targeting vector was linearized with SalI before introduction into R1 ES cells by electroporation. Homologous recombination of a neo allele was confirmed by Southern blotting. Genomic DNA was digested with EcoRV, and the resulting fragments were hybridized with the 3′ probe indicating the 27.5 kb band from the WT allele and the 14 kb one from a neo allele (data not shown). To create a flox allele, a PGK-neo cassette was removed by crossing CAG-FLPe Deleter mice (Kanki et al., 2006).
The genotype of a flox and/or a null allele was determined by polymerase chain reaction (PCR) analysis of genomic DNA with three primers: P1, 5′-AAG-TACACCTGGCAGACATG-3′; P2, 5′-CCTGTCCCATATTTATTCACTGAC-3′; P3, 5′-CTCCTCTTAAAGCTTCTCTA-3′.
P2 and P3 will amplify a 400 bp product from a flox allele. P1 and P3 will amplify a 500 bp product from a null allele but not a 4 kb product from a flox allele by the following cycling conditions: 95°C for 2 minutes, 38 cycles of 95°C for 30 seconds, 58°C for 50 seconds and 72°C for 30 seconds, followed by 72°C for 8 minutes. All reactions included 1.5 mM MgCl2, 0.2 mM dNTPs, 0.3 mM P1, P2 and P3.
Skeletal staining
Skin was removed from E18.5 fetuses and fixed in ethanol for 3 days and then the solution was replaced with acetone. Samples were incubated in 0.02% Alcian Blue, 0.01% Alizarin Red (Sigma) in 75% ethanol at 37°C for 5 days, and then in 1% potassium hydroxide for 2 days.
Histology, immunohistochemistry and western blot analysis
For histological analysis, embryos or embryonic skin samples were fixed in 4% paraformaldehyde, embedded in paraffin and sectioned (10 μm) for staining with Hematoxylin and Eosin or Periodic acid–Schiff (PAS) reagent. PAS staining was performed according to the method of Obinata et al. (Obinata et al., 1991).
The antibodies used for western blotting and immunohistochemistry are described in supplementary material Table S2. For immunofluorescence, the sections were incubated with primary antibodies overnight at 4°C and then with fluorescent secondary antibodies. Sections were mounted with DAPI and photographed with a 510 Meta confocal microscope (Zeiss). For immunohistochemistry, the sections were incubated with primary antibodies using the Vectastain ABC kit (Vector Laboratories) using diaminobenzidine as the enzyme substrate. Mouse-on-Mouse (MOM) detection kit and antigen unmasking solutions (Vector Laboratories) were used if applicable. Images were acquired using a Zeiss axio scope a1 with a AxioCam MRc camera (Zeiss). Western blot procedures were as described by Hwang et al. (Hwang et al., 2011). The blots were probed with primary antibodies and HRP-conjugated secondary antibodies. ECL (Amersham Pharmacia Biotech) reagent was used for detection.
X-gal staining and in situ hybridization
X-gal staining was performed with 1 mg/ml X-gal on frozen sections (30 μm). Radioactive in situ hybridization on paraffin sections was carried out according to Morasso et al. (Morasso et al., 2010). Krtap13 (NM010671) DNA probe was purchased from Thermo Scientific.
Transmission electron microscopy (TEM)
Ultrathin sections were counter-stained with uranyl acetate and lead citrate and examined with a Tecnai TF30 transmission electron microscope.
Barrier function assay and extraction of cornified envelopes
Dye penetration assays were performed as previously described (Hardman et al., 1998). Transepidermal water loss was measured using a Tewameter (Courage+Khazaka, Köln, Germany). Unpaired two-tailed Student's t-test was used to assess significance of the data. CEs were prepared according to Morasso et al. (Morasso et al., 1996).
RNA isolation, real-time PCR and microarray
Fetal skin, epidermis and dermis were collected and kept in Trizol (Invitrogen) until RNA isolation using an RNeasy kit (Qiagen). Real-time PCR was performed in duplicate using the iQTM SYBR Green Supermix (Bio-Rad). The primer sequences for real-time PCR are summarized in supplementary material Table S3. Individual gene expression was normalized against the RPLP0 housekeeping gene. Two-tailed Student's t-test was used to assess significance of the data. Microarray analysis, including data processing, was performed by the National Institutes of Health NIDDK Genomics Core Facility (Chattopadhyay et al., 2009) on independent samples of E18.5 WT and Cyp26b1−/− embryos (n=3 each).
