

Abstract
Nuclear receptors (NRs) comprise one of the most abundant classes of transcriptional regulators of metabolic diseases and have emerged as promising pharmaceutical targets. Small heterodimer partner (SHP; NR0B2) is a unique orphan NR lacking a DNA-binding domain but contains a putative ligand-binding domain. SHP is a transcriptional regulator affecting multiple key biological functions and metabolic processes including cholesterol, bile acid, and fatty acid metabolism, as well as reproductive biology and glucose-energy homeostasis. About half of all mammalian NRs and several transcriptional coregulators can interact with SHP. The SHP-mediated repression of target transcription factors includes at least three mechanisms including direct interference with the C-terminal activation function 2 (AF2) coactivator domains of NRs, recruitment of corepressors, or direct interaction with the surface of NR/transcription factors. Future research must focus on synthetic ligands acting on SHP as a potential therapeutic target in a series of metabolic abnormalities. Current understanding about the pleiotropic role of SHP is examined in this paper, and principal metabolic aspects connected with SHP function will be also discussed.
1. Introduction
Nuclear receptors (NRs) constitute a unique family of ligand-modulated transcription factors. NRs mediate cellular response to small lipophilic endogenous and exogenous ligands [1, 2] and are responsible for sensing a number of hormones, including steroid and thyroid hormones, and act as positive and negative regulators of the expression of specific genes [3–5]. Therefore, NRs play a central role in many aspects of mammalian development, as well as lipid homeostasis, physiology, and metabolism. NRs make up one of the most abundant classes of transcriptional regulators in the body and have emerged as promising pharmaceutical targets.
Classically, NRs consist of several functional domains, that is, a variable N-terminal ligand-independent transactivation domain (which often exhibits a constitutive transcription activation function (AF-1)), a highly conserved DNA-binding domain (DBD) that contains two zinc fingers, a hinge domain (a variable linker region), and a multifunctional C-terminal domain. Furthermore, the C-terminal domain includes the ligand binding (LBD), the dimerization interface, and the ligand-dependent transactivation domain AF-2 [1, 6].
Small heterodimer partner (SHP; NR0B2 for nuclear receptor subfamily 0, group B, member 2; MIM number 604630, 601665) is a member of the mammalian NR superfamily, due to the presence of a putative ligand-binding domain (LBD) [7]. SHP functions as a corepressor through heterodimeric interaction with a wide array of nuclear receptors and repressing their transcriptional activity. SHP achieves its goal via several members of the NR superfamily that are able to regulate SHP expression. However, SHP is also a unique and atypical NR because it lacks the classical DNA-binding domain (DBD), generally present in other NRs [8]. The NR0B family of NRs consists of 2 orphan receptors: SHP and DAX-1 (dosage-sensitive sex reversal adrenal hypoplasia congenita (AHC) critical region on the X chromosome, gene 1). DAX1 is a gene whose mutation causes the X-linked adrenal hypoplasia congenita [9] and is the only family member that lacks a conventional DBD. DAX-1 (NR0B1) is therefore seen as the closest relative of SHP in the NR superfamily [10–12]. Both SHP and DAX-1 appear to be specific to vertebrates. In this respect, no homologous genes have been found in Drosophila melanogaster or Caenorhabditis elegans [12]. Whereas SHP is different from other conventional NRs both structurally and functionally, it acts as a ligand-regulated receptor in metabolic pathways [13]. SHP belongs to the orphan subfamily since there is no known ligand for this receptor, except for some retinoid-related molecules [14]. SHP inhibits transcriptional activation by working on several other nuclear receptors, that is, directly modulating the activities of conventional nuclear receptors by acting as an inducible and tissue-specific corepressor [12, 15]. The discovery of SHP dates back to 1996 [10]; since then, this orphan NR has been identified as a key transcriptional regulator of signaling pathways [8, 16] involving fundamental biological functions and metabolic processes. Such processes include cholesterol, bile acid and fatty acid metabolism, glucose and energy homeostasis, and reproductive biology [17]. Experiments performed by fluorescence in situ hybridization (FISH) analysis of the human metaphase chromosome have shown that SHP is found at a single locus on chromosome 1 at position 1p36.1 and consists of two exons and a single intron spanning approximately 1.8 kb with 257 amino acids in humans [18]. In mice and rats, SHP resides on chromosomes 4 and 5, respectively, both consisting of 260 amino acids. SHP expression is predominantly observed in the liver [10, 18], but it is also detected at lower levels in other tissues, including the pancreas, spleen, small intestine, colon, gallbladder, kidney, adrenal gland, ovary, lung, brainstem, cerebellum, heart, and thymus (Table 1) [19–21].
Table 1.
| LIVER (greater)* | |
| Spleen* | |
| Pancreas* | |
| Central nervous system (brainstem and cerebellum) | |
| Adrenal gland* | |
| Intestine (duodenum*, jejunum*, ileum*, and colon) | |
| Gallbladder, stomach*, kidney*, ovary, lung, prostate, testis, uterus, heart*, thymus, and epididymis |
The genomic structure and human SHP domain structure are depicted in Figure 1 [15]. SHP is indeed able to repress the transcriptional activities of its target NRs and transcriptional regulators through two functional Leu-Xaa-Xaa-Leu-Leu- (LXXLL-) like motifs [22–24]. Such motifs appear to be essential for the interaction with the (activation function 2) AF-2 domains of several sets of NRs [22, 23]. The human SHP is enriched by another 12 amino acids [25–36], and this region between helix 6 and 7 is also involved in the repression of the transactivation of NRs [37].
Figure 1.
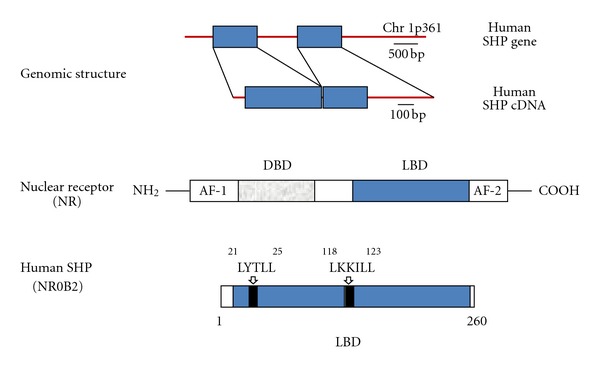
Top: the genomic structure of human SHP. Rectangles represent the two exons with a single intron spanning approximately 1.8 kilobases and located on a single locus on chromosome 1p36.1 [18]. The region 5′ includes ≈600 nucleotides from the transcription start site and is characterized by promoter activity. Bottom: typical nuclear receptor is compared with the domain structure of human SHP. The canonical structure of NR includes the N-terminal activation function 1 (AF1) domain, DNA-binding domain (DBD), ligand-binding domain (LBD), and C-terminal activation function 2 (AF2) domain. SHP lacks the DBD. Two functional LXXLL-related motifs (also named as NR boxes) are typical of the human SHP structural domains. Such motifs are located in the putative N-terminal helix 1 of the LBD and in the C-terminal region of the helix 5. While active NRs exhibit glutamic acid in AF-2, the SHP AF-2 domain is replaced with aspartic acid. Adapted from Chanda et al. [15] and Shulman and Mangelsdorf [130].
About half of all mammalian NRs and several transcriptional coregulators can interact with SHP [12]. Since SHP lacks DNA-binding domain, it exerts the inhibitory effects through protein-protein interaction [10]. SHP expression seems to follow a circadian rhythm in the liver, involving the CLOCK-BMAL1 pathway and suggesting that some of the regulatory functions of SHP and deriving functions must be temporal [19, 20, 38].
Gene expression of SHP is regulated by several factors including NRs, transcription factors, and a number of additional conditions and substances, as extensively reported in Table 2. Also, the central role of SHP is clear since this NR is able to act as a coregulator for wide range of targets, namely, NRs/transcription factors/transcriptional coregulators and few different molecules, as depicted in Table 3. In general, SHP acts as a repressor of the transcriptional activity of the specific interacting partner (via LBD of the partner and NR boxes of SHP) [12, 39–43]. However, it is also demonstrated that SHP is able to upregulate gene transcription, as in the case of PPARα and PPARγ [44–46] and NF-κB [44].
Table 2.
| (1) Nuclear receptors | |
|
| |
| Protein | Model(s)/putative function |
|
| |
| ERα | Uterus, pituitary, kidney, and adrenal gland, HepG2 cell lines/biological effects of estrogens, LDL/HDL metabolism [134]. |
|
| |
| ERRα, β, γ | SHP promoter is activated by the ERRγ, while SHP inhibits ERRγ transactivation (autoregulatory loop). SHP and ERRγ coexpressed in several tissues (e.g., pancreas, kidney, and heart). Role in some forms of moderate obesity? SHP also physically interacts with ERR α and β isoforms (yeast two-hybrid and biochemical assays) [133]. |
|
| |
| FXR | Downregulation of CYP7A1-mediated bile acid biosynthesis by the FXR/SHP/LRH-1 cascade in the liver [64]. |
|
| |
| LXRα | Direct regulation of SHP and repression of CYP7A1-mediated bile acid biosynthesis (in humans not in rodents). Effect on cholesterol homeostasis [135]. |
|
| |
| LRH-1 | Liver/formation of heterodimeric SHP/LRH-1 complex > inactivation of LRH-1 > SHP repression (autoregulatory negative feedback) [64, 65, 136]. Also involved in the CLOCK-BMAL1 circadian activation of SHP [38]. |
|
| |
| PPARγ | Liver/PPARγ decreases gluconeogenic gene expression by the PPARγ/RXRα heterodimer binding to the PPRE in the human SHP promoter. A mechanism explaining the SHP-mediated acute antigluconeogenic effects of PPARγ [137]. |
|
| |
| SF-1 | At least five binding sites for SF-1 detected in the promoter region of SHP. Rat testis and adrenal glands, human fetal adrenal gland [136]. |
|
| |
| (2) Transcription factors | |
|
| |
| Protein | Model(s)/putative function |
|
| |
| CLOCK-BMAL1 | Liver/SHP displays a circadian expression pattern involving CLOCK-BMAL1 (core circadian clock component). Regulation of SHP promoter together with LRH-1 and SHP. Relevance for circadian liver function? [38]. |
|
| |
| E2A proteins (E47, E12, E2/5) | HepG2, HeLa, and CV-1 cells/bHLH transcription factors, the E2A proteins activate human (not mouse) hSHP promoter. E47 and SF-1 stimulate cooperatively SHP promoter. The Id protein inhibits E47 binding to hSHP promoter. A role for tissue-specific gene regulation, B-cell differentiation, tumor suppression? [138]. |
|
| |
| HNF-1α | Liver/modulation of bile acid and liver cholesterol synthesis via the FXR/SHP/LRH-1 complex and effect on CYP7A1 [69]. |
|
| |
| HNF4α | Pancreatic β-cells/decreased expression of SHP may be indirectly mediated by a downregulation of HNF4α. SHP can repress its own transcriptional activation by inhibiting HNF4 α function (feedback autoregulatory loop) and, indirectly (via HNF4 α), HNF1α function. Relevance for pancreatic islet differentiation, insulin secretion, synthesis [116]. |
|
| |
| JNK/c-Jun/AP-1 | Primary rat hepatocytes/bile acid downregulation of CYP7A1-dependent bile acid biosynthesis via the JNK/cJun/AP1 pathway. SHP promoter is a direct target of activated c-Jun binding to AP-1 element [139]. Also, in HL-60 leukemia cells, c-Jun increases the transcriptional activation of the SHP promoter to activate the expression of Shp genes associated with the cascade regulation of monocytic differentiation [140]. |
|
| |
| SMILE | HEK-293T, HepG2, MCF-7, T47D, MDA-MB-435, HeLa, PC-3, C2C12, NIH 3T3, K28, Y-1, and TM4 cell lines/SMILE isoforms (SMILE-L and SMILE-S) regulate the SHP-driven inhibition of ERs transactivation in a cell-type-specific manner [25, 26, 39]. |
|
| |
| SREBP-1 | Liver/effect on human (not mouse) SHP promoter. Cholesterol and bile acid homeostasis, fatty acid synthesis [27]. |
|
| |
| USF-1 | HepG2, H4IIE, and AML12 cells/HGF activates AMPK signaling pathway in hepatocytes, E-box-binding transcription factor USF-1, and binding to the Shp gene promoter. SHP induction of gene expression leads to inhibition of hepatic gluconeogenesis due to SHP-repressed transcription factor HNF4α [28]. |
|
| |
| (3) Transcriptional coregulators | |
|
| |
| Protein | Model(s)/putative function |
|
| |
| RNF31 | NCI-H295R (H295R) adrenocortical carcinoma cell line, COS-7 and HeLa cells/RNF31 interacts with SHP, stabilizes DAX-1, and is required for DAX-1-mediated repression of transcription. Relevant as coregulator of steroidogenic pathways [43]. |
|
| |
| SRC-1 | Murine macrophage cell line RAW 264.7, HeLa, and CV-1 cells/SHP interacts negatively with SRC-1 (a transcription coactivator of nuclear receptors and other transcription factors including NF-κB). See also oxLDL in this table [44]. |
|
| |
| (4) Other SHP inducers | |
|
| |
| Factor | Model(s)/putative function |
|
| |
| Bile acids (final intermediates) | Experiments in HepG2 cells/treatment with chenodeoxycholic acid and late intermediates in the classic pathway of bile acid synthesis: 26-OH-THC (5β-cholestane-3α,7α,12α,26-tetrol), THCA (3α,7α,12α-trihydroxy-5 β-cholestanoic acid), 26-OHDHC (5β-cholestane-3α,7α,26-triol), DHCA (3α,7α-dihydroxy-5β-cholestanoic acid) resulted in 2.4-6.5-fold increase in SHP mRNA expression [132]. Confirmed by Ourlin et al. with the two FXR ligands chenodeoxycholic acid and cholic acid [1]. |
|
| |
| Guggulsterone (plant sterol) | Active extract from Commiphora Mukul. FXR antagonist. In Fisher rats, guggulsterone increased transcription of bile salt export pump (BSEP) mRNA and SHP expression [29]. |
|
| |
| GW4064 (ligand) | Synthetic FXR-selective agonist [29]. In primary cultured human hepatocytes, GW4064 treatment was associated with a marked induction of SHP (≈70-fold) and complete suppression of CYP7A1 [64, 65]. In HepG2 cells, GW4064 (1uM) induced a 3.9-fold increase in SHP mRNA expression. Confirmed by [30]. |
|
| |
| Interleukins (various) | IL-1Ra (−/−) mice/high cytokine levels in IL-1Ra (−/−) mice reduce mRNA expression of CYP7A1 with concurrent upregulation of SHP mRNA expression [31]. SHP significantly expressed in IFN-γ/CH11-resistant HepG2 cells [32]. |
|
| |
| PGC-1α (gene expression inducer) | COS-7 cell lines/PGC-1α mediates the ligand-dependent activation of FXR and transcription of Shp gene. Relevance in mitochondrial oxidative metabolism in brown fat, skeletal muscle, and liver gluconeogenesis [33]. |
|
| |
| PMRT1 (group of protein arginine methyltransferases) | Hepatic cell lines/PRMT1 functions as FXR coactivator and has a role in chromatin remodeling. PRMT1 induces BSEP and SHP and downregulation of NTCP and CYP7A1 (targets of SHP) [30]. |
|
| |
| Procyanidins (polyphenols) | Grape seed procyanidin extract is given orally in male Wistar rats. Increase of liver mRNA levels of small heterodimer partner (SHP) (2.4-fold), cholesterol 7α-hydroxylase (CYP7A1), and cholesterol biosynthetic enzymes with improved lipidogenic profile and atherosclerotic risk [34]. |
|
| |
| (5) Factors/conditions associated with SHP repression | |
|
| |
| β Klotho (type I membrane protein) | In βKlotho (−/−) mice: enhanced bile acid synthesis with attenuation of bile acid-mediated induction of Shp. βKlotho involved in CYP7A1 selective regulation [35]. |
|
| |
| IL-1β (interleukin) | SHP downregulation [36]. |
|
| |
| oxLDL (oxidized low density lipoprotein) | Murine macrophage cell line RAW 264.7, HeLa, and CV-1 cells/oxLDL decreased SHP expression. SHP transcription coactivator of NF-κB which became progressively inert in oxLDL-treated RAW 264.7 cells (see also Table 3). Relevance for differentiation mechanism of resting macrophage cells into foam cells and resulting atherogenesis [44]. |
AP-1: adaptor protein-1; bHLH: basic helix-loop-helix; DAX1: dosage-sensitive sex reversal adrenal hypoplasia congenita critical region on the X chromosome, gene 1; E2A: E2A2 gene products belonging to the basic helix-loop-helix (bHLH) family of transcriptor factors; ERα: estrogen receptorα; ERRγ: estrogen receptor-related receptor-γ; FXR: farnesoid X receptor; HGF: Hepatocyte growth factor; HNF-1α: hepatocyte nuclear factor-1α; HNF4α: hepatocyte nuclear factor-4α; Id: inhibitor of differentiation; IL-1Ra (−/−): interleukin-1 receptor antagonist; JNK: Jun N-terminal kinase; LRH-1: liver receptor homologue-1; LXRα: liver X receptorα; NFκB: nuclear factor-κB; NR: nuclear receptor; NTCP: Na+-taurocholate cotransport peptide; oxLDL: oxidized low-density lipoprotein; PGC-1: PPARγ (peroxisome-proliferator-activated receptor γ) coactivator-1α; PMRT1: protein arginine methyltransferase type 1; PPRE: PPAR response element; RNF31: member of the ring-between-ring (RBR) family of E3 ubiquitin ligases; RXR α: retinoid X receptor; SF-1: steroidogenic factor-1; SHP: small (short) heterodimer partner; hSHP: human small (short) heterodimer partner; SMILE: SHP-interacting leucine zipper protein; SRC-1: steroid receptor coactivator-1; SREBP-1: sterol regulatory element binding protein-1; USF-1: upstream stimulatory factor-1.
