

Abstract
A majority of T cells from chronic inflammatory tissues derived from patients with nasal polyposis were found to express an effector memory phenotype. We report here that these memory T cells failed to activate NF-κB in response to TCR stimulation but responded normally when the proximal TCR signaling molecules were bypassed with PMA and ionomycin. The dysfunction of these cells was associated with a decrease in the phosphorylation of several TCR proximal signaling molecules including ZAP70, Lck and SLP-76. In addition to the disruption in the TCR signaling pathway, the nasal polyp-associated T cells were shown to have a defect in their ability to translocate LAMP-1 to the cell surface. The results presented here establish that the phenotype and anergy of the T cells in the nasal polyp are similar to those which is seen in memory T cells derived from human tumors and other sites of chronic inflammation.
Keywords: tolerance, suppression, anergy, inflammation
Introduction
Nasal polyposis is a chronic inflammatory disease of the upper airways, which can result in significant morbidity for affected patients with nasal blockage and restricted airflow, hyposmia and facial pressure. While nasal polyposis is a relatively common entity, with a prevalence of 0.5–4.3% in the general population, its pathogenesis is poorly understood.
Topical and oral corticosteroids are often first-line therapy for nasal polyposis, but surgical removal is frequently required following incomplete response to medical therapy. Even with surgical removal, nasal polyps commonly recur, and patients with this disorder often undergo repeated surgical procedures. By better understanding the pathologic mechanisms that lead to the chronic inflammatory state seen in nasal polyposis, new and more effective therapies may be developed to attenuate the disease process, targeting the specific immunologic mechanism of the inflammation.
T lymphocytes comprise an important component of the inflammatory cell infiltrate in the nasal polyp microenvironment (Sanchez-Segura et al. 1998; Bernstein et al. 2004). Little is known about the role of T cells in the pathogenesis of this disease. Impaired TCR signaling and downstream effector functions have been observed in T cells derived from other chronic inflammatory microenvironments including those of chronic viral infections (Barber et al. 2006) and human tumors (Broderick et al. 2005; Simpson-Abelson et al. 2009). In these nonmalignant and malignant inflammatory microenvironments, the T cell anergy has been postulated to result from chronic exposure of the T cells to antigen stimulation. By reversing the T cell hyporesponsiveness, it has been possible to reverse the pathology associated with chronic viral infection (Barber et al. 2006) and to kill tumor cells (Broderick et al. 2005; Simpson-Abelson et al. 2009). We suspected that a similar T cell hyporesponsiveness may exist in the microenvironment of the nasal polyp and that, if identified, reversal of this T cell hyporesponsiveness could significantly impact disease outcome. In this report, we show that the T lymphocytes in the chronic inflammatory microenvironment of nasal polyposis express an effector memory T cell phenotype and are hyporesponsive to activation with antibodies to CD3 and CD28.
Materials and methods
Patient samples
Nasal polyp tissue, tonsillar tissue and nasal polyp patient peripheral blood were obtained from the surgical suite at DeGraff Memorial Hospital (North Tonawanda, New York). Polyps were taken from 13 patients undergoing surgery for chronic rhinosinusitis with nasal polyposis. Three of these patients had documented aspirin sensitivity and asthma, consistent with Samter's triad. prior to surgery. Tonsillar tissue was taken from two patients undergoing tonsillectomy for chronic tonsillar hypertrophy. Tissue was transported in DMEM/ F12 medium (Gibco) for preservation until disaggregation. Peripheral blood leukocytes were obtained from five nasal polyp patients and from healthy normal donors and used as controls. All specimens were obtained according to institutional review board-approved protocols.
Histology and Immunohistochemistry
Tissues were fixed in neutral buffered formalin and embedded in paraffin. Eight-micrometer sections were cut and mounted by the State University of New York at Buffalo Histology Service Laboratory. Antihuman antibodies for CD3 and CD45RO were used for immunohistochemical staining, using the LSAB + Peroxidase kit for detection (Dako).
Cell isolation
Single cell suspensions were derived by mechanical disruption of the nasal polyp or tonsillar tissue through a 300-μm gross mesh using a Teflon policeman. Approximately 1–15 × 107 cells could be isolated from a single nasal polyp tissue specimen.
Leukocyte-enriched fractions from peripheral blood were obtained by layering on a Ficoll-Paque PLUS gradient (GE Healthcare) and isolating the PBMC interface. Both fresh and frozen cells were used in activation experiments, and freezing did not result in decreased ability of cells to be activated. All frozen samples were thawed and allowed to recover overnight in complete media prior to activation the following morning.
Flow cytometric phenotyping
Four-color flow cytometry was used to quantify human leukocyte subsets in polyp-derived single cell suspensions. Cells were stained with multiple antibody panels of primary conjugated human-specific antibodies including anti-CD3, anti-CD4, anti-CD8, anti-CD11a, anti-CD25, anti-CD28, anti-CD44, anti-CD45RA, anti-CD45RO, anti-CD62L and anti-CXCR3 (BD Biosciences). Isotype controls were used in all experiments. Cells were blocked with mouse IgG before addition of the panel and were analyzed immediately on a FACSCalibur flow cytometer (BD Biosciences). Data were analyzed with FlowJo software (Treestar).
T cell receptor activation of isolated cells
Anti-CD3/CD28 antibodies were immobilized on maxisorb tubes (Nunc) by incubating 1 μg of anti-CD3 (OKT-3, Bio X-Cell) and 5 μg of anti-CD28 (10 F3, Invitrogen) in 500 μl of PBS at 4°C overnight, as previously reported for human T cell activation (Broderick and Bankert 2006). Following thawing and overnight recovery in complete medium, 5 × 105 cells from samples were incubated in the tubes at 37°C/5% CO2 for the duration of activation (1 h for NF-κB/NFAT translocation, 6 h for CD107a expression).
PMA/ionomycin stimulation of isolated cells
Following thawing and overnight recovery in complete medium, 1 × 106 cells were incubated at 37°C/5% CO2 with 20 ng/ml PMA (Sigma) and 1.5 μM ionomcyin (Sigma) for 1 h and washed in PBS to terminate activation.