RA treatment in pregnant mice
All-trans RA (Sigma) was administered to pregnant CD-1 mice (50 mg/kg of body weight) in corn oil by oral gavage (Okano et al., 2007). Control pregnant mice received corn oil only.
Organotypic culture
Organotypic skin culture was performed as previously described (Kashiwagi et al., 1997; O'Shaughnessy et al., 2007). RA (Sigma) was added at 2.5 μM in DMSO; the control group was administered DMSO only. 30 μl of 1 mM Lucifer Yellow (Sigma) was applied to skin samples and incubated at 37°C for 1 hour before fixation with 4% paraformaldehyde.
Supplementary Material
Acknowledgements
We thank J. Sundberg for providing the Flaky tail mice; M. Uehara for helpful discussions; C. Levy for technical assistance; K. Zaal of the NIAMS Light Imaging Core Facility; R. Leapman of the NIBIB; J. Segre for the loan of the Tewameter.
Footnotes
Funding
This study was supported by an Intramural Research Program of the National Institute of Arthritis and Musculoskeletal and Skin Diseases, National Institutes of Health. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.101550/-/DC1
References
- Abu-Abed S., MacLean G., Fraulob V., Chambon P., Petkovich M., Dollé P. (2002). Differential expression of the retinoic acid-metabolizing enzymes CYP26A1 and CYP26B1 during murine organogenesis. Mech. Dev. 110, 173-177 [DOI] [PubMed] [Google Scholar]
- Asselineau D., Dale B. A., Bernard B. A. (1990). Filaggrin production by cultured human epidermal keratinocytes and its regulation by retinoic acid. Differentiation 45, 221-229 [DOI] [PubMed] [Google Scholar]
- Bergboer J. G., Tjabringa G. S., Kamsteeg M., van Vlijmen-Willems I. M., Rodijk-Olthuis D., Jansen P. A., Thuret J. Y., Narita M., Ishida-Yamamoto A., Zeeuwen P. L., et al. (2011). Psoriasis risk genes of the late cornified envelope-3 group are distinctly expressed compared with genes of other LCE groups. Am. J. Pathol. 178, 1470-1477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell J. L., Jr, Smith M. A., Fisher J. W., Warren D. A. (2004). Dose-response for retinoic acid-induced forelimb malformations and cleft palate: a comparison of computerized image analysis and visual inspection. Birth Defects Res. B Dev. Reprod. Toxicol. 71, 289-295 [DOI] [PubMed] [Google Scholar]
- Chattopadhyay M. K., Chen W., Poy G., Cam M., Stiles D., Tabor H. (2009). Microarray studies on the genes responsive to the addition of spermidine or spermine to a Saccharomyces cerevisiae spermidine synthase mutant. Yeast 26, 531-544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowe D. L. (1993). Retinoic acid mediates post-transcriptional regulation of keratin 19 mRNA levels. J. Cell Sci. 106, 183-188 [DOI] [PubMed] [Google Scholar]
- Cui C. Y., Kunisada M., Esibizione D., Grivennikov S. I., Piao Y., Nedospasov S. A., Schlessinger D. (2007). Lymphotoxin-beta regulates periderm differentiation during embryonic skin development. Hum. Mol. Genet. 16, 2583-2590 [DOI] [PubMed] [Google Scholar]
- de Cid R., Riveira-Munoz E., Zeeuwen P. L., Robarge J., Liao W., Dannhauser E. N., Giardina E., Stuart P. E., Nair R., Helms C., et al. (2009). Deletion of the late cornified envelope LCE3B and LCE3C genes as a susceptibility factor for psoriasis. Nat. Genet. 41, 211-215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delacroix L., Moutier E., Altobelli G., Legras S., Poch O., Choukrallah M. A., Bertin I., Jost B., Davidson I. (2010). Cell-specific interaction of retinoic acid receptors with target genes in mouse embryonic fibroblasts and embryonic stem cells. Mol. Cell. Biol. 30, 231-244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dranse H. J., Sampaio A. V., Petkovich M., Underhill T. M. (2011). Genetic deletion of Cyp26b1 negatively impacts limb skeletogenesis by inhibiting chondrogenesis. J. Cell Sci. 124, 2723-2734 [DOI] [PubMed] [Google Scholar]