Table 3.
| (1) Nuclear receptors | |
|
| |
| Protein | Model(s)/putative function |
|
| |
| AR | The AR/SHP interaction leads to >95% inhibition of AR via the LXXLL motifs. Mechanisms involve inhibition of AR ligand-binding domain and AR N-terminal domain-dependent transactivation and competing with AR coactivators [23]. |
|
| |
| CAR, RAR, TR | HepG2 and JEG-3 cells/early evidence that SHP interacts with several receptor superfamily members and inhibits transactivation. CAR is an NR-inducing CYP2 and CYP3 genes involved in the metabolism of xenobiotics [10, 24]. |
|
| |
| DAX-1 | Human embryonic kidney 293 cells/beside individual homodimerization of DAX1 and SHP, this is the first evidence of DAX1-SHP heterodimerization in the nucleus of mammalian cells. Involvement of the LXXLL motifs and AF-2 domain of DAX1 in this interaction. Distinct functions for SHP (different from transcriptional repressor) are anticipated [141, 142]. |
|
| |
| ER | 293 human embryo kidney cells, Cos7 kidney cells/direct inhibitory binding of SHP to ERs via LXXLL-related motifs to the AF-2 domain [21]. RL95-2 human endometrial carcinoma cells/SHP inhibits the agonist activity of 4-hydroxytamoxifen displaying a potent inhibitory effect for Erα > ERβ. Direct interaction of SHP with ER and inhibition of ER transcriptional activity [143]. Prevention of tamoxifen-induced estrogen agonistic effects and neoplastic changes in the endometrium in women with breast cancer taking tamoxifen? |
|
| |
| ERRγ | HeLa (human cervical carcinoma), CV-1 (green monkey kidney), and HEK 293 (human embryonic kidney) cell lines/SHP inhibits ERRγ transactivation by physical interaction with the 3 members of the ERR subfamily. Interaction is dependent on N-terminal receptor interaction domain of SHP and AF-2 surface of ERRγ. Part of the autoregulatory mechanism of gene expression going through ERRγ/SHP/ERRγ. A potential role in some forms of moderate human obesity during SHP mutations [133]. |
|
| |
| GR | 293 human embryo kidney cells and COS-7 monkey kidney/SHP inhibits the transcriptional activity of GR via the LXXLL motif. Physiological role of SHP in glucocorticoid signaling and gluconeogenesis [22]. See also HNF4 [90] and Foxo1 [115]. |
|
| |
| HNF4 | Human ANG transgenic mice and HepG2 cells treated with bile acids/evidence that bile acids negatively regulate the human ANG gene through the FXR/SHP-mediated process (inhibition of the binding of HNF4 to the ANG promoter) [90]. Mechanisms: SHP binds the AF-2 region and the N-terminal region of HNF4 and inhibits the binding of HNF4 to DNA. Also, modulation of HNF4 activity by SHP has important metabolic effects and interacts with the pathway of gluconeogenesis [47] (see text and Foxo1) [115]. |
|
| |
| LRH-1 | HepG2 cells/SHP interacts directly with the orphan receptor LRH-1 (AF-2 surface) and competes with other coactivators, leading to repression of LRH-1 transcriptional activity [48]. Demonstration that repression of CYP7A1 and bile acid synthesis requires coordinate interaction/transcription of FXR/LRH-1/SHP autoregulatory cascade, essential for maintenance of bile acid-induced negative feedback, and therefore hepatic cholesterol metabolism [65] (see also Figure 2). |
|
| |
| LXRα | In vitro experiments and in vivo human colon Caco-2 cells/SHP directly inhibits the transcriptional activity of LXRα via the AF-2 domain. Relevance for direct downregulation of specific LXR target genes (controlling CYP7A1, ABCA1, ABCG1, ABCG5, ABCG8, CETP, ApoE, SREBP-1c) and therefore cholesterol-bile acid homeostasis [144]. |
|
| |
| Nur77 (NGFI-B) | HepG2 cells/Nur77 plays a key role in apoptosis of many cell types and cancer cells. Evidence that SHP functions to repress the transcriptional function of Nur77 (binding coactivator CBP, see elsewhere in this table). SHP plays a protective role in the Nur77-mediated apoptosis in liver. Mutations in SHP: a role also for affect initiation and progression of inflammatory liver diseases such as alcoholic hepatitis and hepatic viral infections? [32]. |
|
| |
| PPARα | In vitro binding assays and in vivo experiments/the promoter regions of the genes encoding the first two enzymes of the peroxisomal beta-oxidation pathway (AOx, HD), contain transcriptional regulatory sequences (PPRE) bound by the PPARα/RXRα heterodimeric complex. SHP-inhibited transcription by PPARα/RXRα heterodimers from the AOx-PPRE. SHP potentiated transcription by PPARα/RXRα heterodimers from the HD-PPRE (evidence of SHP-dependent upregulation PPARα-mediated gene transcription) [46]. |
|
| |
| PPARγ | In vitro experiments, COS-7 cells/Shp gene expressed also in adipose tissue. SHP induces PPAR activation via C terminus (direct binding to the DBD/hinge region of PPARγ) and inhibition of the repressor activity of NCoR. SHP may act as an endogenous enhancer of PPARγ by competing with NCoR [45]. Mutant SHP proteins display less enhancing activity for PPARy compared with wild-type SHP, and a human model leading to mild obesity and insulin resistance has been described in Japanese during naturally occurring mutations [111] (see also text and Table 3). |
|
| |
| PXR | In vitro experiments, human hepatocytes, mouse model on cholic acid-supplemented diet/SHP act as potent repressor of PXR transactivation. Upon sensing xenobiotics and bile acid precursors, PXR controls CYP3A gene induction and inhibits CYP7α, acting on both bile acid synthesis and catabolism. PXR function might be also inhibited in the presence of cholic acid, chenodeoxycholic acid-dependent SHP upregulation [1]. |
|
| |
| RXR | HepG2 cells/demonstration that SHP acts as a transcriptional repressor for RXR. Full inhibition by SHP requires its direct repressor activity [47]. |
|
| |
| SHP | Human embryonic kidney 293 cells/LXXLL motifs and AF-2 domain are involved in SHP homodimerization in the nucleus (similarly to DAX1-SHP heterodimerization). NR0B family members use similar mechanisms for homodimerization as well as heterodimerization. Distinct functions for SHP (different from transcriptional repressor) are anticipated [141, 142]. |
|
| |
| (2) Transcription factors | |
|
| |
| Protein | Model(s)/putative function |
|
| |
| ARNT | RL95-2 human endometrial carcinoma cells/TCDD binds to AHR (a member of bHLH-PAS family of transcription factors). Studies on physical and functional interaction of SHP with the ligand AHR/ARNT heterodimer showed that SHP inhibits the transcriptional activity of ARNT (not AHR) in vitro. Consequent inhibition of binding of AHR/ARNT to XREs. [41]. Relevance for expression of several genes involved in drug and hormone metabolism [145]. |
|
| |
| BETA2/NeuroD | 293T, COS-7, CV-1 cells/BETA2/NeuroD is a member of tissue-specific class B bHLH proteins and cats as a positive regulator of insulin gene expression [146] and neuronal differentiation [147]. SHP physically interacts and inhibits helix-loop-helix transcription factor BETA2/NeuroD transactivation of an E-box reporter in mouse pancreas islets. The inhibitory effect of SHP requires its C-terminal repression domain, interference with coactivator p300 for binding to BETA2/NeuroD, and direct transcriptional repression function. Relevance for development of the nervous system and the maintenance and formation of pancreatic and enteroendocrine cells [148]. |
|
| |
| C/EBPα | HepG2 hepatoma cells/SHP interacts directly with C/EBPα and represses C/EBPα-driven PEPCK gene transcription. Overall, a role for SHP in regulation of hepatic gluconeogenes is driven by C/EBPα activation in the liver [149]. |
|
| |
| Foxo1 | C57BL/6J mice and HepG2 and HEK293T cells/treatment with chenodeoxycholic acid was associated with FXR-dependent SHP induction, downregulation of gluconeogenic gene expression (G6Pase, PEPCK, FBP1), interaction of the forkhead transcription factor Foxo1 with SHP, and repression of Foxo1-mediated G6Pase transcription (competition with CBP). A similar mechanism is postulated for SHP-driven HNF-4 repression of PEPCK, FBP1 transcription. A mechanism by which bile acids metabolism is linked to gluconeogenic gene expression via an SHP-dependent regulatory pathway [115]. |
|
| |
| HNF3 (Foxa) | HepG2, 293T, NIH3T3, and HeLa cells, primary hepatocytes/SHP physically interacts and inhibits the transcriptional activity of the forkhead transcription factor HNF3 (isoforms α, β, γ). Relevance for SHP-driven regulation of gluconeogenic genes encoding G6Pase, PEPCK, and bile acid synthesis (CYP7A1), via inhibition of DNA-binding of HNF3 [51]. |
|
| |
| Jun D | Two rat models of liver fibrosis and Hepatic Stellate cells (HSC)/promoting the ligand-induced FXR-SHP cascade (by the FXR ligand 6-EDCA, in rat models) and overexpressing SHP in HSC prevented fibrogenic changes in the liver. SHP binds JunD and inhibits DNA binding of adaptor protein (AP)-1 induced by thrombin. FXR ligands as therapeutic agents to treat liver fibrosis? [52]. |
|
| |
| NF-κB | Murine macrophage cell line RAW 264.7/SHP acts as a positive transcription coactivator of NF-κB and essential for NF-κB transactivation by palmitoyl lysophosphatidylcholine (one of the oxLDL constituents). Relevance for differentiation mechanism of resting macrophage cells into foam cells and resulting atherogenesis (see also [44]). |
|
| |
| Smad | HepG2, CV-1, and HeLa cells/SHP represses Smad3-induced transcription by competing for the coactivator p300. SHP therefore represses TGF-β-induced gene expression. Relevance for TGF-β-dependent regulation of cell growth, apoptosis, carcinogenesis, and regeneration following liver injury [40]. SHP-Smad3 interaction similar to SHP-BETA2/NeuroD [148]. |
|
| |
| TRAF6, p65 | Macrophages/a novel function of SHP in innate immunity involving Toll-like receptors (TLRs). SHP negatively regulates TLR signaling to NF-κB. Likely, SHP negatively regulates immune responses initiated by various pathogen-recognition receptors by forming a complex with TRAF6 and effect on TRAF6 ubiquitination. In the cytosol of LPS-stimulated cells. SHP also acts as specific transrepressor of the transcription factor p65 (part of the p50/p65 heterodimer found in NF-κB). An additional role for SHP in sepsis and inflammatory disease? [128, 129]. |
|
| |
| (3) Transcriptional coregulators | |
|
| |
| Protein | Model(s)/putative function |
|
| |
| Brm, BAF155, BAF47, mSin3A, Swi/Snf | HepG2 cells/The CYP7A1 gene was used as a model system. SHP has direct interaction with corepressors at the level of native chromatin. SHP directly interacted and mediated the recruitment of mSin3A-Swi/Snf-Brm chromatin remodelling complex to the CYP7A1 promoter (TATA and BARE II region of the promoter). Also, the mSinA3/HDAC1 corepressor complex is inhibiting transcription by histone deacetylation. SHP also interacted with known proteins belonging to the Swi/Snf complex (BAF155, BAF47). This mechanism explains the complex and subtle SHP-driven inhibition of hepatic bile acid synthesis [50]. |
|
| |
| CBP | HepG2 cells, CV-1 cells/SHP binds coactivator CBP and competes with Nur77. The mechanism explains the repression of the transcriptional function of Nur77, which is fundamental in apoptosis in the liver [32]. |
|
| |
| EID-1 | Cos-7 cells/SHP specifically interacts with EID-1 providing inhibitory mechanisms. EID-1 (a non-HDAC cofactor) acts as inhibitor of the coregulator complex EID1–p300–CBP. Results clarify essential repression mechanisms of SHP involving coinhibitory factors (upstream targets) distinct from NRs corepressor [12, 150]. |
|
| |
| G9a, HDAC-1 | Caco-2, HepG2, HeLa, Cos-1 cells/SHP localized exclusively in nuclease-sensitive euchromatin regions. SHP can functionally interact with HDAC-1 (HDAC of class I) and the euchromatic histone 3 methylase G9a, and the unmodified K9-methylated histone 3 [151]. Additional data on mechanisms involved SHP-driven repressive activity, involving also target genes regulated by G9a and SHP-mediated inhibition of hepatic bile acid synthesis via coordinated chromatin modification at target genes [152]. |
|
| |
| GPS2 | Cos-7, HepG2, Huh7 cells/SHP negatively interacts with GPS2 (a stoichiometric subunit of the NR corepressor, N-Cor) complex, involved in bile acid synthesis and differential coregulation of CYP7A1 and CYP8B1 expression [67]. |
|
| |