Immunofluorescence staining for confocal microscopy
Following activation, cells were affixed to Alcian blue-coated coverslips (Sommer 1977) and incubated in a humid chamber. Cells were stained for visualization by immunofluorescence confocal microscopy using a protocol adapted from Schooley et al. (2003). Briefly, cells were fixed in 2% ultrapure formaldehyde (UPF) and washed with blocking/permeabilization buffer (PBS + 5% normal goat serum + 0.4% Triton X-100). Cells were stained for expression of CD3 using antihuman CD3 antibody (BD Biosciences) followed by goat antimouse Ig-AlexaFluor 568 (Molecular Probes) as the secondary antibody. Cells were then incubated with 2 μg/ml purified anti-NFκB p65 (relA) (Santa Cruz) and washed in blocking buffer (PBS with 5% normal goat serum). Cells were incubated with goat antirabbit Ig-AlexaFluor 488 (Molecular Probes) and TO-PRO-3 and then washed with blocking buffer and PBS before mounting the coverslips on glass slides with VECTASHIELD Mounting Media (Vector). Cells were visualized using a Zeiss LSM 510 Meta Confocal Microscope under a 60× oil immersion objective. Percentages of samples translocating NF-κB were determined by visual examination of at least 100 CD3+ T cells for each condition. Images shown in Figure 2A and B are representative of the data obtained from normal donor PBL stained for specific cellular elements as stated above. Figure 2A demonstrates that from the total cell population present on a coverslip, T cells can be specifically identified by staining for surface expression of CD3 (red). These CD3+ cells are then further examined for the subcellular localization of NF-κB (green). In an unstimulated T cell, the majority of the NF-κB is located in the narrow rim of cytoplasm at the periphery (arrow). After incubation with immobilized antibodies to CD3 and CD28 for 1 h, the majority of the NF-κB is diffusely present throughout the nucleus of the T cells (arrowhead), colocalizing with the TO-PRO3 nuclear stain (blue).
FIG. 2.

Nasal polyp-derived T cells do not translocate NF-κB into the nucleus after TCR stimulation. A Nuclear translocation of NF-κB in normal donor peripheral blood T cells visualized by confocal microscopy. Location of proteins following activation is demonstrated in tricolor merged images of CD3 (red), NF-κB (green) and TO-PRO3 (blue). Arrows indicate NF-κB localized peripherally in unstimulated cell. Arrowheads show colocalization of NF-κB with nuclear stain in a stimulated cell. B Cross-sectional intensity profile of an activated T cell, with peripheral peak CD3 intensity (red) and central peak NF-κB (green) and TO-PRO3 (blue) intensities. C Compared to the baseline unstimulated condition, following activation by 1-h treatment with anti-CD3/CD28, a significant increase in nuclear NF-κB translocation was seen in the majority of CD3+ T cells derived from normal donor PBL (59.5% versus 2.7%, n = 12, p < 0.001), polyp patient PBL (65.4% versus 2.6%, n = 5, p < 0.001) and tonsillar tissue cell suspensions (54.4% versus 4.1%, n = 2, p < 0.001). Stimulated polyp-derived T cells show no significant increase in nuclear NF-κB over the unstimulated condition (10.8% versus 7.5%, n = 5, NS). Asterisk indicates p < 0.001 compared with stimulated patient PBL, normal donor PBL or tonsil-derived T cells.
Immunofluorescent labeling for assessment of NF-κB and NFAT nuclear translocation using multispectral imaging flow cytometry
Nasal polyp cell suspension or normal donor peripheral blood was thawed, allowed to recover in complete medium overnight and stimulated for 1 h in anti-CD3- or anti-CD28-coated tubes. Following activation, the cells were placed in polypropylene tubes (BD Biosciences) for immunofluorescence staining. Cells were fixed in 4% UPF and washed using 2% FBS in PBS. Cells were incubated with purified anti-NF-κB p65 (rel A) (Santa Cruz) or anti-NFAT-FITC (BD Biosciences) in PBS containing 0.1% Triton/3% FBS. For NF-κB staining, the secondary antibody was FITC-conjugated F(ab′)2 donkey antirabbit IgG (Jackson ImmunoResearch). Cells were blocked with normal mouse Ig and stained with mouse antihuman CD3-PE (BD Biosciences) or CD3-PE-TR (Invitrogen). Cells were washed and resuspended in 2% UPF. DRAQ5 or DAPI was added before acquisition.
Quantitative assessment of NF-κB and NFAT translocation using ImageStream multispectral imaging flow cytometry
The method of quantifying nuclear translocation has been previously described (George et al. 2004; 2006). Briefly, a single cell is separated into spectrally distinct, spatially correlated images. Images are collected on the Image StreamX (Amnis Corporation) and are analyzed using Image Stream Data Exploration and Analysis Software (IDEAS).
Translocation of NF-κB or NFAT was assessed using the similarity score between the nuclear image and NF-κB/NFAT image for each cell. After compensation, similarity analysis is performed by identifying single, in focus, DRAQ5+ (or DAPI+), live, CD3+ cells. Single cells are identified based on their small area by high aspect ratio (to eliminate debris and clumps). Live cells were identified by low brightfield contrast and low nuclear bright detail intensity or by live/dead violet stain (Invitrogen).
The similarity score is a log-transformed Pearson's correlation coefficient (ρ) of individual pixel intensities within the nuclear image and NF-κB/NFAT image (George et al. 2006). The greater the similarity scores the greater the degree of nuclear translocation of the transcription factor.
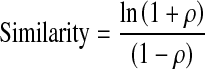 |
Fisher's discriminant ratio (Rd) is used to quantify the degree of NF-κB/NFAT translocation between two sample distributions using their similarity scores. For NF-κB translocation, the Rd value is calculated according to Eq. 1A below (where S = stimulated sample; U = unstimulated control):
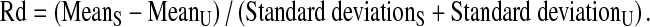 |
1A |
In contrast to NF-κB, the distributions generated for NFAT with anti-CD3/CD28 stimulation have been found to be bimodal, with two normal distribution peaks; these populations are referred to as Peak A and Peak B. Since the Rd values in Peak A were in the same range as those observed in unstimulated peak, there is a subpopulation (Peak B) in which stimulation induced translocation. To quantify NFAT translocation, the Rd value was determined according to Eq. 1B below (where SB = stimulated peak B and U = unstimulated control):
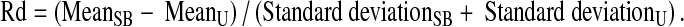 |
1B |
At least 5,000 cells per condition were collected per sample.
Flow cytometry for surface expression of CD107a
Cells were placed in anti-CD3- or anti-CD28-coated tubes for 6 h of stimulation. Ten microliters of anti-CD107a FITC (BD Biosciences) and 1 μl of monensin solution (eBioscience) per 5 × 105 cells were present during activation. Postactivation, cells were placed into polystyrene tubes, spun, washed, blocked with normal mouse IgG and stained on ice with fluorochrome-conjugated antibodies for CD45, CD4, CD8 and CD107a (BD Biosciences). Cells were washed, fixed using 2% UPF and analyzed using a FACSCalibur flow cytometer (BD Biosciences). One hundred thousand events were collected per condition. Data were analyzed with FlowJo (Treestar).