- Elias P. M. (2005). Stratum corneum defensive functions: an integrated view. J. Invest. Dermatol. 125, 183-200 [DOI] [PubMed] [Google Scholar]
- Fallon P. G., Sasaki T., Sandilands A., Campbell L. E., Saunders S. P., Mangan N. E., Callanan J. J., Kawasaki H., Shiohama A., Kubo A., et al. (2009). A homozygous frameshift mutation in the mouse Flg gene facilitates enhanced percutaneous allergen priming. Nat. Genet. 41, 602-608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng S., Mezentsev A., Kalachikov S., Raith K., Roop D. R., Panteleyev A. A. (2006). Targeted ablation of Arnt in mouse epidermis results in profound defects in desquamation and epidermal barrier function. J. Cell Sci. 119, 4901-4912 [DOI] [PubMed] [Google Scholar]
- Hardman M. J., Sisi P., Banbury D. N., Byrne C. (1998). Patterned acquisition of skin barrier function during development. Development 125, 1541-1552 [DOI] [PubMed] [Google Scholar]
- Hardman M. J., Moore L., Ferguson M. W., Byrne C. (1999). Barrier formation in the human fetus is patterned. J. Invest. Dermatol. 113, 1106-1113 [DOI] [PubMed] [Google Scholar]
- Hwang J., Kita R., Kwon H. S., Choi E. H., Lee S. H., Udey M. C., Morasso M. I. (2011). Epidermal ablation of Dlx3 is linked to IL-17-associated skin inflammation. Proc. Natl. Acad. Sci. USA 108, 11566-11571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imakado S., Bickenbach J. R., Bundman D. S., Rothnagel J. A., Attar P. S., Wang X. J., Walczak V. R., Wisniewski S., Pote J., Gordon J. S., et al. (1995). Targeting expression of a dominant-negative retinoic acid receptor mutant in the epidermis of transgenic mice results in loss of barrier function. Genes Dev. 9, 317-329 [DOI] [PubMed] [Google Scholar]
- Jackson B., Tilli C. M., Hardman M. J., Avilion A. A., MacLeod M. C., Ashcroft G. S., Byrne C. (2005). Late cornified envelope family in differentiating epithelia--response to calcium and ultraviolet irradiation. J. Invest. Dermatol. 124, 1062-1070 [DOI] [PubMed] [Google Scholar]
- Johannesson M., Ståhlberg A., Ameri J., Sand F. W., Norrman K., Semb H. (2009). FGF4 and retinoic acid direct differentiation of hESCs into PDX1-expressing foregut endoderm in a time- and concentration-dependent manner. PLoS ONE 4, e4794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanki H., Suzuki H., Itohara S. (2006). High-efficiency CAG-FLPe deleter mice in C57BL/6J background. Exp. Anim. 55, 137-141 [DOI] [PubMed] [Google Scholar]
- Kashiwagi M., Kuroki T., Huh N. (1997). Specific inhibition of hair follicle formation by epidermal growth factor in an organ culture of developing mouse skin. Dev. Biol. 189, 22-32 [DOI] [PubMed] [Google Scholar]
- Koch P. J., de Viragh P. A., Scharer E., Bundman D., Longley M. A., Bickenbach J., Kawachi Y., Suga Y., Zhou Z., Huber M., et al. (2000). Lessons from loricrin-deficient mice: compensatory mechanisms maintaining skin barrier function in the absence of a major cornified envelope protein. J. Cell Biol. 151, 389-400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammer E. J., Chen D. T., Hoar R. M., Agnish N. D., Benke P. J., Braun J. T., Curry C. J., Fernhoff P. M., Grix A. W., Jr, Lott I. T., et al. (1985). Retinoic acid embryopathy. N. Engl. J. Med. 313, 837-841 [DOI] [PubMed] [Google Scholar]
- Lee D. D., Stojadinovic O., Krzyzanowska A., Vouthounis C., Blumenberg M., Tomic-Canic M. (2009). Retinoid-responsive transcriptional changes in epidermal keratinocytes. J. Cell. Physiol. 220, 427-439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe L. A., Yamada S., Kuehn M. R. (2000). HoxB6-Cre transgenic mice express Cre recombinase in extra-embryonic mesoderm, in lateral plate and limb mesoderm and at the midbrain/hindbrain junction. Genesis 26, 118-120 [DOI] [PubMed] [Google Scholar]