| SIRT1 | HepG2, HEK293T (293T), and HeLa cells/SIRT1 is a HDAC of class III. SHP recruits SIRT1 (activating deacetylase activity of SIRT1) to repress LRH1 transcriptional activity as well as inhibition LRH1 target gene promoter activity and mRNA levels. A novel mechanism is described for SHP repressive action and control of bile acid homeostasis. SIRT1 in working concertedly with NRs and affecting chromatin remodeling in target gene promoters [42]. |
|
| |
| SMRT/NcoR | Hepatoma cell lines/studies on the role of SHP in CAR-mediated transactivation of the CYP2B gene. SHP might interact with subunits of functionally distinct coregulator complexes, including HDAC3-N-CoR-SMRT [24, 120]. |
|
| |
| (4) Others | |
|
| |
| Factor | Model(s)/putative function |
|
| |
| miRNA-206 | SHP−/− mice/SHP as an important transcriptional activator of miRNA-206 gene expression via a cascade dual inhibitory mechanism involving AP1 but also YY1 and ERRγ. Relevance for multiple steps involving cellular development, proliferation, and differentiation [153]. |
|
| |
| RNA Pol II | Caco-2 cells/within the pathway of SHP-LXR interaction, it is shown that SHP can interact in vitro with RNA polymerase II but not with TFIID and TFIIE transcription initiation factor II D (TFIID), general transcription factor II E (TFIIE) (components of the basal transcription machinery). A further mechanism by which SHP could inhibit both basal and induced transactivation [144]. |
ABCA1, ABCG1, ABCG5, and ABCG8: ATP-binding cassette transporters; AP1: transcription factor activator protein 1; AHR: aryl hydrocarbon receptor (AHR); ARNT: aryl hydrocarbon receptor (AHR)/AHR nuclear translocator protein; ANG: angiotensin; AOx, acyl-CoA oxidase; ApoE: apolipoprotein E; bHLH-PAS: basic helix–loop–helix–PAS; AR: androgen receptor; BAFs: Brm- or Brg-1-associated factors; BARE: bile acid response element; Brm: human Brahma; CAR: constitutive androstane receptor; CBP: CREB-binding protein; C/EBPα: CCAAT/enhancer-binding protein α; CETP: cholesteryl ester transfer protein; CREB: coactivator cAMP-response element-binding protein; CYP7A1: cholesterol-7-α-hydroxylase; DAX1: dosage-sensitive sex reversal adrenal hypoplasia congenita critical region on the X chromosome: gene 1; DBD: DNA-binding domain; 6-ECDCA, 6-ethylchenodeoxycholic acid; EID1: E1A-like inhibitor of differentiation 1; ER: estrogen receptor; ERRγ: estrogen receptor-related receptor-γ; FBP1: fructose-1,6-bisphosphatase; FXR: farnesoid X receptor; G6Pase: glucose-6-phosphase; GR: glucocorticoid receptor; GPS2: G protein pathway suppressor 2; HD: enoyl-CoA hydratase/3-hydroxyacyl-CoA dehydrogenase; HDACs: histone deacetylases; HDAC-1: histone deacetylase-1; HDAC-1: histone deacetylase-3; JunD: predominat Jun family protein; HNF3/Foxa: hepatocyte nuclear factor-3; HNF4: hepatocyte nuclear factor-4; LPS: lipopolysaccharides; LXRα: liver X receptorα; LRH-1: liver receptor homologue-1; miRNAs (miR): microRNAs; NcoR: nuclear receptor corepressor; NF-κB: nuclear factor-κB; Nur77: nuclear growth factor I-B; PEPCK: phosphoenolpyruvate carboxykinase; PPRE: peroxisome proliferator-response elements; PXR: pregnane X receptors RAR: retinoid acid receptor; RNA Pol II: RNA polymerase II; RXR: retinoid X receptor; SIRT1: sirtuin1; SREBP-1c: sterol regulatory element-binding protein-1c; TCDD, 2,3,7,8-tetrachlorodibenzo-p-dioxin; TFIID: transcription initiation factor II D (TFIID); TFIIE: transcription factor II E; TGF-β: transforming growth factor-β; TLRs: Toll-like receptors; TR: thyroid receptor; TRAF6: TNF-receptor-associated factor-6; XRE, xenobiotic response element; YY1: Ying Yang 1.
Both N-terminal NR interaction domain and C-terminal domain of SHP are important for repression [47, 48]. Overall, the SHP-mediated repression of target transcription factors occurs by at least three distinct transcriptional repression mechanisms (Figure 2).
Figure 2.
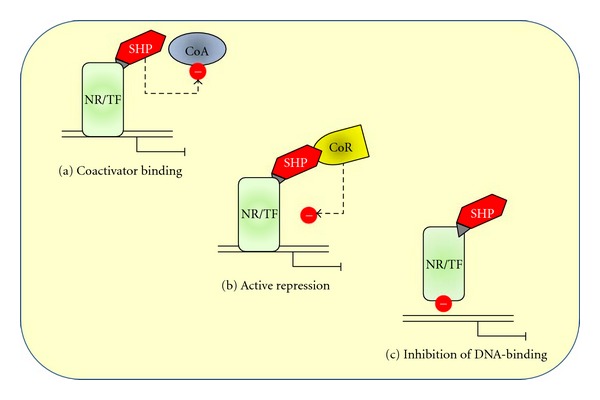
The SHP-mediated repression of target transcription factors occurs by at least three distinct transcriptional repression mechanism: (a) direct interference with the AF-2 coactivator domain of NRs (competition for coactivator binding, leading to the repression of NR-mediated transcriptional activity); (b) recruitment of corepressors, resulting in active repression; (c) direct interaction with the surface of NR or transcription factor, resulting in the blockade of DNA binding and the consequent inhibition of its transcriptional activity. See text for details. The dotted arrows and (-) symbols indicate inhibition. CoA: coactivator; CoR: corepressor; NR: nuclear receptor; SHP: small heterodimer factor; TF: transcription factor. Modified after [12, 15, 131].
A first mechanism involves direct interference with the AF-2 coactivator domain of NRs (competition for coactivator binding, leading to the repression of NR-mediated transcriptional activity). This is the case for the inhibition of estrogen receptor α (ERα) and estrogen receptor β (ERβ) [49].
A second mechanism for the SHP-mediated repression involves the recruitment of corepressors including direct interactions among mammalian homolog of the Saccharomyces cerevisiae transcriptional corepressor Sin3p (mSin3A), human Brahma (Brm), SWItch/Sucrose NonFermentable (SWI/SNF) complexes leading to the repression of cholesterol 7α-hydroxylase (CYP7A1) [50]).
A third mechanism of inhibition of SHP involves the direct interaction with the surface of NR or transcription factor, resulting in the blockade of DNA binding and the consequent inhibition of its transcriptional activity. This is the case for RAR-RXR heterodimers [10], PXR-RXR binding to DNA by SHP [1], interaction with hepatocyte nuclear factor (HNF4), or Jun family of the activator protein 1 (AP-1) transcription factor complex (JunD) [51, 52].
All three mechanisms might occur sequentially or alternatively according to type of cells and promoters [12].
Clearly, information on factors that increase or decrease SHP expression and that are regulated by SHP is essential for understanding the regulatory effects of this orphan NR. Few years of research have not been enough to identify a true ligand. Interestingly, it is suggested that targeting posttranslational modifications of SHP may be an effective therapeutic strategy. Selected groups of genes could be controlled to cure a vast range of metabolic and SHP-related diseases [53]. Overall, the huge amount of information on SHP function is currently available, making this NR essential in a number of functions involving cholesterol and bile acid metabolism, lipogenesis, glucose metabolism, steroid hormone biosynthesis, xenobiotic homeostasis/metabolism, and cell cycle.
In particular, the ability of SHP in interacting with different metabolic signaling pathways including bile acids and lipid homeostasis, fat mass, adipocytes, and obesity will be reviewed here.
2. Bile Acids and Lipid Homeostasis
The wide ability of SHP to target multiple genes in diverse signaling pathways points to the key role of SHP in various biological processes, including the metabolism of bile salts, glucose, and fatty acids. Both unique structure and functional properties account for the complexity of SHP signaling. Studies suggest that loss of SHP might positively affect cholesterol and bile acid homeostasis in pathophysiologically relevant conditions [54]. Bile acids (BAs) are amphipatic cholesterol metabolites which are synthesized in the liver, secreted into bile, stored in the gallbladder, and secreted postprandially into the duodenum. BAs are synthesized from cholesterol, and this pathway provides the elimination of excess cholesterol in the body [55]. Moreover, BAs should be seen as physiological detergents which, in the small intestine, are essential for the absorption, transport, and distribution of lipophilic molecules, including dietary lipids, steroids, and lipid-soluble vitamins. In the intestine, BAs undergo extensive metabolism by the intestinal microflora. A high efficient system is the enterohepatic circulation of BAs [55, 56], where more than 90–95% of BAs are returned to the liver from the terminal ileum via the portal vein. Thus, the concentration of BAs in serum, liver, and intestine is tightly regulated to prevent damage to enterohepatic tissues due to their strong detergent moiety [57–59]. The major rate-limiting step in biosynthetic pathway of BAs in humans is initiated by cholesterol 7α-hydroxylase (CYP7A1), the microsomal P450 liver enzyme, to produce two primary BAs, cholic acid, and chenodeoxycholic acid, essential in the overall balance of cholesterol homeostasis. Sterol 12α hydroxylase (CYP8B1) catalyzes the synthesis of cholic acid, a step which determines the cholic acid to CDCA ratio in the bile [60]. Secondary bile acids (deoxycholic acid and lithocholic acid) and tertiary bile acids (ursodeoxycholic acid) in humans are produced following intestinal dehydroxylation of primary bile acids by intestinal bacteria [58, 61].
Regulation of BA biosynthesis is highly coordinated and is mediated by key NRs including the orphan receptor, liver receptor homologue-1 (LRH1; NR5A2), the hepatocyte nuclear factor 4α (HNF4α), SHP, and the bile acid receptor farnesoid X receptor (FXR; NR1H4). Thus, the activation of FXR initiates a feedback regulatory loop via induction of SHP, which suppresses LRH-1- and HNF4α-dependent expression of the two major pathway enzymes cholesterol 7hydroxylase (CYP7A1) and sterol 12 hydroxylase (CYP8B1).
The BA feedback regulation primarily occurs since BAs act as transcriptional regulators for the expression of the gene encoding CYP7A1. Both cholic acid and chenodeoxycholic acid function as endogenous ligands for the nuclear bile acid receptor FXR [62]. FXR expression is high in the intestine and liver, the two sites where BAs reach high concentrations to activate FXR. The transcription by FXR includes heterodimerization with retinoid X receptors (RXRs) in the cytoplasm, translocation into the nucleus, and binding to DNA response elements in the regulatory regions of target genes [63]. When the bind of BAs to FXR, SHP transcription is increased [60, 64, 65], this alteration leads to the inhibition of LRH-1 activity or HNF4α on the BA response elements (BAREs) of CYP7A1 and CYP8B1 promoters [64, 65]. In this scenario, BA synthesis is downregulated by a precise feedback regulatory mechanism, which represents the major pathway under normal physiological conditions [64–66] (Figure 3). LRH1 is also a well-known activator of Shp gene transcription [64, 65], and this step leads to an autoregulatory loop of gene expression by SHP [42]. This step also includes the G protein pathway suppressor 2 (GPS2) interacting with FXR, LRH-1, and HNF4α to regulate CYP7A1 and CYP8B1 expression in human hepatocytes [67] (Table 3). A critical role in maintaining cholesterol homeostasis for CYP7A1 has been recently advocated in a model of in Cyp7a1-tg mice [68].
Figure 3.
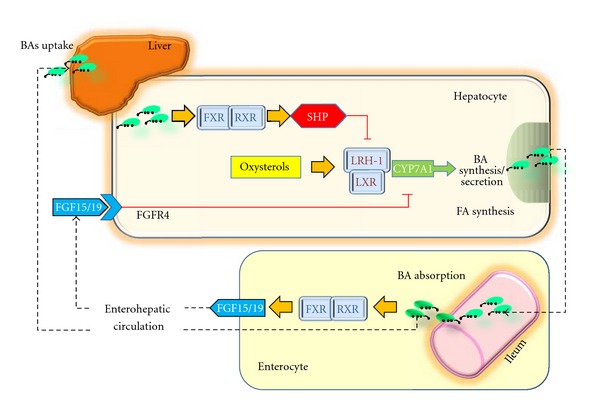
The potential molecular mechanisms of crosstalk between nuclear receptors LXR and FXR–SHP–LRH-1 regulatory cascade in the liver and intestine. Bile acids act as ligands for FXR, which regulates transcription by binding as a heterodimer with RXRs. This step results in increased SHP expression. SHP in turn inhibits LRH-1, preventing the activation of target genes that participate in bile acid and fatty acid synthesis. In the absence of bile acids, LRH-1 acts together with LXR to stimulate bile acid synthesis [64, 65, 132]. The important pathways in the intestine that contribute to modulation of bile acid synthesis are also depicted (see text for details). There is a bile-acid-mediated activation of intestinal FXR and, as a result, the release of FGF15 in the small intestine. The secreted FGF15 by the intestine circulates to the liver, likely through the portal circulation or lymph flow [81] and induces the activation of FGFR4 in the liver. The FGF15/FGFR4 pathway synergizes with SHP in vivo to repress CYP7A1 expression [57]. Bas: bile acids; FGF: fibroblast growth factor; FGFR4: FGF receptor; FXR: farnesoid X receptor; LRH-1: liver receptor homologue-1; LXR: liver X receptor; RXR: retinoid X receptors; SHP: short heterodimer partner. Adapted from Ory [66] and Inagaki et al. [57].