Phosflow
To assess both signaling molecule expression and phosphorylation status, cells were suspended in ice cold PBS + 1% FCS at 1.2 × 106 cells/ml and placed into polypropylene flow tubes. Samples were centrifuged at 4°C and resuspended in residual buffer. To assess T cell phosphorylation status, samples were activated with 1 μg of anti-CD3 (Bio X-Cell) and 1 μg of anti-CD28 (Invitrogen) per 1.2 × 106 cells and crosslinked on ice with 2 μg goat antimouse Ig (BD Biosciences). Samples were then washed, resuspended in warm media and activated in 37°C water bath for 1 or 3 min. Samples were incubated with warmed Cytofix buffer (BD Biosciences) after activation. Samples for both signaling molecule expression and phosphorylation status were washed and permeabilized on ice with Perm Buffer III (BD Biosciences). Samples were washed with Stain Buffer (BD Biosciences), transferred to polystyrene flow tubes, blocked with mouse Ig and stained at room temperature with antibodies to signaling molecules and cell surface markers including SLP76-Alexa Fluor 488, phospho-SLP76-Alexa Fluor 488, ZAP70-PE, phospho-ZAP70-Alexa Fluor 647, Lck-Alexa Fluor 647, phospho-Lck-PE, CD3ζ-PE and phospho-CD3ζ-PE (BD Biosciences), CD45RO-PerCP-Cy5.5 and either CD45RA-FITC or CD45RA-APC (BD Biosciences). Data were collected on a FACS Calibur flow cytometer (BD Biosciences) and analyzed using FlowJo (Treestar). Two hundred thousand events or more were collected per sample. Changes in phosphorylation of signaling molecules following TCR stimulation were determined by calculating the ratio of mean fluorescence intensity (MFI) of CD45RO + cells from stimulated versus unstimulated populations of normal donor PBL or nasal polyp cell suspensions.
Statistical analysis
Statistical analysis was performed using Prism software (GraphPad). Statistical analysis of NF-κB translocation by confocal microscopy employed a one-way ANOVA test, while the significance of the differences in multispectral imaging, CD107a expression and protein phosphorylation were analyzed using Student's t-test. A p value of <0.05 was considered statistically significant.
Results
Phenotyping of nasal polyp-infiltrating lymphocytes
Nasal polyposis is a disease that is characterized by chronic inflammation that typically includes T cells that are predominantly CD8+ with a minority being CD4+ (Sanchez-Segura et al. 1998; Bernstein et al. 2004). Immunohistochemical staining of nasal polyp tissues reveals the presence of CD3+ T cells (Fig. 1A) that are predominantly CD45RO + (Fig. 1B). Single cell suspensions from six nasal polyp specimens were analyzed phenotypically by flow cytometry. The majority of CD3+, CD45RO + T cells derived from the polyps are positive for CD44, CD11a, CXCR3 and CD28 (Fig. 1C), and negative for CD62L and CD25. This phenotype is consistent with that of the effector memory T cell (Tem) subset of T cells. Tem are long-lived cells that have previously encountered and responded to their cognitive antigen and are typically found in the periphery of sites of chronic inflammation. This subset of T cells has also been shown to be present in human tumor microenvironments, and these tumor-associated T cells have been shown to be hyporesponsive to activation via the T cell antigen receptor (Broderick et al. 2005; Simpson-Abelson et al. 2009; Agrawal et al. 1998). This finding led us to investigate whether the T cells derived from the chronic inflammatory microenvironment of nasal polyps are similarly impaired in their ability to be activated.
FIG. 1.

T cells derived from nasal polyp tissue exhibit an effector memory phenotype. Immunohistochemical staining of nasal polyp tissue shows significant populations of A CD3+ cells and B CD45RO + cells. C Polyp-derived lymphocytes, gated on the CD3+/CD45RO + population are positive for CD44, CXCR3, CD11a and CD28 and are negative for CD62L and CD25. Gray shaded histograms represent surface marker-specific antibodies, while outline histograms represent isotype controls. Histograms show a representative example of six independent experiments.
T cells in nasal polyp tissue show diminished nuclear translocation of NF-κB
To assess the response of nasal polyp-derived T cells to activation via the T cell receptor, we assayed five different nasal polyp specimens by immunofluorescence confocal microscopy for their ability to translocate NF-κB into the nucleus in response to anti-CD3/CD28. Measuring NF-κB translocation by confocal microscopy has made it possible to quantify the activation potential of T cells in a mixed population of cells at the single cell level (see “Materials and methods”). As shown in Figure 2A, peripheral blood T cells are identified as CD3+ (red fluorescence) cells, and the localization of NF-κB (green fluorescence) prior to stimulation is shown to be present in the cytosol, clearly separate from the location of nuclear staining (blue fluorescence). Following activation of peripheral blood T cells, most of the NF-κB translocates and is present primarily within the nucleus, evidenced by colocalization of NF-κB (green) and nuclear (blue) stains. Figure 2B shows a cross-sectional fluorescence intensity profile of an activated peripheral blood T cell, in which peak CD3 intensity (red) is seen at the periphery of the cell, and peak NF-κB (green) and nuclear stain (blue) intensities are both seen in the center of the cell.
The ability of T cells to respond to stimulation with immobilized anti-CD3 and anti-CD28 was quantified by determining the percentage of cells in which the NF-κB is within the nucleus. In the baseline unstimulated state, few T cells from normal donor PBL, nasal polyp tissue, tonsillar tissue or nasal polyp patient PBL showed nuclear localization of NF-κB (Fig. 2C). There was no significant difference between the baseline nuclear localization of NF-κB in nasal polyp-derived CD3+ T cells versus the CD3+ T cells of any of the three sources of control specimens. Following 1-h activation with anti-CD3/CD28, a significant increase in nuclear NF-κB translocation was seen in the majority of T cells derived from the PBL of normal donors (59.5%, n = 12, p < 0.001), the PBL of patients with nasal polyps (65.4%, n = 5, p < 0.001) and tonsillar tissue cell suspensions (54.4%, n = 2, p < 0.001). In contrast, the majority of the T cells derived from nasal polyp tissue specimens failed to translocate NF-κB to the nucleus after anti-CD3/CD28 stimulation (10.8%, n = 5). These polyp-derived T cells exhibited no statistically significant difference in NF-κB translocation in the stimulated versus unstimulated state. The staining pattern of the majority of the polyp-derived T cells exposed to the activation protocol was the same as shown for unstimulated T cells derived from normal donor peripheral blood (see Fig. 2A; arrow).
T cells derived from the nasal polyp are impaired in their ability to translocate NF-κB as determined by multispectral imaging flow cytometry
We employed multispectral imaging flow cytometry to verify our confocal microscopy results showing that NF-κB nuclear translocation is impaired in stimulated T cells from nasal polyp tissue. Multispectral imaging is a high throughput, objective, quantitative analysis of nuclear translocation that allows for the extensive phenotypic characterization of thousands of cells (George et al. 2006) and has been used by various laboratories to quantifiably measure nuclear translocation (George et al. 2006; Migueles et al. 2008; Medeiros et al. 2007).