- M'Boneko V., Merker H. J. (1988). Development and morphology of the periderm of mouse embryos (days 9-12 of gestation). Acta Anat. (Basel) 133, 325-336 [DOI] [PubMed] [Google Scholar]
- Marshall D., Hardman M. J., Nield K. M., Byrne C. (2001). Differentially expressed late constituents of the epidermal cornified envelope. Proc. Natl. Acad. Sci. USA 98, 13031-13036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzalupo S., Coulombe P. A. (2001). A reporter transgene based on a human keratin 6 gene promoter is specifically expressed in the periderm of mouse embryos. Mech. Dev. 100, 65-69 [DOI] [PubMed] [Google Scholar]
- Mischke D., Korge B. P., Marenholz I., Volz A., Ziegler A. (1996). Genes encoding structural proteins of epidermal cornification and S100 calcium-binding proteins form a gene complex (“epidermal differentiation complex”) on human chromosome 1q21. J. Invest. Dermatol. 106, 989-992 [DOI] [PubMed] [Google Scholar]
- Moniaga C. S., Egawa G., Kawasaki H., Hara-Chikuma M., Honda T., Tanizaki H., Nakajima S., Otsuka A., Matsuoka H., Kubo A., et al. (2010). Flaky tail mouse denotes human atopic dermatitis in the steady state and by topical application with Dermatophagoides pteronyssinus extract. Am. J. Pathol. 176, 2385-2393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morasso M. I. (2010). Detection of gene expression in embryonic tissues and stratified epidermis by in situ hybridization. Methods Mol. Biol. 585, 253-260 [DOI] [PubMed] [Google Scholar]
- Morasso M. I., Markova N. G., Sargent T. D. (1996). Regulation of epidermal differentiation by a Distal-less homeodomain gene. J. Cell Biol. 135, 1879-1887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan P., Chin S. S., Wang D., Liu S., Sinha S., Garrett-Sinha L. A. (2010). Ets1 blocks terminal differentiation of keratinocytes and induces expression of matrix metalloproteases and innate immune mediators. J. Cell Sci. 123, 3566-3575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niederreither K., Dollé P. (2008). Retinoic acid in development: towards an integrated view. Nat. Rev. Genet. 9, 541-553 [DOI] [PubMed] [Google Scholar]
- Noji S., Nohno T., Koyama E., Muto K., Ohyama K., Aoki Y., Tamura K., Ohsugi K., Ide H., Taniguchi S., et al. (1991). Retinoic acid induces polarizing activity but is unlikely to be a morphogen in the chick limb bud. Nature 350, 83-86 [DOI] [PubMed] [Google Scholar]
- O'Shaughnessy R. F., Akgûl B., Storey A., Pfister H., Harwood C. A., Byrne C. (2007). Cutaneous human papillomaviruses down-regulate AKT1, whereas AKT2 up-regulation and activation associates with tumors. Cancer Res. 67, 8207-8215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obinata A., Akimoto Y., Hirano H., Endo H. (1991). Short term retinol treatment in vitro induces stable transdifferentiation of chick epidermal cells into mucus-secreting cells. Dev. Genes Evol. 200, 289-295 [DOI] [PubMed] [Google Scholar]
- Okano J., Suzuki S., Shiota K. (2007). Involvement of apoptotic cell death and cell cycle perturbation in retinoic acid-induced cleft palate in mice. Toxicol. Appl. Pharmacol. 221, 42-56 [DOI] [PubMed] [Google Scholar]
- Palmer C. N., Irvine A. D., Terron-Kwiatkowski A., Zhao Y., Liao H., Lee S. P., Goudie D. R., Sandilands A., Campbell L. E., Smith F. J., et al. (2006). Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat. Genet. 38, 441-446 [DOI] [PubMed] [Google Scholar]
- Presland R. B., Boggess D., Lewis S. P., Hull C., Fleckman P., Sundberg J. P. (2000). Loss of normal profilaggrin and filaggrin in flaky tail (ft/ft) mice: an animal model for the filaggrin-deficient skin disease ichthyosis vulgaris. J. Invest. Dermatol. 115, 1072-1081 [DOI] [PubMed] [Google Scholar]