The hepatocyte nuclear factor-1α (HNF1α), which haploinsufficiency causes the Maturity-onset diabetes of the young type 3 (MODY3), also appears to modulate SHP expression via the FXR pathway. In this respect, HNF1α (−/−) mice displayed a defect in bile acid transport, increased bile acid and liver cholesterol synthesis, and impaired HDL metabolism [69].
A role for SHP in mediating the recruitment of mSin3A-Swi/Snf to the CYP7A1 promoter, with chromatin remodeling and gene repression, has been described. In HepG2 cells, Kemper et al. [50] have shown that bile acid treatment resulted in SHP-mediated recruitment of transcriptional coregulators mSin3A and Swi/Snf complex to the promoter, chromatin remodeling, and gene repression (Table 3). This is an additional mechanism involving transformation of nucleosome conformation for the repression by SHP of genes activated by various NRs. In line with such results, increased synthesis and accumulation of BAs occurs in SHP (−/−) mice, due to the loss of SHP repression and consequent derepression of the rate-limiting CYP7A1 and cholesterol 12α-hydroxylase (CYP8B1) (the rate-determining enzyme of the alternative but minor BA synthesis pathway) in the biosynthetic pathway [70–72].
Mechanisms independent of the FXR/SHP/LRH pathway might also exist, since BAs feeding to SHP (−/−) mice reduced the levels of CYP7A1 mRNA to similar levels of control mice [70, 71]. Such SHP-independent and alternative pathways include the protein kinase C/Jun N-terminal kinase (PKC/JNK) pathway [73], the FXR/FGFR4 (FGF receptor 4) pathway [57, 74], the cytokine/JNK pathway [75], the pregnane X receptor (PXR) mediated pathway [76], and the JNK/c-Jun signaling pathway [77].
Another study demonstrated, in SHP (−/−) mice on a background of 129 strain, the protection against hypercholesterolemia in three different models: an atherogenic diet, hypothyroidism, and SHP (−/−) mice intercrossed with LDLR (−/−) mice (to generate SHP/LDLR double (−/−) mice in a mixed 129-C57BL/6 background). When fed an atherogenic diet, the latter strain was almost completely resistant to diet-mediated increases in triglyceride, very low-density lipoprotein (VLDL) cholesterol, and low-density lipoprotein (LDL) cholesterol but had an increase in high-density lipoprotein (HDL) cholesterol as compared with LDLR (−/−) mice. Such results point to the protection against dyslipidemia following the inhibition of hepatic SHP expression, although no antagonist ligands have yet been identified for SHP [78]. We have recently examined biliary lipid secretion and cholesterol gallstone formation in male SHP (−/−) and (+/+) mice before and during the feeding of a lithogenic diet for 56 days [79]. Deletion of the Shp gene significantly increased hepatic bile salt synthesis, and doubled the increase of biliary bile salt outputs in SHP (−/−) mice than in (+/+) mice. The intestinal bile acid pool size was significantly greater in SHP (−/−) mice than in (+/+) mice. These increased BAs are efficacious ligands of FXR and can stimulate the expression of intestinal fibroblast growth factor 15 (FGF15) in mice through the FXR signaling pathway, which is consistent with the expanded bile acid pool size in SHP (−/−) mice. At 14 days on the lithogenic diet, fasting gallbladder volume was significantly larger in SHP (+/+) mice than in (−/−) mice [80].
Indeed, FGF15/19 (mouse and human orthologs, resp.) is another FXR gene target in the intestine and appears to contribute to the fine tuning of bile acid synthesis in the liver. Thus, a model for FXR-mediated repression of bile acid synthesis should also take into account the bile acid-mediated activation of intestinal FXR and FGF15 in the small intestine (while the FXR-SHP pathway is activated in the liver). According to the most plausible view, FGF15 acts as a hormone to signal between intestine and liver. The secreted FGF15 by the intestine circulates to the liver, likely through the portal circulation or lymph flow [81], and induces the activation of FGFR4 in the liver. As shown in Figure 3, the FGF15/FGFR4 pathway synergizes with SHP in vivo to repress CYP7A1 expression [57]. In humans, a similar mechanism should involve the FGF19. Of note, activation of FXR transcription in the intestine protected the liver from cholestasis in mice by inducing FGF15 expression and reducing the hepatic pool of BA. This suggests a potential approach to reverse cholestasis in patients [82]. Hepatic fatty acid homeostasis is also regulated by SHP since regulating these genes involves in fatty acid uptake, synthesis, and export [83–87]. In a study exploring global gene expression profiling combined with chromatin immunoprecipitation assays in transgenic mice constitutively expressing SHP in the liver, overexpression of SHP in the liver was associated with the depletion of the hepatic bile acid pool and a concomitant accumulation of triglycerides in the liver [84]. By contrast, fat accumulation induced by a high-cholesterol or high-fat diet is prevented by the deletion of SHP [88, 89]. The pleiotropic role of SHP can also be found in the case of nonalcoholic liver steatosis since OB/SHP double (−/−) mice (a model of severe obesity and insulin resistance) became resistant to liver steatosis and showed improved insulin sensitivity [86].
Another interesting role for SHP emerged after it was found that BAs negatively regulate the human angiotensinogen (ANG) gene. ANG is the precursor of vasoactive octapeptide angiotensin II, and BAs act through the SHP pathway by preventing hepatocyte nuclear factor-4 (HNF4) from binding to the human ANG promoter [90].
3. Fat Mass, Adipocytes, and Obesity
SHP appears to play a central role in obesity. Human obesity is considered a polygenic disorder characterized by partly known abnormal molecular mechanisms resulting in increased fat mass, with an imbalance between the energy acquired from nutrients that dissipated as heat (i.e., thermogenesis). In this respect, weight stability requires a balance between calories consumed and calories expended [91]. In adipose tissue depots, two main types of adipocytes exist, that is, brown adipocytes and white adipocytes. In several animal species, some adipose tissue sites mainly include brown adipocytes (BATs) and the other contains mainly white adipocytes (WATs). BAT dissipates chemical energy to produce heat either as a defense against cold [92] or as energy expenditure to compensate food intake [93, 94]. The unusual function of BAT might be better understood by considering that they share a common origin with myocytes [95, 96], and BAT was indeed considered something in between muscle and adipose tissue [95]. BAT is deemed as the major site for sympathetic (adrenergic) mediated adaptive thermogenesis; this pathway involves the uncoupling protein-1 (UCP1). WAT is mainly implicated in the regulation of lipid storage and catabolism but also in the synthesis and secretion of adipokines [97–100]. While the percentage of young men with BAT is high, the activity of BAT is reduced in men who are overweight or obese [101]. Thermogenesis unequivocally exists in both humans and animals, and BAT is the major site of thermogenesis which can be increased by environmental factors (i.e., adaptive thermogenesis). In both human and animal species, dietary composition, chronic cold exposure, and exercise may increase thermogenesis [102]. As far as adipose tissue biology is concerned, SHP seems to play a distinct regulatory function in WAT, as compared with BAT. A number of experiments have focused on animal models of obesity and subtle molecular changes. SHP-deficient mice are protected against high-fat-diet-induced obesity [89].
Peroxisome proliferator-activated receptor (PPAR) γ coactivator-1 (PGC-1) family members are multifunctional transcriptional coregulators. PGC-1 acts as a molecular switch in several metabolic pathways. In particular, PGC-1α and PGC-1β regulate mitochondrial biogenesis, adaptive thermogenesis, fatty acid and glucose metabolism, fiber-type switching in skeletal muscle, peripheral circadian clock, and development of the heart [103]. In particular, SHP functions as a negative regulator of energy production in BAT [89] because SHP is a negative regulator of PGC-1α expression in BAT. In turn, PCG-1α is a coactivator of uncoupling protein 1 (UCP1) which plays a major role in energy dissipation as heat in multilocular BAT of different animal species and humans [104–106]. Fat-specific (BAT) SHP-overexpressed transgenic mice had increased body weight and adiposity. Energy metabolism, however, was increased, and BAT cold exposure function was enhanced with activation of thermogenic genes and mitochondrial biogenesis (enhanced β1-AR gene expression and PGC1α). Compared with wild-type mice on a high-fat diet, SHP overexpression was associated with enhanced diet-induced obesity phenotype with weight gain, increased adiposity, and severe glucose intolerance. An additional feature of SHP transgenic mice was a decreased diet-induced adaptive thermogenesis, increased intake of food, and decreased physical activity [107]. This leads to the conclusion that, although expressed at low levels in fat, activation of SHP in adipocytes has a strong effect on weight gain and diet-induced obesity [107]. Moreover, if mechanisms linked to energy metabolism and the development of obesity are considered, SHP has distinct roles in WAT and BAT. As previously mentioned, while SHP deletion in obese leptin-deficient mice (ob/ob) prevented the development of nonalcoholic fatty liver and improved peripheral insulin sensitivity [86], SHP deletion did not overcome the severe obesity caused by leptin deficiency. A significant protective effect from obesity by SHP deficiency was likely associated with the low basal level of SHP expressed in fat. Adipogenesis appears to be influenced by SHP: when SHP was overexpressed in 3T3-L1 preadipocytes, cell differentiation was inhibited, as well as the accumulation of neutral lipids within the cells. Thus, SHP may act as a molecular switch governing adipogenesis. In particular, SHP appears to be a potent adipogenic suppressor, and preadipocytes are kept in an undifferentiated state through the inhibition of the adipogenic transcription factors and stimulators [108]. Further studies will address whether the loss of SHP function results in inhibition of lipid accumulation in adipocytes, similar to what is observed in hepatocytes. In a future clinical setting, treatment of obesity might also include drugs able to mimic or stimulate the effects of SHP. Mutations in the Shp gene have also been reported in patients with lipodystrophy carrying four different polymorphisms [109].
SHP mutations may not be considered a common cause of severe obesity. A number of important clinical studies have examined this issue (Table 4); however, Hung et al. [110] in UK examined the relationships between genetic variation in SHP and weight at birth, adiposity, and insulin levels in three different populations (the Genetics of Obesity Study) GOOS, the Avon Longitudinal Study of Parents and Children (ALSPAC), and the Ely studies). In the 329 cases of severe early-onset obesity (GOOS study), two novel and rare missense mutations (R34G and R36G) were identified which might in part contribute to obesity in the probands. Furthermore, two common polymorphisms, namely, G171A (12% of subjects with higher birth weight) and −195CTGAdel (16% of subjects with lower birth weight) were found. In the ALSPAC cohort of 1,079 children, the G171A variant was associated with increased body mass index and waist circumference together with higher insulin secretion 30 minutes after glucose load. Thus, whereas mutations in the Shp gene cannot be seen as a common cause of severe human obesity, genetic variation in the Shp gene locus may influence birth weight and have effects on body size. The effect might ultimately involve insulin secretion by the negative regulation between SHP and the hepatocyte nuclear factor-4α (HFN-4α), a transcription factor involved in differentiation and function of pancreatic β-cells [110].
Table 4.
Studies on the association between SHP (NR0B2) genetic variation and birth weight, high BMI obesity, and fasting insulin diabetes.
| Author | Country | Study populations/mutation | Subjects number | Mutation(s) | Association with birth weight increase | Association with BMI/ obesity | Association with increased insulin levels | Association with diabetes | Conclusions |
|---|---|---|---|---|---|---|---|---|---|
| Nishigori et al. [111] | Japan | Young-onset type 2 diabetes | 274 | In 7 subjects, 5 different mutations (H53fsdel10, L98fsdel9insAC, R34X, A195S, R213C) and 1 apparent polymorphism (R216H) (all in a heterozygous state) | Yes | Yes | — | No | Shp genetic variation: most common monogenic determinant of obesity and increased birth weight in Japanese |
|
| |||||||||
| Hung et al. [110] | UK | GOOS (severe early-onset obesity) | 329 | R34G and R36C Missense mutations | Yes | No (selection of extreme obesity: stronger effect from other major gene?) | Yes | — | Genetic variation in the SHP locus may influence birth weight and have effects on BMI, possibly through effects on insulin secretion |
| G171A (12%) | Yes | — | — | ||||||
| -195CTGAdel (16%) common polymorphisms | No (lower birth weight) | No (lower fasting levels) | |||||||
| UK | ALSPAC (cohort of children) | 1,079 | G171A | No | Yes (higher BMI and waist circumference at 7 yrs) | Yes (higher fasting levels and 30-min response) | — | Subtle effects in heterozygosity, stronger effects in homozygosity | |
| -195CTGAdel | No (lower BMI) | ||||||||
| UK | Ely Study (Caucasian adults) | 600 | G171A | Data not available | Yes (BMI increased) | No | — | ||
| -195CTGAdel | Yes (female: higher BMI), No (male: lower BMI) |
||||||||
|
| |||||||||
| Mitchell et al. [113] | UK | Young-onset type 2 diabetes, obesity, birth weight | 1,927 | Birth weight: the only child homozygous for the A allele had a birth weight ≥4 kg | No | No | — | No | Mutations in SHP < UK than in Japanese obese type 2 subjects |
| G171A coding polymorphism in 14.1% of UK subjects | |||||||||
| The A allele (G/A genotype) not associated with obesity or increased birth weight | |||||||||
| Obesity: no association if G/A genotype; yes (?) (if A/A homozygotes) | Yes (?) | Homozygous for the rare A allele: predisposed to moderate obesity and possibly increased birth weight | |||||||
|
| |||||||||
| Echwald et al. [114] | Denmark | Early-onset obesity (men) | 750 | 2 silent variants c.65C4T [p. Y22Y], c.339G4A [p. P113P] | — | — | — | Very low prevalence of functional SHP variants associated with obesity among Danes | |
| 3 missense variants c.100C4G [p. R34G], c.278G4A [p. G93D], c.415C4A [p. P139H] | Yes (only among obese) | A role for G171A polymorphism low penetrance SHP variants) for obesity risk in Europe? | |||||||
| G171A polymorphism (8.9%) | No (P = 0.07 versus obese) | Major differences in prevalence and impact of SHP variants between Danish and Japanese obese | |||||||
| Nonobese controls | 795 | No variants G171A polymorphism (7.1) | |||||||
| Functional analyses in MIN6-m9 and HepG2 cell lines | 93D mutant protein: reduced in vitro inhibition of the HNF4α transactivation of the HNF-1α promoter expression | ||||||||
Note: SHP is expressed in the liver, pancreas, spleen, small intestine, and adrenal gland in humans [18] and inhibits the transcriptional activity of hepatocyte nuclear factor-4 α (HNF4α). ALSPAC: Avon Longitudinal Study of Parents and Children; GOOS: Genetics of Obesity Study; HNF4α: hepatocyte nuclear factor-4α.