In agreement with our confocal results, we determined that the T cells derived from four nasal polyp specimens were deficient in their ability to translocate NF-κB when stimulated with anti-CD3/CD28, compared to the T cells derived from the normal donor peripheral blood. As shown in the overlays in Figure 3A and B, the unstimulated and anti-CD3/CD28-stimulated samples for the nasal polyps completely overlay one another in contrast to normal donor peripheral blood in which the stimulated sample was shifted to the right of the unstimulated sample, indicating translocation of NF-κB into the nucleus. This is also seen in Figure 3C, in which the Rd values for activated T cells derived from the nasal polyp were significantly lower than that of those from normal donor peripheral blood.
FIG. 3.

Nasal polyp-derived T cells show impaired ability to translocate NF-κB as determined by multispectral imaging flow cytometry. Following stimulation for 1 h with anti-CD3/CD28, T cells derived from the nasal polyp were deficient in their ability to translocate NF-κB compared to T cells from normal donor PBL. A In normal donor PBL, the stimulated similarity distribution plot (black) for NF-κB is shifted to the right of the unstimulated plot (gray), demonstrating translocation. B The similarity distribution plots for the unstimulated and stimulated samples in the nasal polyp completely overlay, indicating the cells were unable to translocate NF-κB with stimulation. C The Rd values after activation for the T cells derived from normal donor PBL were significantly greater than for T cells derived from nasal polyps (n = 4, p < 0.05).
Having validated our confocal microscopy NF-κB translocation results using multispectral imaging flow cytometry, we set out to assess NFAT translocation using this technology in T cells derived from both nasal polyps and normal donor peripheral blood.
T cells derived from the nasal polyp translocate NFAT
Like NF-κB, NFAT in normal T cells translocates into the nucleus upon anti-CD3/CD28 stimulation. The proximal TCR signaling pathway for NF-κB and NFAT is similar up to the activation of phospholipase C (PLC-γ). Once PLC-γ is activated it catalyzes the conversion of phosphatidylinositol-bisphosphate (PIP2) to diacylglycerol (DAG) and inositol-1,4,5 trisphosphate (IP3). DAG is necessary for the activation of protein kinase C (PKCθ) and the subsequent translocation of NF-κB, whereas IP3 induces the release of Ca2+ and subsequent dephosphorylation and translocation of NFAT into the nucleus (Nel and Slaughter 2002). When assessing NFAT translocation using multispectral imaging flow cytometry, we determined that the T cells derived from the three nasal polyp specimens tested were capable of translocating NFAT to the same degree as the T cells from normal donor peripheral blood (Fig. 4). Thus, the arrest in the CD3/CD28 signaling pathway of nasal polyp-derived T cells appears to be selective to the NF-κB pathway.
FIG. 4.

T cells from nasal polyps are able to translocate NFAT, as assessed by multispectral imaging flow cytometry. Following stimulation with anti-CD3/CD28, T cells derived from the nasal polyp translocated NFAT to the same degree as the T cells derived from normal donor peripheral blood. Data are presented from three independent experiments.
Nasal polyp-infiltrating T cells are still activatable with PMA/ionomycin
To localize the point in the CD3/CD28 pathway where the nasal polyp T cell signaling arrest is occurring, lymphocytes from normal donor PBL, nasal polyp tissue, patient PBL and tonsil were stimulated with PMA, a diacylglycerol analog, and ionomycin, a calcium ionophore. Together, these substances bypass the proximal TCR signaling cascade and initiate downstream events resulting in the translocation of NF-κB into the nucleus.
Over 95% of CD3+ T lymphocytes from all sources translocated NF-κB in response to PMA/ionomycin (Fig. 5). NF-κB translocation in nasal polyp T cells was not significantly different from that in T cells from normal donor PBL, patient PBL or tonsil at baseline or following activation with PMA and ionomycin. This finding demonstrates that nasal polyp-derived T cells remain viable and that the distal TCR signaling cascade is intact in these T cells. These results suggest that the hyporesoponsiveness of T cells in the nasal polyp to activation is due to events that occur within the proximal TCR signaling cascade.
FIG. 5.

Direct activation of the distal TCR signaling cascade results in translocation of NF-κB into the nucleus of nasal polyp-derived T cells. Following activation with PMA and ionomycin, the percentage of CD3+ T cells translocating NF-κB is not significantly different in nasal polyp tissue (95.6%, n = 5) versus normal donor PBL (97.2%, n = 8), patient PBL (96.7%, n = 3), or tonsillar tissue (95.5%, n = 2).
T lymphocytes from nasal polyp tissue display impaired phosphorylation of proximal TCR signaling cascade molecules
Because PMA and ionomycin activation demonstrated that the distal TCR signaling cascade was intact in nasal polyp-infiltrating T cells, we turned our focus to the more proximal signaling cascade. Using Phosflow analysis, we monitored the phosphorylation status of the proximal TCR signaling cascade molecules. The tyrosine phosphorylation of signaling molecules CD3ζ, ZAP70, Lck and SLP-76 was assessed in the T cell populations from four nasal polyp samples and from four normal donor peripheral blood samples. When compared to CD45RO + T cells from normal donor PBL, nasal polyp-derived CD45RO + T cells showed significantly decreased phosphorylation of ZAP70 (1.8 versus 2.8, n = 4, p < 0.05), SLP-76 (3.0 versus 6.1, n = 4, p < 0.05) and Lck (1.9 versus 3.0, n = 4, p < 0.05) following brief stimulation with anti-CD3/CD28 (Fig. 6), as measured by fold change in MFI from the unstimulated state. The decrease in the phosphorylation of the CD3ζ chain of the TCR complex in T cells from nasal polyps approached, but did not reach, statistical significance (1.6 versus 5.6, n = 3). While there was some variability in proximal SLP76 and CD3ζ phosphorylation between different control PBL samples, this did not translate into significant variability in downstream activation potential of the cells, i.e., the percentage of cells able to translocate NF-κB or express surface CD107a show little variation between control samples (see Figs. 2C and 8).
FIG. 6.

Phosphorylation of signaling molecules in the proximal TCR signaling pathway is impaired in nasal polyp-derived T cells. Compared to CD45RO + T cells from normal donor PBL, polyp-derived CD45RO + T cells show decreased phosphorylation of ZAP70 (1.8 versus 2.8, n = 4, p < 0.05), SLP-76 (3.0 versus 6.1, n = 4, p < 0.05), Lck (1.9 versus 3.0, n = 4, p < 0.05) and CD3ζ (1.6 versus 5.6, n = 3, NS) following stimulation with anti-CD3/CD28, as measured by change in mean fluorescence intensity. Independent experiments comparing normal donor peripheral blood T cells (closed symbols) to polyp-derived T cells (open symbols) are represented by different symbols, and means are represented by horizontal lines.
FIG. 8.

Nasal polyp-derived CD8+ T cells show impaired upregulation of CD107a surface expression. A At baseline, there are few unstimulated CD8+ T cells from both normal donor PBL and nasal polyp tissue expressing CD107a (2.48% versus 2.06%). Following a 6-h activation with anti-CD3/CD28, CD8+ T cells from nasal polyp tissue show decreased CD107a expression (8.9%, n = 4) compared with CD8+ T cells from normal donor PBL (31.2%, n = 4, p < 0.01). B Representative of four experiments of CD107a expression in CD8+ T cells from normal donor PBL versus nasal polyp cell suspension.