- Presland R. B., Tomic-Canic M., Lewis S. P., Dale B. A. (2001). Regulation of human profilaggrin promoter activity in cultured epithelial cells by retinoic acid and glucocorticoids. J. Dermatol. Sci. 27, 192-205 [DOI] [PubMed] [Google Scholar]
- Rogers M. A., Langbein L., Winter H., Ehmann C., Praetzel S., Schweizer J. (2002). Characterization of a first domain of human high glycine-tyrosine and high sulfur keratin-associated protein (KAP) genes on chromosome 21q22.1. J. Biol. Chem. 277, 48993-49002 [DOI] [PubMed] [Google Scholar]
- Ross S. A., McCaffery P. J., Drager U. C., De Luca L. M. (2000). Retinoids in embryonal development. Physiol. Rev. 80, 1021-1054 [DOI] [PubMed] [Google Scholar]
- Saitou M., Sugai S., Tanaka T., Shimouchi K., Fuchs E., Narumiya S., Kakizuka A. (1995). Inhibition of skin development by targeted expression of a dominant-negative retinoic acid receptor. Nature 374, 159-162 [DOI] [PubMed] [Google Scholar]
- Sandilands A., Sutherland C., Irvine A. D., McLean W. H. (2009). Filaggrin in the frontline: role in skin barrier function and disease. J. Cell Sci. 122, 1285-1294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharschmidt T. C., Man M. Q., Hatano Y., Crumrine D., Gunathilake R., Sundberg J. P., Silva K. A., Mauro T. M., Hupe M., Cho S., et al. (2009). Filaggrin deficiency confers a paracellular barrier abnormality that reduces inflammatory thresholds to irritants and haptens. J. Allergy Clin. Immunol. 124, 496-506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segre J. A. (2006). Epidermal barrier formation and recovery in skin disorders. J. Clin. Invest. 116, 1150-1158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevilla L. M., Nachat R., Groot K. R., Klement J. F., Uitto J., Djian P., Määttä A., Watt F. M. (2007). Mice deficient in involucrin, envoplakin, and periplakin have a defective epidermal barrier. J. Cell Biol. 179, 1599-1612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegenthaler G., Tomatis I., Chatellard-Gruaz D., Jaconi S., Eriksson U., Saurat J. H. (1992). Expression of CRABP-I and -II in human epidermal cells. Alteration of relative protein amounts is linked to the state of differentiation. Biochem. J. 287, 383-389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano P. (1999). Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet. 21, 70-71 [DOI] [PubMed] [Google Scholar]
- Takaishi M., Takata Y., Kuroki T., Huh N. (1998). Isolation and characterization of a putative keratin-associated protein gene expressed in embryonic skin of mice. J. Invest. Dermatol. 111, 128-132 [DOI] [PubMed] [Google Scholar]
- Uehara M., Yashiro K., Takaoka K., Yamamoto M., Hamada H. (2009). Removal of maternal retinoic acid by embryonic CYP26 is required for correct Nodal expression during early embryonic patterning. Genes Dev. 23, 1689-1698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umesono K., Murakami K. K., Thompson C. C., Evans R. M. (1991). Direct repeats as selective response elements for the thyroid hormone, retinoic acid, and vitamin D3 receptors. Cell 65, 1255-1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yashiro K., Zhao X., Uehara M., Yamashita K., Nishijima M., Nishino J., Saijoh Y., Sakai Y., Hamada H. (2004). Regulation of retinoic acid distribution is required for proximodistal patterning and outgrowth of the developing mouse limb. Dev. Cell 6, 411-422 [DOI] [PubMed] [Google Scholar]
- Yuspa S. H., Harris C. C. (1974). Altered differentiation of mouse epidermal cells treated with retinyl acetate in vitro. Exp. Cell Res. 86, 95-105 [DOI] [PubMed] [Google Scholar]
- Yuspa S. H., Ben T., Lichti U. (1983). Regulation of epidermal transglutaminase activity and terminal differentiation by retinoids and phorbol esters. Cancer Res. 43, 5707-5712 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.