A possibility is that decreased SHP expression or function results in increased HFN-4α activity with a cascade of events, including fetal hyperinsulinemia, and increased birth weight. At a later stage, sustained hyperinsulinemia might be responsible of insulin resistance and obesity of the adult [110].
Mutations in the Shp gene were also associated with influence on birth weight, mild obesity, and insulin levels in the study by Nishigori et al. on 274 Japanese subjects [111]. Mutations in several genes encoding transcription factors of the hepatocyte nuclear factor (HNF) cascade are associated with maturity-onset diabetes of the young (MODY). MODY is a monogenic form of early-onset diabetes mellitus (defective insulin secretion with normal body weight), and SHP is deemed as a plausible candidate MODY gene; this is because SHP is able to inhibit the transcriptional activity of the hepatocyte nuclear factor-4α (HFN-4α), a key member of the MODY regulatory network. Thus, further studies have looked for segregation of SHP mutations with MODY in a cohort of Japanese patients with early-onset diabetes. In this context, variants in SHP appeared to cosegregate with increased body mass index in families, thus contributing to obesity among Japanese subjects. Also, increased risk of morbidity was observed in another study from Japan, examining patients with type 2 diabetes and SHP mutations [112].
Major differences, however, might exist in the prevalence and function of SHP variants in different populations. Of note, the results from other Caucasian cohorts did not confirm the association between SHP mutation and obesity [113, 114]. Echwald et al. conducted an elegant study on the prevalence of SHP variants by single-strand conformational polymorphism and heteroduplex analysis among 750 Danish obese men with early-onset obesity [114]. As control, a cohort of 795 nonobese control subjects was genotyped using PCR-RFLP. Functional analyses of the identified coding region variants were performed in both MIN6-m9 and HepG2 cell lines. Five novel variants were identified (including 3 missense variants (c.100C>G [p.R34G], c.278G>A [p.G93D], and c.415C>A [p.P139H]) and 2 silent variants (c.65C>T [p.Y22Y] and c.339G>A [p.P113P])). The previously reported [111] c.512G>C [p.G171A] common polymorphism was identified; however, the prevalence of functional SHP variants associated with obesity was considerably lower among Danish subjects (1 out of 750 obese, none of control subjects), compared to the prevalence observed in Japan by Nishigori et al. [111]. Mitchell at al. [113] investigated SHP variants in 1927 UK subjects according to type 2 diabetes, obesity, and birth weight. Although reporting a raised body mass index among homozygous carriers of the 171A variant (<1%), this polymorphism was unlikely to be associated with all three conditions in Caucasians. Taken together, the above-mentioned studies suggest that the 171A variant might contribute only to subsets of polygenic obesity.
4. Other Functions of SHP
The existence of multiple interactions of SHP with NRs, transcription factors and transcriptional cofactors (Tables 2 and 3) points to the pleiotropic and central role of SHP in the body.
SHP has been hypothesized to act in glucose homeostasis via complex pathways involving the inhibition of glucocorticoid receptors (GR) in mammalian cells and the inhibition of PGC-1 gene, a coactivator of NRs important for gluconeogenic gene expression and the PGC-1-regulated phospho(enol)pyruvate carboxykinase (PEPCK) promoter. Such steps underscore a physiologically relevant role for SHP in modulating hepatic glucocorticoid action [22]. Following the bile acid-induced induction, SHP inhibited a number of other pathways, including the HNF4α-mediated transactivation of the PEPCK and fructose biphosphate (FBP) promoters, as well as the transactivation of the glucose-6-phosphatase (G6Pase) promoter mediated by Foxo1 [115]. The interaction between SHP inhibitory function and the 3 isoforms (α, β, and γ) of the hepatocyte nuclear factor-3 (HNF4) points to the regulatory role of SHP on gluconeogenesis [51]. A role for SHP in insulin secretion pathway has also been reported. Mutations in hepatocyte nuclear factor 1α (HNF-1α) is associated with maturity-onset diabetes of the young type 3. This condition depends on impaired insulin secretory response in pancreatic beta cells.
Indeed, loss of HNF-1α function in HNF-1α (−/−) mice resulted in altered expression of genes involved in glucose-stimulated insulin secretion, but also insulin synthesis, and beta-cell differentiation. Pancreatic islets of HNF-1α (−/−) mice showed a distinctive reduction of SHP expression and a downregulation of the HNF4α gene expression. Since SHP appears to repress its own transcriptional activation following heterodimerization with HNF4α, a feedback autoregulatory loop between SHP and HNF4α has been hypothesized [116]. Also, SHP likely functions as a negative regulator of pancreatic islet insulin secretion. SHP (−/−) mice were characterized by hypoinsulinemia, increased glucose-dependent response of islets, increased peripheral insulin sensitivity, and increased glycogen stores [117]. The role played by SHP in the regulation of hepatic gluconeogenesis has also emerged in a number of additional experiments. For example, the liver of SHP (−/−) mice showed increased glycogen stores [117], while hepatic Shp gene expression (induced by the antidiabetic biguanide drug metformin) was associated with inhibition of hepatic gluconeogenesis. Induction of SHP was achieved via AMP-activated protein kinase (AMPK) and associated with downregulation of essential gluconeogenic enzyme genes, that is, phosphoenolpyruvate carboxykinase (PEPCK), glucose-6-phosphatase (G6Pase) [118], and fructose-1,6-bisphosphatase (FBP1) [119].
PGC-1 gene is a coactivator of NRs, and this step is relevant for gluconeogenic gene expression. Yamagata et al. [119] showed that bile acid (chenodeoxycholic acid) was able to induce the downregulation of PGC-1 gene, and this mechanism involved forkhead transcription factors (Foxo1, Foxo3a, Foxo4) via a SHP-dependent manner.
Drug metabolism and detoxification might be regulated by SHP. This is also the case for excess BAs: the pregnane X receptor (PXR) induces CYP3A and inhibits CYP7α, both involved in biochemical pathways leading to the conversion of cholesterol into primary BAs, whereas CYP3A is also involved in the detoxification of toxic secondary bile acid derivatives. SHP acts as a potent repressor of PXR transactivation, and this finding suggests that PXR can act on both bile acid synthesis and elimination detoxification [1]. Additional mechanisms involved in the SHP-dependent control of pathways of drug metabolism have been identified. The expression of genes involved with the metabolism of xenobiotics might be regulated by SHP in the spleen acting on (aryl hydrocarbon receptor (AHR)/AHR nuclear translocator (ARNT)) AHR/ARNT heterodimers which, in turn, bind to xenobiotic response elements (XREs) at the level of specific DNA sequences [41]. A number of genes involved in hormone and drug metabolism would be expressed (i.e., UGT16, ALDH3, CYP1A1, CYP1A2, CYP1B1, etc.). SHP also appears to downregulate the constitutive-androstane-receptor- (CAR-) mediated CYP2B1 gene expression, induced by phenobarbital to form the CAR/RXR heterodimer which, in turn, binds to 2 DR-4 sites to form the phenobarbital responsive unit in the CYP2B gene [120] (Table 3). One role of SHP in steroidogenesis has been identified in the testes with influence on testosterone synthesis and germ cell differentiation [121] and in the intestine for glucocorticoid synthesis [122].
A role for SHP in cell proliferation and apoptosis signaling is emerging. Depending on the cell type, SHP seems to have both inhibitory and stimulatory effects on apoptosis. However, the manipulation of SHP through the synthetic ligands adamantyl-substituted retinoid-related (ARR) compounds 6-[3-(1-adamantyl)-4-hydroxyphenyl]-2-naphthalenecarboxylic acid (CD437/AHPN) and 4-[3-(1-adamantyl)-4-hydroxyphenyl]-3-chlorocinnamic acid (3-Cl-AHPC) induces apoptosis of a number of malignant cells (i.e., leukemia and breast carcinoma) both in vitro and in vivo [123, 124]. The complex mechanism implies binding of ARR and 3-Cl-AHPC to SHP with formation of a corepressor complex containing Sin3A and nuclear receptor corepressor (N-CoR) which activate local control of mitochondrial function and apoptosis, with a limiting function on tumorigenesis [17, 123] (Table 3). SHP appears to be also involved in DNA methylation and acting as a tumor suppressor, at least in the human and mouse livers [125–127]. Whether manipulation of SHP will be helpful in the treatment of hepatic and other gastrointestinal cancers is still a matter of research. The recent finding that SHP negatively regulates TLR signaling to NF-κB has raised the interest for the role of SHP in mechanisms governing innate immunity. SHP appears to negatively regulate the expression of genes encoding inflammatory molecules. Of note, direct binding of NF-κB seems to occur in resting cells, while binding of SHP to TRAF6 occurs in LPS-stimulated cells [128, 129].
5. Conclusions and Perspectives
A remarkable number of metabolic functions in the body appear to be regulated by the orphan unique NR, small heterodimer partner SHP, which targets a complex set of genes in multiple pathways as a transcriptional corepressor (Figure 4). Pathways include fatty acid metabolism, glucose homeostasis, and drug-hormone detoxification. When looking at complex mechanisms leading to some important lipidopathies, that is, obesity and liver steatosis, enlightening data about the regulatory function of SHP are provided by studies using Shp-deleted and Shp-overexpressed animal models. Most likely, a condition of Shp deficiency might counteract lipid accumulation and improve plasma lipoprotein profiles. Further studies are urgently needed to confirm that such an important metabolic regulatory mechanism of SHP is true and has high translational value. To date, however, no synthetic antagonists or agonists for SHP are available, and one should keep in mind that rather divergent and somewhat elusive data have been observed regarding the loss of SHP function in humans and rodents. Thus, careful examination of subtle SHP intrinsic functions is essential to dissect potential modulatory pathways of SHP for a variety of metabolic abnormalities but also in tumorigenesis. Moreover, identifying specific endogenous ligands and synthetic agonists of SHP will pave the way to for therapeutic intervention. The effect of synthetic ligands on SHP modulation in hepatocytes and adipocytes, for example, might represent therapeutic tools for the treatment of constituents of the metabolic syndrome, namely, hypercholesterolemia, overweight obesity, and liver steatosis.
Figure 4.
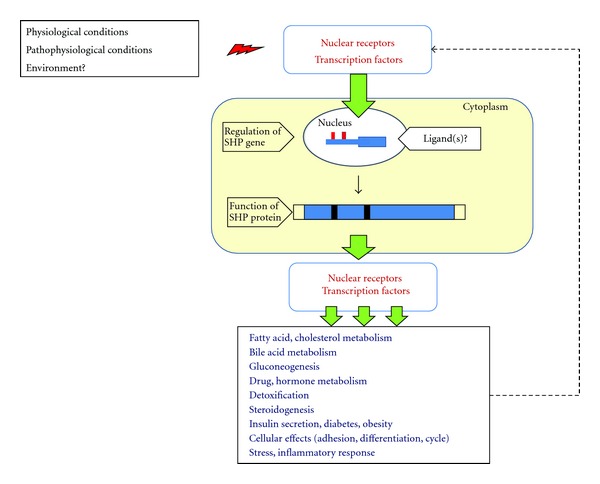
Schematic diagram of the function and gene regulation of SHP. Different conditions will lead to activation of nuclear receptors and/or transcription factors able to regulate Shp gene expression in the nucleus and protein synthesis in the cytoplasm. The protein acts as a transcriptional corepressor of a number of other nuclear receptors and transcription factors involved in a wide series of regulatory pathways. The potential role of a feedback mechanism and of ligand(s) is hypothesized.
Conflict of Interests
The authors declare that there is no conflict of interests.
Acknowledgments
This work was supported in part by research grants DK54012 and DK73917 (D. Q.-H. Wang) from the National Institutes of Health (US Public Health Service), FIRB 2003 RBAU01RANB002 (P. Portincasa) from the Italian Ministry of University and Research, and ORBA10ROPA from the University of Bari. P. Portincasa was a recipient of the short-term mobility grant 2005 from the Italian National Research Council (CNR). L. Bonfrate was a recipient of the travel grant for young investigators from the European Society for Clinical Investigation, 2011. Due to space limitations, the authors apologize to those whose publications related to the discussed issues could not be cited.
Abbreviations
- AF1:
Activation function 1
- AF2:
Activation function 2
- AHR/ARNT:
Aryl hydrocarbon receptor (AHR)/AHR nuclear translocator (ARNT)
- ALDH3:
Human aldehyde dehydrogenase 3 gene
- AMPK:
AMP-activated protein kinase
- ARR:
Adamantyl-substituted retinoid-related molecules
- β1-AR:
β1-adrenergic receptor
- BAREs:
Bile acids response elements
- BAT:
Brown adipose tissue
- Brm:
Human Brahma (a catalytic component of the SWI/SNF-related chromatin-remodeling complex)
- CAR:
Constitutive androstane receptor
- CD437/AHPN:
6-[3-(1-Adamantyl)-4-hydroxyphenyl]-2-naphthalenecarboxylic acid
- 3-Cl-AHPC:
4-[3-(1-Adamantyl)-4-hydroxyphenyl]-3-chlorocinnamic acid
- CYP3A:
Hepatic cytochrome P-4503A
- CYP1A1:
Cytochrome P450, family 1, subfamily A, polypeptide 1
- CYP1A2:
Cytochrome P450, family 1, subfamily A, polypeptide 2
- CYP1B1:
Cytochrome P450, family 1, subfamily B, polypeptide 1
- CYP2B1:
Cytochrome P450, subfamily IIB
- CYP7A1:
Cholesterol 7 alpha-hydroxylase
- CYP8B1:
Cholesterol 12alpha-hydroxylase
- DBD:
DNA-binding domain
- DAX-1:
Dosage-sensitive sex reversal adrenal hypoplasia congenita (AHC) critical region on the X chromosome, gene 1
- DR-4:
Nuclear receptor half-site repeat
- ER:
Estrogen receptor
- FBP1:
Fructose-1,6-bisphosphatase
- FGF:
Fibroblast growth factor
- FGFR:
Fibroblast growth factor receptor
- FISH:
Fluorescence in situ hybridization
- Foxo:
Forkhead transcription factors
- FXR:
Farnesoid X receptor
- G6Pase:
Glucose-6-phosphatase
- GPCR:
G-protein-coupled Receptor
- GPS2:
G protein pathway suppressor 2
- GR:
Glucocorticoid receptors
- HDL:
High-density lipoprotein
- HNF:
Hepatocyte nuclear factor
- JNK:
Jun N-terminal kinase
- JUN-D:
Jun family of the activator protein 1 (AP-1) transcription factor complex
- LBD:
Ligand-binding domain
- LDL:
Low-density lipoprotein
- LDLR:
Low-density lipoprotein receptor
- LRH1:
Liver receptor homologue 1
- 3T3-L1:
Mouse embryonic fibroblast adipose-like cell line.