In these studies, we focused on examining the differences in phosphorylation for the CD45RO + T cells from the nasal polyp tissue versus CD45RO + T cells from normal donor peripheral blood because these cells included the effector memory T cell population that characterize the majority of nasal polyp-derived T cells. However, impaired signaling molecule phosphorylation was not specific to the effector memory T cell subset, as we also observed decreased phosphorylation in the CD45RA + subset of nasal polyp T cells versus CD45RA + cells from normal donor peripheral blood (data not shown).
The impaired phosphorylation of proximal TCR signaling cascade molecules in nasal polyp T cells is not due to decreased expression of the signaling molecules themselves
After demonstrating that there was impaired phosphorylation of proximal signaling molecules in nasal polyp-derived T cells, it was important to determine if there was a decreased expression of the signaling molecules themselves. Using intracellular flow cytometry, we examined the expression of the CD3ζ chain, ZAP70, Lck and SLP-76 in CD45RO + T cells derived from three different nasal polyp tissue samples versus CD45RO + T cells derived from three samples of normal donor peripheral blood (Fig. 7). The MFI for CD3ζ and ZAP70 was not significantly different in the polyp-derived T cells versus the normal donor peripheral blood T cells. While there was a statistically significant decrease in the MFI for Lck and SLP-76 in the polyp-derived T cells versus T cells from normal donor PBL, the absolute differences in expression of these signaling molecules were small.
FIG. 7.

Only minor differences in signaling molecule expression are observed between nasal polyp-derived CD45RO + T cells and normal donor PBL-derived CD45RO + T cells. In the polyp-derived CD45RO + T cells versus the normal donor peripheral blood CD45RO + T cells, the MFI for CD3ζ (11.6 versus 18.0, NS) and ZAP70 (5.7 versus 6.1, NS) were not significantly different. While there was a statistically significant decrease in the MFI for Lck (39.5 versus 48.6, p < 0.05) and SLP-76 (9.4 versus 11.2, p < 0.05) in the polyp-derived T cells versus T cells from normal donor PBL, the absolute differences in expression of these signaling molecules were small. Results are based upon three independent experiments.
In response to activation, the translocation of LAMP-1 to the cell surface is abrogated in nasal polyp-derived CD8+ T cells
After demonstrating impaired signaling in the nasal polyp-derived T cells, we set out to determine if these cells could perform a known effector function. Cytotoxicity is an important effector function of CD8+ T cells and is characterized by fusion of cytotoxic granule membranes with the cell membrane, followed by release of granule contents including perforin and granzyme into the extracellular space. CD107a (LAMP-1) is a molecule present on cytotoxic granule membranes and CD107a mobilization to the cell surface is a marker of CD8+ T cell degranulation following stimulation (Betts et al. 2003).
Following a 6-h stimulation with anti-CD3/CD28, CD8+ T cells from normal donor PBL showed a marked increase in percent of cells with detectable CD107a compared to the unstimulated condition (Fig. 8, 31.2% versus 1.9%, n = 4). In contrast, nasal polyp-infiltrating CD8+ T cells derived from four separate patients showed significantly impaired expression of CD107a following a 6-h TCR stimulation (8.9%, n = 4, p < 0.01). While CD107a mobilization is considered to be a functional downstream marker of T cell activation and is widely used as a reliable marker of activation (Migueles et al. 2008; Li and Pauza 2011; Wong et al. 2007; Agnellini et al. 2007), there are no definitive studies that directly link the CD107a translocation to NF-κB or NFAT activation.
Our collective results establish that the T cells present within the chronic inflammatory microenvironment of nasal polyps are hyporesponsive to activation via the TCR and, phenotypically, are functionally similar to T cells present within human tumor microenvironments.
Discussion
In this work, we have shown that nasal polyp-derived T cells are impaired in their ability to phosphorylate signaling molecules in the proximal TCR signaling cascade, to translocate the transcription factor NF-κB into the cell nucleus and to fuse cytotoxic granule membranes with the cell membrane, an integral step in T cell cytotoxicity. Indeed, the only endpoint of TCR signaling that remains intact in our observations has been translocation of NFAT into the cell nucleus. This finding suggests a similarity between our hyporesponsive T cells derived from nasal polyp tissue and previously-defined anergic T cells, which demonstrate preserved ability to activate the NFAT transcription factor in the absence of activation of the AP-1 or NF-κB signaling pathway (Dagur et al. 2010; Sundstedt et al. 1996; Macian et al. 2002).
The proximal TCR signaling molecules examined in this study (ZAP70, SLP-76, Lck and CD3ζ) are common to both the NF-κB and NFAT signaling pathways. Therefore, with the impaired phosphorylation of these molecules in nasal polyp-derived T cells, it was not anticipated that only the translocation of NF-κB would be deficient, leaving the movement of NFAT into the cell nucleus intact in response to activation with anti-CD3/CD28. Yet this normal NFAT translocation does not rule out defects in the phosphorylation of upstream signaling molecules, as we observed decreased, but not absent, phosphorylation of these molecules in polyp-derived CD45RO + T cells. There may be different activation thresholds in the TCR signaling cascade, below which the NFAT pathway can still be activated, while the NF-κB pathway is severely impaired. Another possible explanation for our finding is that diacylglycerol (DAG) kinases may be upregulated in nasal polyp-derived T cells. Others have previously found that in anergic T cells, signaling incorporating calcium flux and NFAT are disproportionately increased over other signals. This is a consequence of DAG kinases which convert DAG to phosphatidic acid, making DAG less available and, thereby, suppressing the translocation of NF-κB into the nucleus without blocking NFAT translocation (Zha et al. 2006). In addition to these possible explanations of discordant NF-κB and NFAT translocation in nasal polyp-derived T cells, it should be recognized that much of the knowledge of TCR signaling in general is based on work done in naïve T cells. The signaling pathways in memory T cells such as the CD45RO + cells we focused on have not been well examined. Any differences in these cells may resolve the issue of our seemingly contradictory findings of impaired proximal signaling molecule phosphorylation and normal NFAT nuclear translocation.
While there have been several different causes of T cell hyporesponsiveness identified in previous models, our initial attempts in the nasal polyp have not identified a specific molecular cause of the observed T cell hyporesponsiveness. Loss of expression of the CD28 costimulatory molecule has been proposed as a cause of T cell senescence in multiple inflammatory diseases (Vallejo et al. 2004; Martens et al. 1997; Moosig et al. 1998; Schirmer et al. 2002). However, we have shown here that the CD28 costimulatory molecule is still expressed in nasal polyp-derived memory T cells (see Fig. 1) and, therefore, cannot account for the loss of T cell signaling.
Our findings of impaired proximal TCR signaling molecule phosphorylation in the absence of significant downregulation of expression of the molecules themselves mirrors what has been previously reported in models of T cell hyporesponsiveness. While it has been suggested by others that defective TCR signaling in tumor microenvironments was associated with reduced expression of proximal signaling molecules including CD3ζ, ZAP-70 and Lck (Jarnicki et al. 1996; Maccalli et al. 1999), subsequent studies have revealed that while the phosphorylation/activation of several proximal signaling molecules was impaired in in vivo models of T cell dysfunction, there was no significant deficits in expression of these signaling molecules (Sloan-Lancaster et al. 1994; Chiodetti et al. 2006). The results obtained with nasal polyp T cells (see Fig. 6) largely agree with these findings.
Upregulation of coinhibitory molecules CTLA-4 and PD-1 in chronically stimulated T effector cells has been implicated in dysfunctional T cell signaling (Mueller and Ahmed 2009; Peggs et al. 2008) and expression of multiple inhibitory receptors has been associated with T cell exhaustion in a mouse model of chronic viral infection (Blackburn et al. 2009). However, we have not identified a significant expression of inhibitory molecules on the nasal polyp T cell surface such as PD-1, CTLA-4, LAG3 and CD180 (unpublished data) and consider this to be an unlikely explanation for the failure of the nasal polyp T cells to fully respond to TCR stimulation.
The T cell hyporesponsiveness that we have observed is restricted to T lymphocytes infiltrating the nasal polyp tissue itself. As peripheral blood T cells from nasal polyp patients demonstrate a preserved ability to translocate NF-κB into the cell nucleus (see Fig. 2C), our findings are not evidence of a systemic immune dysfunction in nasal polyp patients.
The T cell hyporesponsiveness seen in the nasal polyp appears to be specific to this and other chronic inflammatory microenvironments, both malignant and nonmalignant (George et al. 2004; Agrawal et al. 1998; Broderick et al. 2006). The ability of tonsil-infiltrating T cells to translocate NF-κB in response to TCR stimulation (see Fig. 2C) suggests that the hyporesponsiveness seen in nasal polyp T cells is not simply a characteristic of tissue-infiltrating T cells and is not an artifact caused by our tissue disruption protocol, as the tonsillar tissue tested was handled in the same manner as all nasal polyps tested.
We propose that the observed hyporesponsive state of the T cells in the nasal polyp microenvironment is a consequence of persistent antigen stimulation generated from one or more as yet unidentified pathogenic microbes or an immune response to a self tissue. Persistent antigen stimulation has, in fact, been shown to lead to T cell hyporesponsiveness in the setting of several chronic viral infections (Jelley-Gibbs et al. 2005; Ahmed and Gray 1996; Letvin and Walker 2003; Feunou et al. 2003; Hu et al. 1996) and mycobacterial infections (Dagur et al. 2010). The possibility of an autoimmune response driving the pathology of nasal polyps may be further supported by evidence of HLA I and HLA II linkages in nasal polyposis (Molnar-Gabor et al. 2000; Luxenberger et al. 2000; Ramírez-Anguiano et al. 2006).
The ability of memory T cells to become nonresponsive to persistent stimulation is considered important for at least two reasons. First, this would prevent significant tissue damage resulting from an uncontrolled T cell proliferation, and an associated overproduction of inflammatory cytokines and other biologically active factors. Perhaps equally important, the shutdown of the activation of memory T cells would prevent the loss of potentially important cells from the T cell repertoire. In the absence of such control, one would expect that whenever T cells were faced with a chronic stimulation they would be driven to a terminal senescence, resulting in an unacceptable loss of T cell clones which would ultimately diminish the ability of the adaptive arm of the immune system to respond to pathogenic organisms. The mechanism or mechanisms by which this important T cell conservation is mediated has not yet been completely defined for all situations in which T cells are chronically exposed to stimulatory conditions.
In contrast to previous work from our laboratory in the lung tumor environment (Broderick et al. 2006; Broderick and Bankert 2006), preliminary results with the nasal polyp-associated T cells show that hyporesponsiveness in these T cells is not reversed with blockade of TGF-β or with exogenous IL-12 treatment (unpublished data). This suggests that there are likely multiple different mechanisms by which memory T cells are prevented from an undesirable continuous response to persistent stimulation. Given the significant decrease in the phosphorylation of several of the most proximal TCR signaling molecules that was observed in the polyp T cells (see Fig. 6A) one possible T cell regulatory mechanism that should be considered is that which has been reported for the carcinoembryonic antigen-related cell adhesion molecule CEACAM1 (Gray-Owen and Blumberg 2006). CEACAM1 has been shown to inhibit T cells during active immune responses and in immune-mediated diseases (Gray-Owen and Blumberg 2006) including cancer (Markel et al. 2006). CEACAM1 functions as a coinhibitory molecule that, following activation, results in a series of well-defined molecular events that culminates in the SHP1-dependent dephosphorylation of the most proximal T cell signaling molecules, thereby, blocking downstream activation (Gray-Owen and Blumberg 2006).
With an understanding of how T cells become unresponsive in the face of chronic stimulation, it should be possible to design protocols to reverse the anergy. As the reactivation of quiescent T cells has been shown to reverse the pathology of varied conditions including malignancy and chronic viral infection (Agrawal et al. 1998; Barber et al. 2006), it will be important to determine ways to reverse the T cell hyporesponsiveness of nasal polyp-infiltrating T cells. By reactivating these T cells we may be able to rid the tissue of the putative causative pathogenic microbes, thereby, reversing the chronic inflammatory state driven by an innate immune response to unidentified microbes and their products. In addition, continued advancements in characterizing the immunology of this inflammatory microenvironment may someday result in a targeted, logical therapy for this difficult-to-treat chronic disease.
Acknowledgments
We would like to thank Dr. Wade J. Sigurdson for his expertise with the confocal microscope as well as Jenni L. Loyall and Liza Pope for their technical assistance. We thank Dr. Stephen Brooks for his helpful discussion of this work and Dr. Paul Wallace for his expert advice. We would also like to thank Thaddeus C. George of Amnis Corporation, and Dr. Hans Minderman and Kieran O'Loughlin of Roswell Park Cancer Institute for their expertise and assistance in ImageStream acquisition and analysis. This work was supported in part by the National Institute of Health Grant R56AI079188, RO1-CA108970 and RO1-CA131407, and by the Ralph Hochstetter Medical Research Fund in honor of Dr. Henry C. and Bertha H. Buswell (HKL).