- LXXLL:
Leu-Xaa-Xaa-Leu-Leu
- mSin3A:
Mammalian homolog of the Saccharomyces cerevisiae transcriptional corepressor Sin3p
- N-CoR:
Nuclear receptor corepressor
- NRs:
Nuclear receptors
- PEPCK:
Phosphoenolpyruvate carboxykinase
- PGC-1:
Peroxisome proliferator-activated receptor-γ (PPAR-γ) coactivator-1
- PKC:
Protein kinase C
- PXR:
Pregnane X receptor
- RAR:
Retinoid acid receptor
- RNA Pol II:
RNA polymerase II
- RNF31
Member of the ring-between-ring (RBR) family of E3 ubiquitin ligases
- RXR:
Retinoid X receptor
- SHP:
Small heterodimer partner
- SMILE:
SHP-interacting leucine zipper protein
- SMRT:
Silencing mediator of retinoid and thyroid hormone receptor
- SRC-1:
Steroid receptor coactivator-1
- SWI/SNF:
SWItch/Sucrose NonFermentable (yeast nucleosome remodeling complex composed of several proteins which are products of the SWI and SNF genes)
- UCP1:
Uncoupling protein-1
- USF-1:
Upstream stimulatory factor-1
- TFIID:
Transcription initiation factor II D
- TFIIE:
Transcription factor II E
- UGT-16:
Uridine 5′-diphospho (UDP) glucuronosyl transferase family member
- VLDL:
Very-low-density lipoprotein
- XREs:
Xenobiotic response elements
- WAT:
White adipose tissue.
References
- 1.Ourlin JC, Lasserre F, Pineau T, et al. The small heterodimer partner interacts with the pregnane X receptor and represses its transcriptional activity. Molecular Endocrinology. 2003;17(9):1693–1703. doi: 10.1210/me.2002-0383. [DOI] [PubMed] [Google Scholar]
- 2.Gronemeyer H, Gustafsson JÅ, Laudet V. Principles for modulation of the nuclear receptor superfamily. Nature Reviews Drug Discovery. 2004;3(11):950–964. doi: 10.1038/nrd1551. [DOI] [PubMed] [Google Scholar]
- 3.Mangelsdorf DJ, Thummel C, Beato M, et al. The nuclear receptor super-family: the second decade. Cell. 1995;83(6):835–839. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mangelsdorf DJ, Evans RM. The RXR heterodimers and orphan receptors. Cell. 1995;83(6):841–850. doi: 10.1016/0092-8674(95)90200-7. [DOI] [PubMed] [Google Scholar]
- 5.Pardee K, Necakov AS, Krause H. Nuclear receptors: small molecule sensors that coordinate growth, metabolism and reproduction. Subcellular Biochemistry. 2011;52:123–153. doi: 10.1007/978-90-481-9069-0_6. [DOI] [PubMed] [Google Scholar]
- 6.Gronemeyer H, Moras D. How to finger DNA. Nature. 1995;375(6528):190–191. doi: 10.1038/375190a0. [DOI] [PubMed] [Google Scholar]
- 7.Lalli E, Sassone-Corsi P. DAX-1, an unusual orphan receptor at the crossroads of steroidogenic function and sexual differentiation. Molecular Endocrinology. 2003;17(8):1445–1453. doi: 10.1210/me.2003-0159. [DOI] [PubMed] [Google Scholar]
- 8.Lee YS, Chanda D, Sim J, Park YY, Choi HS. Structure and function of the atypical orphan nuclear receptor small heterodimer partner. International Review of Cytology. 2007;261:117–158. doi: 10.1016/S0074-7696(07)61003-1. [DOI] [PubMed] [Google Scholar]
- 9.Muscatelli F, Strom TM, Walker AP, et al. Mutations in the DAX-1 gene give rise to both X-linked adrenal hypoplasia congentia and hypogonadotropic hypogonadism. Nature. 1994;372(6507):672–676. doi: 10.1038/372672a0. [DOI] [PubMed] [Google Scholar]
- 10.Seol W, Choi HS, Moore DD. An orphan nuclear hormone receptor that lacks a DNA binding domain and heterodimerizes with other receptors. Science. 1996;272(5266):1336–1339. doi: 10.1126/science.272.5266.1336. [DOI] [PubMed] [Google Scholar]
- 11.Zanaria E, Muscatelli F, Bardoni B, et al. An unusual member of the nuclear hormone receptor superfamily responsible for X-linked adrenal hypoplasia congenita. Nature. 1994;372(6507):635–641. doi: 10.1038/372635a0. [DOI] [PubMed] [Google Scholar]
- 12.Båvner A, Sanyal S, Gustafsson J-Å, Treuter E. Transcriptional corepression by SHP: molecular mechanisms and physiological consequences. Trends in Endocrinology and Metabolism. 2005;16(10):478–488. doi: 10.1016/j.tem.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 13.Miao J, Choi S-E, Seok SM, et al. Ligand-dependent regulation of the activity of the orphan nuclear receptor, small heterodimer partner (SHP), in the repression of bile acid biosynthetic CYP7A1 and CYP8B1 genes. Molecular Endocrinology. 2011;25(7):1159–1169. doi: 10.1210/me.2011-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Farhana L, Dawson MI, Leid M, et al. Adamantyl-substituted retinoid-related molecules bind small heterodimer partner and modulate the Sin3A repressor. Cancer Research. 2007;67(1):318–325. doi: 10.1158/0008-5472.CAN-06-2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chanda D, Park JH, Choi HS. Molecular basis of endocrine regulation by orphan nuclear receptor small heterodimer partner. Endocrine Journal. 2008;55(2):253–268. doi: 10.1507/endocrj.k07e-103. [DOI] [PubMed] [Google Scholar]
- 16.Giguere V. Orphan nuclear receptors: from gene to function. Endocrine Reviews. 1999;20(5):689–725. doi: 10.1210/edrv.20.5.0378. [DOI] [PubMed] [Google Scholar]
- 17.Zhang Y, Hagedorn CH, Wang L. Role of nuclear receptor SHP in metabolism and cancer. Biochimica et Biophysica Acta. 2011;1812(8):893–908. doi: 10.1016/j.bbadis.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee HK, Lee YK, Park SH, et al. Structure and expression of the orphan nuclear receptor SHP gene. Journal of Biological Chemistry. 1998;273(23):14398–14402. doi: 10.1074/jbc.273.23.14398. [DOI] [PubMed] [Google Scholar]
- 19.Bookout AL, Jeong Y, Downes M, Yu RT, Evans RM, Mangelsdorf DJ. Anatomical profiling of nuclear receptor expression reveals a hierarchical transcriptional network. Cell. 2006;126(4):789–799. doi: 10.1016/j.cell.2006.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang X, Downes M, Yu RT, et al. Nuclear receptor expression links the circadian clock to metabolism. Cell. 2006;126(4):801–810. doi: 10.1016/j.cell.2006.06.050. [DOI] [PubMed] [Google Scholar]
- 21.Johansson L, Båvner A, Thomsen JS, Färnegårdh M, Gustafsson JÅ, Treuter E. The orphan nuclear receptor SHP utilizes conserved LXXLL-related motifs for interactions with ligand-activated estrogen receptors. Molecular and Cellular Biology. 2000;20(4):1124–1133. doi: 10.1128/mcb.20.4.1124-1133.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Borgius LJ, Steffensen KR, Gustafsson JÅ, Treuter E. Glucocorticoid signaling is perturbed by the atypical orphan receptor and corepressor SHP. Journal of Biological Chemistry. 2002;277(51):49761–49766. doi: 10.1074/jbc.M205641200. [DOI] [PubMed] [Google Scholar]
- 23.Gobinet J, Auzou G, Nicolas JC, Sultan C, Jalaguier S. Characterization of the interaction between androgen receptor and a new transcriptional inhibitor, SHP. Biochemistry. 2001;40(50):15369–15377. doi: 10.1021/bi011384o. [DOI] [PubMed] [Google Scholar]
- 24.Seol W, Chung M, Moore DD. Novel receptor interaction and repression domains in the orphan receptor SHP. Molecular and Cellular Biology. 1997;17(12):7126–7131. doi: 10.1128/mcb.17.12.7126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xie YB, Nedumaran B, Choi HS. Molecular characterization of SMILE as a novel corepressor of nuclear receptors. Nucleic Acids Research. 2009;37(12):4100–4115. doi: 10.1093/nar/gkp333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xie YB, Park JH, Kim DK, et al. Transcriptional corepressor SMILE recruits SIRT1 to inhibit nuclear receptor estrogen receptor-related receptor γ transactivation. Journal of Biological Chemistry. 2009;284(42):28762–28774. doi: 10.1074/jbc.M109.034165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim HJ, Kim JY, Kim JY, et al. Differential regulation of human and mouse orphan nuclear receptor small heterodimer partner promoter by sterol regulatory element binding protein-1. Journal of Biological Chemistry. 2004;279(27):28122–28131. doi: 10.1074/jbc.M313302200. [DOI] [PubMed] [Google Scholar]
- 28.Chanda D, Li T, Song K-H, et al. Hepatocyte growth factor family negatively regulates hepatic gluconeogenesis via induction of orphan nuclear receptorsmall heterodimer partner in primary hepatocytes. Journal of Biological Chemistry. 2009;284(42):28510–28521. doi: 10.1074/jbc.M109.022244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cui J, Huang L, Zhao A, et al. Guggulsterone is a farnesoid X receptor antagonist in coactivator association assays but acts to enhance transcription of bile salt export pump. Journal of Biological Chemistry. 2003;278(12):10214–10220. doi: 10.1074/jbc.M209323200. [DOI] [PubMed] [Google Scholar]
- 30.Rizzo G, Renga B, Antonelli E, Passeri D, Pellicciari R, Fiorucci S. The methyl transferase PRMT1 functions as co-activator of farnesoid X receptor (FXR)/9-cis retinoid X receptor and regulates transcription of FXR responsive genes. Molecular Pharmacology. 2005;68(2):551–558. doi: 10.1124/mol.105.012104. [DOI] [PubMed] [Google Scholar]
- 31.Isoda K, Sawada S, Ayaori M, et al. Deficiency of interleukin-1 receptor antagonist deteriorates fatty liver and cholesterol metabolism in hypercholesterolemic mice. Journal of Biological Chemistry. 2005;280(8):7002–7009. doi: 10.1074/jbc.M412220200. [DOI] [PubMed] [Google Scholar]
- 32.Yeo MG, Yoo YG, Choi HS, Pak YK, Lee MO. Negative cross-talk between Nur77 and small heterodimer partner and its role in apoptotic cell death of hepatoma cells. Molecular Endocrinology. 2005;19(4):950–963. doi: 10.1210/me.2004-0209. [DOI] [PubMed] [Google Scholar]
- 33.Kanaya E, Shiraki T, Jingami H. The nuclear bile acid receptor FXR is activated by PGC-1α in a ligand-dependent manner. Biochemical Journal. 2004;382(3):913–921. doi: 10.1042/BJ20040432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Del Bas JM, Fernández-Larrea J, Blay M, et al. Grape seed procyanidins improve atherosclerotic risk index and induce liver CYP7A1 and SHP expression in healthy rats. FASEB Journal. 2005;19(3):479–481. doi: 10.1096/fj.04-3095fje. [DOI] [PubMed] [Google Scholar]
- 35.Ito S, Fujimori T, Furuya A, Satoh J, Nabeshima Y, Nabeshima Y-I. Impaired negative feedback suppression of bile acid synthesis in mice lacking βKlotho. Journal of Clinical Investigation. 2005;115(8):2202–2208. doi: 10.1172/JCI23076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Evans MJ, Lai K, Shaw LJ, Harnish DC, Chadwick CC. Estrogen receptor α inhibits IL-1β induction of gene expression in the mouse liver. Endocrinology. 2002;143(7):2559–2570. doi: 10.1210/endo.143.7.8919. [DOI] [PubMed] [Google Scholar]
- 37.Park YY, Kim HJ, Kim JY, et al. Differential role of the loop region between helices H6 and H7 within the orphan nuclear receptors small heterodimer partner and DAX-1. Molecular Endocrinology. 2004;18(5):1082–1095. doi: 10.1210/me.2003-0339. [DOI] [PubMed] [Google Scholar]
- 38.Oiwa A, Kakizawa T, Miyamoto T, et al. Synergistic regulation of the mouse orphan nuclear receptor SHP gene promoter by CLOCK-BMAL1 and LRH-1. Biochemical and Biophysical Research Communications. 2007;353(4):895–901. doi: 10.1016/j.bbrc.2006.12.131. [DOI] [PubMed] [Google Scholar]
- 39.Xie YB, Lee OH, Nedumaran B, et al. SMILE, a new orphan nuclear receptor SHP-interacting protein, regulates SHP-repressed estrogen receptor transactivation. Biochemical Journal. 2008;416(3):463–473. doi: 10.1042/BJ20080782. [DOI] [PubMed] [Google Scholar]
- 40.Suh JH, Huang J, Park YY, et al. Orphan nuclear receptor small heterodimer partner inhibits transforming growth factor-β signaling by repressing Smad3 transactivation. Journal of Biological Chemistry. 2006;281(51):39169–39178. doi: 10.1074/jbc.M605947200. [DOI] [PubMed] [Google Scholar]
- 41.Klinge CM, Jernigan SC, Risinger KE, et al. Short heterodimer partner (SHP) orphan nuclear receptor inhibits the transcriptional activity of aryl hydrocarbon receptor (AHR)/AHR nuclear translocator (ARNT) Archives of Biochemistry and Biophysics. 2001;390(1):64–70. doi: 10.1006/abbi.2001.2366. [DOI] [PubMed] [Google Scholar]
- 42.Chanda D, Xie YB, Choi HS. Transcriptional corepressor shp recruits sirt1 histone deacetylase to inhibit LRH-1 transactivation. Nucleic Acids Research. 2010;38(14):4607–4619. doi: 10.1093/nar/gkq227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ehrlund A, Anthonisen EH, Gustafsson N, et al. E3 ubiquitin ligase RNF31 cooperates with DAX-1 in transcriptional repression of steroidogenesis. Molecular and Cellular Biology. 2009;29(8):2230–2242. doi: 10.1128/MCB.00743-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim YS, Han CY, Kim SW, et al. The orphan nuclear receptor small heterodimer partner as a novel coregulator of nuclear factor-κB in oxidized low density lipoprotein-treated macrophage cell line RAW 264.7. Journal of Biological Chemistry. 2001;276(36):33736–33740. doi: 10.1074/jbc.M101977200. [DOI] [PubMed] [Google Scholar]
- 45.Nishizawa H, Yamagata K, Shimomura I, et al. Small heterodimer partner, an orphan nuclear receptor, augments peroxisome proliferator-activated receptor γ transactivation. Journal of Biological Chemistry. 2002;277(2):1586–1592. doi: 10.1074/jbc.M104301200. [DOI] [PubMed] [Google Scholar]