Contributor Information
Heather K. Lehman, Email: hkm@buffalo.edu
Michelle R. Simpson-Abelson, Email: MRA45@pitt.edu
Thomas F. Conway, Jr., Email: tfconway@buffalo.edu
Raymond J. Kelleher, Jr., Email: rjk6@buffalo.edu
Joel M. Bernstein, Email: jbernste@buffalo.edu
Richard B. Bankert, Phone: +1-716-8292701, FAX: +1-716-8292662, Email: rbankert@buffalo.edu
References
- Agnellini P, Wolint P, Rehr M, Cahenzli U, Karrer U, Oxenius A. Impaired NFAT nuclear translocation results in split exhaustion of virus-specific CD8+ T cell functions during chronic viral infection. Proc. Natl. Acad. Sci. USA. 2007;104:4564–4570. doi: 10.1073/pnas.0610335104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal S, Marquet J, Delfau-Larue MH, Copie-Bergman C, Jouault H, Reyes F, Bensussan A, Farcet JP. CD3 hyporesponsiveness and in vitro apoptosis are features of T cells from both malignant and nonmalignant secondary lymphoid organs. J Clin Invest. 1998;102:1715–1723. doi: 10.1172/JCI3784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed R, Gray D. Immunological memory and protective immunity: understanding their relation. Science. 1996;272:54–60. doi: 10.1126/science.272.5258.54. [DOI] [PubMed] [Google Scholar]
- Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- Bernstein JM, Ballow M, Rich G, Allen C, Swanson M, Dmochowski J. Lymphocyte subpopulations and cytokines in nasal polyps: is there a local immune system in the nasal polyp? Otolaryngol Head Neck Surg. 2004;130:526–535. doi: 10.1016/j.otohns.2003.12.022. [DOI] [PubMed] [Google Scholar]
- Betts MR, Brenchley JM, Price DA, Rosa SC, Douek DC, Roederer M, Koup RA. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J Immunol Methods. 2003;281:65–78. doi: 10.1016/S0022-1759(03)00265-5. [DOI] [PubMed] [Google Scholar]
- Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, Betts MR, Freeman GJ, Vignali DA, Wherry EJ. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol. 2009;10:29–37. doi: 10.1038/ni.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broderick L, Bankert RB. Membrane-associated TGF-beta1 inhibits human memory T cell signaling in malignant and nonmalignant inflammatory microenvironments. J Immunol. 2006;177:3082–3088. doi: 10.4049/jimmunol.177.5.3082. [DOI] [PubMed] [Google Scholar]
- Broderick L, Brooks SP, Takita H, Baer AN, Bernstein JM, Bankert RB. IL-12 reverses anergy to T cell receptor triggering in human lung tumor-associated memory T cells. Clin Immunol. 2006;118:159–169. doi: 10.1016/j.clim.2005.09.008. [DOI] [PubMed] [Google Scholar]
- Broderick L, Yokota SJ, Reineke J, Mathiowitz E, Stewart CC, Barcos M, Kelleher RJ, Jr, Bankert RB. Human CD4+ effector memory T cells persisting in the microenvironment of lung cancer xenografts are activated by local delivery of IL-12 to proliferate, produce IFN-gamma, and eradicate tumor cells. J Immunol. 2005;174:898–906. doi: 10.4049/jimmunol.174.2.898. [DOI] [PubMed] [Google Scholar]
- Chiodetti L, Choi S, Barber DL, Schwartz RH. Adaptive tolerance and clonal anergy are distinct biochemical states. J Immunol. 2006;176:2279–2291. doi: 10.4049/jimmunol.176.4.2279. [DOI] [PubMed] [Google Scholar]
- Dagur PK, Sharma B, Kumar G, Khan NA, Katoch VM, Sengupta U, Joshi B. Mycobacterial antigen(s) induce anergy by altering TCR- and TCR/CD28-induced signalling events: insights into T-cell unresponsiveness in leprosy. Mol Immunol. 2010;47:943–952. doi: 10.1016/j.molimm.2009.11.009. [DOI] [PubMed] [Google Scholar]
- Feunou P, Poulin L, Habran C, Moine A, Goldman M, Braun MY. CD4 + CD25+ and CD4 + CD25− T cells act respectively as inducer and effector T suppressor cells in superantigen-induced tolerance. J Immunol. 2003;171:3475–3484. doi: 10.4049/jimmunol.171.7.3475. [DOI] [PubMed] [Google Scholar]
- George TC, Basiji DA, Hall BE, Lynch DH, Ortyn WE, Perry DJ, Seo MJ, Zimmerman CA, Morrissey PJ. Distinguishing modes of cell death using the ImageStream multispectral imaging flow cytometer. Cytometry A. 2004;59:237–245. doi: 10.1002/cyto.a.20048. [DOI] [PubMed] [Google Scholar]
- George TC, Fanning SL, Fitzgerald-Bocarsly P, Medeiros RB, Highfill S, Shimizu Y, Hall BE, Frost K, Basiji D, Ortyn WE, et al. Quantitative measurement of nuclear translocation events using similarity analysis of multispectral cellular images obtained in flow. J Immunol Methods. 2006;311:117–129. doi: 10.1016/j.jim.2006.01.018. [DOI] [PubMed] [Google Scholar]
- Gray-Owen SD, Blumberg RS. CEACAM1: contact-dependent control of immunity. Nat Rev Immunol. 2006;6:433–446. doi: 10.1038/nri1864. [DOI] [PubMed] [Google Scholar]
- Hu HL, Cornwell WD, Rogers TJ, Lin YS. In vivo analysis of a superantigen-induced T cell suppressor factor. Cell Immunol. 1996;167:285–292. doi: 10.1006/cimm.1996.0037. [DOI] [PubMed] [Google Scholar]
- Jarnicki AG, Fitzpatrick DR, Robinson BW, Bielefeldt-Ohmann H. Altered CD3 chain and cytokine gene expression in tumor infiltrating T lymphocytes during the development of mesothelioma. Cancer Lett. 1996;103:1–9. doi: 10.1016/0304-3835(96)04178-X. [DOI] [PubMed] [Google Scholar]
- Jelley-Gibbs DM, Dibble JP, Filipson S, Haynes L, Kemp RA, Swain SL. Repeated stimulation of CD4 effector T cells can limit their protective function. J Exp Med. 2005;201:1101–1112. doi: 10.1084/jem.20041852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letvin NL, Walker BD. Immunopathogenesis and immunotherapy in AIDS virus infections. Nat Med. 2003;9:861–866. doi: 10.1038/nm0703-861. [DOI] [PubMed] [Google Scholar]
- Li H, Pauza DC. Interplay of T-cell receptor and interleukin-2 signalling in Vγ2Vδ2 T-cell cytotoxicity. Immunology. 2011;132:96–103. doi: 10.1111/j.1365-2567.2010.03343.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luxenberger W, Posch U, Berghold A, Hofmann T, Lang-Loidolt D. HLA patterns in patients with nasal polyposis. Eur Arch Otorhinolaryngol. 2000;257:137–139. doi: 10.1007/s004050050210. [DOI] [PubMed] [Google Scholar]