- 46.Kassam A, Capone JP, Rachubinski RA. The short heterodimer partner receptor differentially modulates peroxisome proliferator-activated receptor α-mediated transcription from the peroxisome proliferator-response elements of the genes encoding the peroxisomal β-oxidation enzymes acyl-CoA oxidase and hydratase-dehydrogenase. Molecular and Cellular Endocrinology. 2001;176(1-2):49–56. doi: 10.1016/s0303-7207(01)00475-0. [DOI] [PubMed] [Google Scholar]
- 47.Lee YK, Dell H, Dowhan DH, Hadzopoulou-Cladaras M, Moore DD. The orphan nuclear receptor SHP inhibits hepatocyte nuclear factor 4 and retinoid X receptor transactivation: two mechanisms for repression. Molecular and Cellular Biology. 2000;20(1):187–195. doi: 10.1128/mcb.20.1.187-195.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee YK, Moore DD. Dual mechanisms for repression of the monomeric orphan receptor liver receptor homologous protein-1 by the orphan small heterodimer partner. Journal of Biological Chemistry. 2002;277(4):2463–2467. doi: 10.1074/jbc.M105161200. [DOI] [PubMed] [Google Scholar]
- 49.Johansson L, Thomsen JS, Damdimopoulos AE, Spyrou G, Gustafsson JA, Treuter E. The orphan nuclear receptor SHP inhibits agonist-dependent transcriptional activity of estrogen receptors ERα and ERβ . Journal of Biological Chemistry. 1999;274(1):345–353. doi: 10.1074/jbc.274.1.345. [DOI] [PubMed] [Google Scholar]
- 50.Kemper JK, Kim H, Miao J, Bhalla S, Bae Y. Role of an mSin3A-Swi/Snf chromatin remodeling complex in the feedback repression of bile acid biosynthesis by SHP. Molecular and Cellular Biology. 2004;24(17):7707–7719. doi: 10.1128/MCB.24.17.7707-7719.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim JY, Kim HJ, Kim KT, et al. Orphan nuclear receptor small heterodimer partner represses hepatocyte nuclear factor 3/foxa transactivation via inhibition of its DNA binding. Molecular Endocrinology. 2004;18(12):2880–2894. doi: 10.1210/me.2004-0211. [DOI] [PubMed] [Google Scholar]
- 52.Fiorucci S, Antonelli E, Rizzo G, et al. The nuclear receptor SHP mediates inhibition of hepatic stellate cells by FXR and protects against liver fibrosis. Gastroenterology. 2004;127(5):1497–1512. doi: 10.1053/j.gastro.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 53.Kanamaluru D, Xiao Z, Fang S, et al. Arginine methylation by PRMT5 at a naturally occurring mutation site Is critical for liver metabolic regulation by small heterodimer partner. Molecular and Cellular Biology. 2011;31(7):1540–1550. doi: 10.1128/MCB.01212-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hoeke MO, Plass JRM, Heegsma J, et al. Low retinol levels differentially modulate bile salt-induced expression of human and mouse hepatic bile salt transporters. Hepatology. 2009;49(1):151–159. doi: 10.1002/hep.22661. [DOI] [PubMed] [Google Scholar]
- 55.Sherlock S, Dooley J. Diseases of the Liver and Biliary System. Oxford, UK: Blackwell Science; 2002. [Google Scholar]
- 56.Chiang JY. Regulation of bile acid synthesis. Frontiers in Bioscience. 1998;3:d176–d193. doi: 10.2741/a273. [DOI] [PubMed] [Google Scholar]
- 57.Inagaki T, Choi M, Moschetta A, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metabolism. 2005;2(4):217–225. doi: 10.1016/j.cmet.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 58.Diogo CV, Grattagliano I, Oliveira PJ, Bonfrate L, Portincasa P. Re-wiring the circuit: mitochondria as a pharmacological target in liver disease. Current Medicinal Chemistry. 2011;18(35):5448–5465. doi: 10.2174/092986711798194432. [DOI] [PubMed] [Google Scholar]
- 59.Grattagliano I, Russmann S, Diogo C, et al. Mitochondria in chronic liver disease. Current Drug Targets. 2011;12(6):879–893. doi: 10.2174/138945011795528877. [DOI] [PubMed] [Google Scholar]
- 60.Chiang JYL. Regulation of bile acid synthesis: pathways, nuclear receptors, and mechanisms. Journal of Hepatology. 2004;40(3):539–551. doi: 10.1016/j.jhep.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 61.Jelinek DF, Andersson S, Slaughter CA, Russell DW. Cloning and regulation of cholesterol 7α-hydroxylase, the rate-limiting enzyme in bile acid biosynthesis. Journal of Biological Chemistry. 1990;265(14):8190–8197. [PMC free article] [PubMed] [Google Scholar]
- 62.Parks DJ, Blanchard SG, Bledsoe RK, et al. Bile acids: natural ligands for an orphan nuclear receptor. Science. 1999;284(5418):1365–1368. doi: 10.1126/science.284.5418.1365. [DOI] [PubMed] [Google Scholar]
- 63.Laffitte BA, Kast HR, Nguyen CM, Zavacki AM, Moore DD, Edwards PA. Identification of the DNA binding specificity and potential target genes for the farnesoid X-activated receptor. Journal of Biological Chemistry. 2000;275(14):10638–10647. doi: 10.1074/jbc.275.14.10638. [DOI] [PubMed] [Google Scholar]
- 64.Goodwin B, Jones SA, Price RR, et al. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Molecular Cell. 2000;6(3):517–526. doi: 10.1016/s1097-2765(00)00051-4. [DOI] [PubMed] [Google Scholar]
- 65.Lu TT, Makishima M, Repa JJ, et al. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptos. Molecular Cell. 2000;6(3):507–515. doi: 10.1016/s1097-2765(00)00050-2. [DOI] [PubMed] [Google Scholar]
- 66.Ory DS. Nuclear receptor signaling in the control of cholesterol homeostasis: have the orphans found a home? Circulation Research. 2004;95(7):660–670. doi: 10.1161/01.RES.0000143422.83209.be. [DOI] [PubMed] [Google Scholar]
- 67.Sanyal S, Båvner A, Haroniti A, et al. Involvement of corepressor complex subunit GPS2 in transcriptional pathways governing human bile acid biosynthesis. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(40):15665–15670. doi: 10.1073/pnas.0706736104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li T, Matozel M, Boehme S, et al. Overexpression of cholesterol 7α-hydroxylase promotes hepatic bile acid synthesis and secretion and maintains cholesterol homeostasis. Hepatology. 2011;53(3):996–1006. doi: 10.1002/hep.24107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shih DQ, Bussen M, Sehayek E, et al. Hepatocyte nuclear factor-1α is an essential regulator of bile acid and plasma cholesterol metabolism. Nature Genetics. 2001;27(4):375–382. doi: 10.1038/86871. [DOI] [PubMed] [Google Scholar]
- 70.Wang L, Lee YK, Bundman D, et al. Redundant pathways for negative feedback regulation of bile acid production. Developmental Cell. 2002;2(6):721–731. doi: 10.1016/s1534-5807(02)00187-9. [DOI] [PubMed] [Google Scholar]
- 71.Kerr TA, Saeki S, Schneider M, et al. Loss of nuclear receptor SHP impairs but does not eliminate negative feedback regulation of bile acid synthesis. Developmental Cell. 2002;2(6):713–720. doi: 10.1016/s1534-5807(02)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Khurana S, Raufman J-P, Pallone TL. Bile acids regulate cardiovascular function. Clinical and Translational Science. 2011;4(3):210–218. doi: 10.1111/j.1752-8062.2011.00272.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Stravitz RT, Rao YP, Vlahcevic ZR, et al. Hepatocellular protein kinase C activation by bile acids: implications for regulation of cholesterol 7α-hydroxylase. American Journal of Physiology. 1996;271(2):G293–G303. doi: 10.1152/ajpgi.1996.271.2.G293. [DOI] [PubMed] [Google Scholar]
- 74.Holt JA, Luo G, Billin AN, et al. Definition of a novel growth factor-dependent signal cascade for the suppression of bile acid biosynthesis. Genes and Development. 2003;17(13):1581–1591. doi: 10.1101/gad.1083503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.De Fabiani E, Mitro N, Anzulovich AC, Pinelli A, Galli G, Crestani M. The negative effects of bile acids and tumor necrosis factor-α on the transcription of cholesterol 7α-hydroxylase gene (CYP7A1) converge to hepatic nuclear factor-4: a novel mechanism of feedback regulation of bile acid synthesis mediated by nuclear receptors. Journal of Biological Chemistry. 2001;276(33):30708–30716. doi: 10.1074/jbc.M103270200. [DOI] [PubMed] [Google Scholar]
- 76.Li T, Chiang JYL. Mechanism of rifampicin and pregnane X receptor inhibition of human cholesterol 7α-hydroxylase gene transcription. American Journal of Physiology. 2005;288(51):G74–G84. doi: 10.1152/ajpgi.00258.2004. [DOI] [PubMed] [Google Scholar]
- 77.Li T, Jahan A, Chiang JYL. Bile acids and cytokines inhibit the human cholesterol 7α-hydroxylase gene via the JNK/c-Jun pathway in human liver cells. Hepatology. 2006;43(6):1202–1210. doi: 10.1002/hep.21183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hartman HB, Lai KD, Evans MJ. Loss of small heterodimer partner expression in the liver protects against dyslipidemia. Journal of Lipid Research. 2009;50(2):193–203. doi: 10.1194/jlr.M800323-JLR200. [DOI] [PubMed] [Google Scholar]
- 79.Wang HH, Portincasa P, Mendez-Sanchez N, Uribe M, Wang DQH. Effect of ezetimibe on the prevention and dissolution of cholesterol gallstones. Gastroenterology. 2008;134(7):2101–2110. doi: 10.1053/j.gastro.2008.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang HH, Portincasa P, Liu M, Wang DQH. Rapid postprandial gallbladder refilling and increased turnover of bile prevent cholesterol crystallization in small heterodimer partner (SHP) knockout mice. DDW 2011, Chicago, USA. Gastroenterology. 2011;140(5):S67–S68. [Google Scholar]
- 81.Bjorkhem I, Blomstrand R, Lewenhaupt A, Svensson L. Effect of lymphatic drainage on 7α-hydroxylation of cholesterol in rat liver. Biochemical and Biophysical Research Communications. 1978;85(2):532–540. doi: 10.1016/0006-291x(78)91196-8. [DOI] [PubMed] [Google Scholar]
- 82.Modica S, Petruzzelli M, Bellafante E, et al. Selective activation of nuclear bile acid receptor FXR in the intestine protects mice against cholestasis. Gastroenterology. 2011;142(2):355–365. doi: 10.1053/j.gastro.2011.10.028. [DOI] [PubMed] [Google Scholar]
- 83.Watanabe M, Houten SM, Wang L, et al. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. Journal of Clinical Investigation. 2004;113(10):1408–1418. doi: 10.1172/JCI21025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Boulias K, Katrakili N, Bamberg K, Underhill P, Greenfield A, Talianidis I. Regulation of hepatic metabolic pathways by the orphan nuclear receptor SHP. EMBO Journal. 2005;24(14):2624–2633. doi: 10.1038/sj.emboj.7600728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Matsukuma KE, Wang L, Bennett MK, Osborne TF. A key role for orphan nuclear receptor liver receptor homologue-1 in activation of fatty acid synthase promoter by liver X receptor. Journal of Biological Chemistry. 2007;282(28):20164–20171. doi: 10.1074/jbc.M702895200. [DOI] [PubMed] [Google Scholar]
- 86.Huang J, Iqbal J, Saha PK, et al. Molecular characterization of the role of orphan receptor small heterodimer partner in development of fatty liver. Hepatology. 2007;46(1):147–157. doi: 10.1002/hep.21632. [DOI] [PubMed] [Google Scholar]
- 87.Delerive P, Galardi CM, Bisi JE, Nicodeme E, Goodwin B. Identification of liver receptor homolog-1 as a novel regulator of apolipoprotein AI gene transcription. Molecular Endocrinology. 2004;18(10):2378–2387. doi: 10.1210/me.2004-0132. [DOI] [PubMed] [Google Scholar]
- 88.Wang L, Han Y, Kim CS, Lee YK, Moore DD. Resistance of SHP-null mice to bile acid-induced liver damage. Journal of Biological Chemistry. 2003;278(45):44475–44481. doi: 10.1074/jbc.M305258200. [DOI] [PubMed] [Google Scholar]
- 89.Wang L, Liu J, Saha P, et al. The orphan nuclear receptor SHP regulates PGC-1α expression and energy production in brown adipocytes. Cell Metabolism. 2005;2(4):227–238. doi: 10.1016/j.cmet.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 90.Shimamoto Y, Ishida J, Yamagata K, et al. Inhibitory effect of the small heterodimer partner on hepatocyte nuclear factor-4 mediates bile acid-induced repression of the human angiotensinogen gene. Journal of Biological Chemistry. 2004;279(9):7770–7776. doi: 10.1074/jbc.M310577200. [DOI] [PubMed] [Google Scholar]
- 91.Mozaffarian D, Hao T, Rimm EB, Willett WC, Hu FB. Changes in diet and lifestyle and long-term weight gain in women and men. New England Journal of Medicine. 2011;364(25):2392–2404. doi: 10.1056/NEJMoa1014296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kajimura S, Seale P, Spiegelman BM. Transcriptional control of brown fat development. Cell Metabolism. 2010;11(4):257–262. doi: 10.1016/j.cmet.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rothwell NJ, Stock MJ. Effect of chronic food restriction on energy balance, thermogenic capacity, and brown-adipose-tissue activity in the rat. Bioscience Reports. 1982;2(8):543–549. doi: 10.1007/BF01314214. [DOI] [PubMed] [Google Scholar]
- 94.Rothwell NJ, Stock MJ. Effects of feeding a palatable “cafeteria” diet on energy balance in young and adult lean (+/?) Zucker rats. British Journal of Nutrition. 1982;47(3):461–471. doi: 10.1079/bjn19820058. [DOI] [PubMed] [Google Scholar]