- Maccalli C, Pisarra P, Vegetti C, Sensi M, Parmiani G, Anichini A. Differential loss of T cell signaling molecules in metastatic melanoma patients' T lymphocyte subsets expressing distinct TCR variable regions. J Immunol. 1999;163:6912–6923. [PubMed] [Google Scholar]
- Macian F, Garcia-Cozar F, Im SH, Horton HF, Byrne MC, Rao A. Transcriptional mechanisms underlying lymphocyte tolerance. Cell. 2002;109:719–731. doi: 10.1016/S0092-8674(02)00767-5. [DOI] [PubMed] [Google Scholar]
- Markel G, Seidman R, Stern N, Cohen-Sinai T, Izhaki O, Katz G, Besser M, Treves AJ, Blumberg RS, Loewenthal R, et al. Inhibition of human tumor-infiltrating lymphocyte effector functions by the homophilic carcinoembryonic cell adhesion molecule 1 interactions. J Immunol. 2006;177:6062–6071. doi: 10.4049/jimmunol.177.9.6062. [DOI] [PubMed] [Google Scholar]
- Martens PB, Goronzy JJ, Schaid D, Weyand CM. Expansion of unusual CD4+ T cells in severe rheumatoid arthritis. Arthritis Rheum. 1997;40:1106–1114. doi: 10.1002/art.1780400615. [DOI] [PubMed] [Google Scholar]
- Medeiros RB, Burbach BJ, Mueller KL, Srivastava R, Moon JJ, Highfill S, Peterson EJ, Shimizu Y. Regulation of NF-kappaB activation in T cells via association of the adapter proteins ADAP and CARMA1. Science. 2007;316:754–758. doi: 10.1126/science.1137895. [DOI] [PubMed] [Google Scholar]
- Migueles SA, Osborne CM, Royce C, Compton AA, Joshi RP, Weeks KA, Rood JE, Berkley AM, Sacha JB, Cogliano-Shutta NA, et al. Lytic granule loading of CD8+ T cells is required for HIV-infected cell elimination associated with immune control. Immunity. 2008;29:1009–1021. doi: 10.1016/j.immuni.2008.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molnar-Gabor E, Endreffy E, Rozsasi A. HLA-DRB1, -DQA1, and -DQB1 genotypes in patients with nasal polyposis. Laryngoscope. 2000;110:422–425. doi: 10.1097/00005537-200003000-00017. [DOI] [PubMed] [Google Scholar]
- Moosig F, Csernok E, Wang G, Gross WL. Costimulatory molecules in Wegener's granulomatosis (WG): lack of expression of CD28 and preferential up-regulation of its ligands B7-1 (CD80) and B7-2 (CD86) on T cells. Clin Exp Immunol. 1998;114:113–118. doi: 10.1046/j.1365-2249.1998.00695.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller SN, Ahmed R. High antigen levels are the cause of T cell exhaustion during chronic viral infection. Proc Natl Acad Sci USA. 2009;106:8623–8628. doi: 10.1073/pnas.0809818106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nel AE, Slaughter N. T-cell activation through the antigen receptor. Part 2: role of signaling cascades in T-cell differentiation, anergy, immune senescence, and development of immunotherapy. J Allergy Clin Immunol. 2002;109:901–915. doi: 10.1067/mai.2002.124965. [DOI] [PubMed] [Google Scholar]
- Peggs KS, Quezada SA, Allison JP. Cell intrinsic mechanisms of T-cell inhibition and application to cancer therapy. Immunol Rev. 2008;224:141–165. doi: 10.1111/j.1600-065X.2008.00649.x. [DOI] [PubMed] [Google Scholar]
- Ramírez-Anguiano J, Yamamoto-Furusho JK, Barquera R, Beltrán O, Granados J. Association of HLA-DR3 and HLA-DR4 with sinonasal polyposis in Mexican Mestizos. Otolaryngol Head Neck Surg. 2006;135:90–93. doi: 10.1016/j.otohns.2006.01.009. [DOI] [PubMed] [Google Scholar]
- Sanchez-Segura A, Brieva JA, Rodriguez C. T lymphocytes that infiltrate nasal polyps have a specialized phenotype and produce a mixed TH1/TH2 pattern of cytokines. J Allergy Clin Immunol. 1998;102:953–960. doi: 10.1016/S0091-6749(98)70333-1. [DOI] [PubMed] [Google Scholar]
- Schirmer M, Goldberger C, Wurzner R, Duftner C, Pfeiffer KP, Clausen J, Neumayr G, Falkenbach A. Circulating cytotoxic CD8(+) CD28(−) T cells in ankylosing spondylitis. Arthritis Res. 2002;4:71–76. doi: 10.1186/ar386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schooley K, Zhu P, Dower SK, Qwarnstrom EE. Regulation of nuclear translocation of nuclear factor-kappaB relA: evidence for complex dynamics at the single-cell level. Biochem J. 2003;369:331–339. doi: 10.1042/BJ20020253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson-Abelson MR, Purohit VS, Pang WM, Iyer V, Odunsi K, Demmy TL, Yokota SJ, Loyall JL, Kelleher RJ, Jr, Balu-Iyer S, Bankert RB. IL-12 delivered intratumorally by multilamellar liposomes reactivates memory T cells in human tumor microenvironments. Clin Immunol. 2009;132:71–82. doi: 10.1016/j.clim.2009.03.516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan-Lancaster J, Shaw AS, Rothbard J, Allen PM. Partial T cell signaling: altered phospho-zeta and lack of zap70 recruitment in APL-induced T cell anergy. Cell. 1994;79:913–922. doi: 10.1016/0092-8674(94)90080-9. [DOI] [PubMed] [Google Scholar]
- Sommer J. To cationize glass. J Cell Biol. 1977;75:745a. [Google Scholar]
- Sundstedt A, Sigvardsson M, Leanderson T, Hedlund G, Kalland T, Dohlsten M. In vivo anergized CD4+ T cells express perturbed AP-1 and NF-kappa B transcription factors. Proc Natl Acad Sci USA. 1996;93:979–984. doi: 10.1073/pnas.93.3.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallejo AN, Weyand CM, Goronzy JJ. T-cell senescence: a culprit of immune abnormalities in chronic inflammation and persistent infection. Trends Mol Med. 2004;10:119–124. doi: 10.1016/j.molmed.2004.01.002. [DOI] [PubMed] [Google Scholar]
- Wong RM, Scotland RR, Lau RL, Wang C, Korman AJ, Kast WM, Weber JS. Programmed death-1 blockade enhances expansion and functional capacity of human melanoma antigen-specific CTLs. Intl Immunol. 2007;19:1223–1234. doi: 10.1093/intimm/dxm091. [DOI] [PubMed] [Google Scholar]
- Zha Y, Marks R, Ho AW, Peterson AC, Janardhan S, Brown I, Praveen K, Stang S, Stone JC, Gajewski TF. T cell anergy is reversed by active Ras and is regulated by diacylglycerol kinase-α. Nature Immunol. 2006;7:1166–1173. doi: 10.1038/ni1394. [DOI] [PubMed] [Google Scholar]