- 95.Cannon B, Nedergaard J. Developmental biology: neither fat nor flesh. Nature. 2008;454(7207):947–948. doi: 10.1038/454947a. [DOI] [PubMed] [Google Scholar]
- 96.Seale P, Bjork B, Yang W, et al. PRDM16 controls a brown fat/skeletal muscle switch. Nature. 2008;454(7207):961–967. doi: 10.1038/nature07182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Klingenspor M. Cold-induced recruitment of brown adipose tissue thermogenesis. Experimental Physiology. 2003;88(1):141–148. doi: 10.1113/eph8802508. [DOI] [PubMed] [Google Scholar]
- 98.Trayhurn P. Adipose tissue in obesity—an inflammatory issue. Endocrinology. 2005;146(3):1003–1005. doi: 10.1210/en.2004-1597. [DOI] [PubMed] [Google Scholar]
- 99.Trayhurn P, Wood IS. Signalling role of adipose tissue: adipokines and inflammation in obesity. Biochemical Society Transactions. 2005;33(5):1078–1081. doi: 10.1042/BST0331078. [DOI] [PubMed] [Google Scholar]
- 100.Wood IS, Wang B, Jenkins JR, Trayhurn P. The pro-inflammatory cytokine IL-18 is expressed in human adipose tissue and strongly upregulated by TNFα in human adipocytes. Biochemical and Biophysical Research Communications. 2005;337(2):422–429. doi: 10.1016/j.bbrc.2005.09.068. [DOI] [PubMed] [Google Scholar]
- 101.Van Marken Lichtenbelt WD, Vanhommerig JW, Smulders NM, et al. Cold-activated brown adipose tissue in healthy men. New England Journal of Medicine. 2009;360(15):1500–1508. doi: 10.1056/NEJMoa0808718. [DOI] [PubMed] [Google Scholar]
- 102.Lowell BB, Spiegelman BM. Towards a molecular understanding of adaptive thermogenesis. Nature. 2000;404(6778):652–660. doi: 10.1038/35007527. [DOI] [PubMed] [Google Scholar]
- 103.Liu C, Lin JD. PGC-1 coactivators in the control of energy metabolism. Acta Biochimica et Biophysica Sinica. 2011;43(4):248–257. doi: 10.1093/abbs/gmr007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Stuart JA, Cadenas S, Jekabsons MB, Roussel D, Brand MD. Mitochondrial proton leak and the uncoupling protein 1 homologues. Biochimica et Biophysica Acta. 2001;1504(1):144–158. doi: 10.1016/s0005-2728(00)00243-7. [DOI] [PubMed] [Google Scholar]
- 105.Garruti G, Ricquier D. Analysis of uncoupling protein and its mRNA in adipose tissue deposits of adult humans. International Journal of Obesity. 1992;16(5):383–390. [PubMed] [Google Scholar]
- 106.Bouillaud F, Villarroya F, Hentz E, Raimbault S, Cassard AM, Ricquier D. Detection of brown adipose tissue uncoupling protein mRNA in adult patients by a human genomic probe. Clinical Science. 1988;75(1):21–27. doi: 10.1042/cs0750021. [DOI] [PubMed] [Google Scholar]
- 107.Tabbi-Anneni I, Cooksey R, Gunda V, et al. Overexpression of nuclear receptor SHP in adipose tissues affects diet-induced obesity and adaptive thermogenesis. American Journal of Physiology. 2010;298(5):E961–E970. doi: 10.1152/ajpendo.00655.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Song G, Park K, Wang L. Gene expression profiling reveals a diverse array of pathways inhibited by nuclear receptor SHP during adipogenesis. International Journal of Clinical and Experimental Pathology. 2009;2(3):275–285. [PMC free article] [PubMed] [Google Scholar]
- 109.Cao H, Hegele RA. Identification of polymorphisms in the human SHP1 gene. Journal of Human Genetics. 2002;47(8):445–447. doi: 10.1007/s100380200062. [DOI] [PubMed] [Google Scholar]
- 110.Hung CCC, Farooqi IS, Ong K, et al. Contribution of variants in the small heterodimer partner gene to birthweight, adiposity, and insulin levels: mutational analysis and association studies in multiple populations. Diabetes. 2003;52(5):1288–1291. doi: 10.2337/diabetes.52.5.1288. [DOI] [PubMed] [Google Scholar]
- 111.Nishigori H, Tomura H, Tonooka N, et al. Mutations in the small heterodimer partner gene are associated with mild obesity in Japanese subjects. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(2):575–580. doi: 10.1073/pnas.021544398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Enya M, Horikawa Y, Kuroda E, et al. Mutations in the small heterodimer partner gene increase morbidity risk in Japanese type 2 diabetes patients. Human Mutation. 2008;29(11):E271–E277. doi: 10.1002/humu.20865. [DOI] [PubMed] [Google Scholar]
- 113.Mitchell SMS, Weedon MN, Owen KR, et al. Genetic variation in the small heterodimer partner gene and young-onset type 2 diabetes, obesity, and birth weight in U.K. subjects. Diabetes. 2003;52(5):1276–1279. doi: 10.2337/diabetes.52.5.1276. [DOI] [PubMed] [Google Scholar]
- 114.Echwald SM, Andersen KL, Sørensen TIA, et al. Mutation analysis of NROB2 among 1545 danish men identifies a novel c.278G > a (p.G93D) variant with reduced functional activity. Human Mutation. 2004;24(5):381–387. doi: 10.1002/humu.20090. [DOI] [PubMed] [Google Scholar]
- 115.Yamagata K, Daitoku H, Shimamoto Y, et al. Bile acids regulate gluconeogenic gene expression via small heterodimer partner-mediated repression of hepatocyte nuclear factor 4 and Foxo1. Journal of Biological Chemistry. 2004;279(22):23158–23165. doi: 10.1074/jbc.M314322200. [DOI] [PubMed] [Google Scholar]
- 116.Shih DQ, Screenan S, Munoz KN, et al. Loss of HNF-1α function in mice leads to abnormal expression of genes involved in pancreatic islet development and metabolism. Diabetes. 2001;50(7-12):2472–2480. doi: 10.2337/diabetes.50.11.2472. [DOI] [PubMed] [Google Scholar]
- 117.Wang L, Huang J, Saha P, et al. Orphan receptor small heterodimer partner is an important mediator of glucose homeostasis. Molecular Endocrinology. 2006;20(11):2671–2681. doi: 10.1210/me.2006-0224. [DOI] [PubMed] [Google Scholar]
- 118.Kim YD, Park KG, Lee YS, et al. Metformin inhibits hepatic gluconeogenesis through AMP-activated protein kinase-dependent regulation of the orphan nuclear receptor SHP. Diabetes. 2008;57(2):306–314. doi: 10.2337/db07-0381. [DOI] [PubMed] [Google Scholar]
- 119.Yamagata K, Yoshimochi K, Daitoku H, Hirota K, Fukamizu A. Bile acid represses the peroxisome proliferator-activated receptor-γ coactivator-1 promoter activity in a small heterodimer partner-dependent manner. International Journal of Molecular Medicine. 2007;19(5):751–756. [PubMed] [Google Scholar]
- 120.Bae Y, Kemper JK, Kemper B. Repression of CAR-mediated transactivation of CYP2B genes by the orphan nuclear receptor, short heterodimer partner (SHP) DNA and Cell Biology. 2004;23(2):81–91. doi: 10.1089/104454904322759894. [DOI] [PubMed] [Google Scholar]
- 121.Volle DH, Duggavathi R, Magnier BC, et al. The small heterodimer partner is a gonadal gatekeeper of sexual maturation in male mice. Genes and Development. 2007;21(3):303–315. doi: 10.1101/gad.409307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mueller M, Atanasov A, Cima I, Corazza N, Schoonjans K, Brunner T. Differential regulation of glucocorticoid synthesis in murine intestinal epithelial versus adrenocortical cell lines. Endocrinology. 2007;148(3):1445–1453. doi: 10.1210/en.2006-0591. [DOI] [PubMed] [Google Scholar]
- 123.Zhang Y, Wang L. Nuclear receptor small heterodimer partner in apoptosis signaling and liver cancer. Cancers. 2011;3(1):198–212. doi: 10.3390/cancers3010198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zhang Y, Soto J, Park K, et al. Nuclear receptor SHP, a death receptor that targets mitochondria, induces apoptosis and inhibits tumor growth. Molecular and Cellular Biology. 2010;30(6):1341–1356. doi: 10.1128/MCB.01076-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Park YY, Choi HS, Lee JS. Systems-level analysis of gene expression data revealed NR0B2/SHP as potential tumor suppressor in human liver cancer. Molecules and Cells. 2010;30(5):485–491. doi: 10.1007/s10059-010-0136-6. [DOI] [PubMed] [Google Scholar]
- 126.Zhang Y, Xu P, Park K, Choi Y, Moore DD, Wang L. Orphan receptor small heterodimer partner suppresses tumorigenesis by modulating cyclin D1 expression and cellular proliferation. Hepatology. 2008;48(1):289–298. doi: 10.1002/hep.22342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.He N, Park K, Zhang Y, Huang J, Lu S, Wang L. Epigenetic inhibition of nuclear receptor small heterodimer partner is associated with and regulates hepatocellular carcinoma growth. Gastroenterology. 2008;134(3):793–802. doi: 10.1053/j.gastro.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 128.Yuk J-M, Shin D-M, Lee H-M, et al. The orphan nuclear receptor SHP acts as a negative regulator in inflammatory signaling triggered by Toll-like receptors. Nature Immunology. 2011;12(8):742–751. doi: 10.1038/ni.2064. [DOI] [PubMed] [Google Scholar]
- 129.Beyaert R. SHP works a double shift to control TLR signaling. Nature Immunology. 2011;12(8):725–727. doi: 10.1038/ni.2075. [DOI] [PubMed] [Google Scholar]
- 130.Shulman AI, Mangelsdorf DJ. Retinoid X receptor heterodimers in the metabolic syndrome. New England Journal of Medicine. 2005;353(6):604–615. doi: 10.1056/NEJMra043590. [DOI] [PubMed] [Google Scholar]
- 131.Wang L. Role of small heterodimer partner in lipid homeostasis and its potential as a therapeutic target for obesity. Clinical Lipidology. 2010;5(4):445–448. [Google Scholar]
- 132.Nishimaki-Mogami T, Une M, Fujino T, et al. Identification of intermediates in the bile acid synthetic pathway as ligands for the farnesoid X receptor. Journal of Lipid Research. 2004;45(8):1538–1545. doi: 10.1194/jlr.M400102-JLR200. [DOI] [PubMed] [Google Scholar]
- 133.Sanyal S, Kim JY, Kim HJ, et al. Differential regulation of the orphan nuclear receptor Small Heterodimer Partner (SHP) gene promoter by orphan nuclear receptor ERR isoforms. Journal of Biological Chemistry. 2002;277(3):1739–1748. doi: 10.1074/jbc.M106140200. [DOI] [PubMed] [Google Scholar]
- 134.Lai K, Harnish DC, Evans MJ. Estrogen receptor α regulates expression of the orphan receptor small heterodimer partner. Journal of Biological Chemistry. 2003;278(38):36418–36429. doi: 10.1074/jbc.M303913200. [DOI] [PubMed] [Google Scholar]
- 135.Goodwin B, Watson MA, Kim H, Miao J, Kemper JK, Kliewer SA. Differential regulation of rat and human CYP7A 1 by the nuclear oxysterol receptor liver X receptor-α . Molecular Endocrinology. 2003;17(3):386–394. doi: 10.1210/me.2002-0246. [DOI] [PubMed] [Google Scholar]
- 136.Lee YK, Parker KL, Choi HS, Moore DD. Activation of the promoter of the orphan receptor SHP by orphan receptors that bind DNA as monomers. Journal of Biological Chemistry. 1999;274(30):20869–20873. doi: 10.1074/jbc.274.30.20869. [DOI] [PubMed] [Google Scholar]
- 137.Kim H-i, Koh Y-K, Kim T-H, et al. Transcriptional activation of SHP by PPAR-γ in liver. Biochemical and Biophysical Research Communications. 2007;360(2):301–306. doi: 10.1016/j.bbrc.2007.05.171. [DOI] [PubMed] [Google Scholar]
- 138.Kim HJ, Kim JY, Park YY, Choi HS. Synergistic activation of the human orphan nuclear receptor SHP gene promoter by basic helix-loop-helix protein E2A and orphan nuclear receptor SF-1. Nucleic Acids Research. 2003;31(23):6860–6872. doi: 10.1093/nar/gkg906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Gupta S, Stravitz RT, Dent P, Hylemon PB. Down-regulation of cholesterol 7α-hydroxylase (CYP7A1) gene expression by bile acids in primary rat hepatocytes is mediated by the c-Jun N-terminal kinase pathway. Journal of Biological Chemistry. 2001;276(19):15816–15822. doi: 10.1074/jbc.M010878200. [DOI] [PubMed] [Google Scholar]
- 140.Choi YH, Park MJ, Kim KW, Lee HC, Choi YH, Cheong J. The orphan nuclear receptor SHP is involved in monocytic differentiation, and its expression is increased by c-Jun. Journal of Leukocyte Biology. 2004;76(5):1082–1088. doi: 10.1189/jlb.1203658. [DOI] [PubMed] [Google Scholar]
- 141.Iyer AK, Zhang YH, McCabe ERB. Dosage-sensitive sex reversal adrenal hypoplasia congenita critical region on the X chromosome, gene 1 (DAX1) (NR0B1) and Small Heterodimer Partner (SHP) (NR0B2) form homodimers individually, as well as DAX1-SHP heterodimers. Molecular Endocrinology. 2006;20(10):2326–2342. doi: 10.1210/me.2005-0383. [DOI] [PubMed] [Google Scholar]
- 142.Iyer AK, Zhang YH, McCabe ERB. LXXLL motifs and AF-2 domain mediate SHP (NR0B2) homodimerization and DAX1 (NR0B1)-DAX1A heterodimerization. Molecular Genetics and Metabolism. 2007;92(1-2):151–159. doi: 10.1016/j.ymgme.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Klinge CM, Jernigan SC, Risinger KE. The agonist activity of tamoxifen is inhibited by the short heterodimer partner orphan nuclear receptor in human endometrial cancer cells. Endocrinology. 2002;143(3):853–867. doi: 10.1210/endo.143.3.8676. [DOI] [PubMed] [Google Scholar]
- 144.Brendel C, Schoonjans K, Botrugno OA, Treuter E, Auwerx J. The small heterodimer partner interacts with the liver X receptor α and represses its transcriptional activity. Molecular Endocrinology. 2002;16(9):2065–2076. doi: 10.1210/me.2001-0194. [DOI] [PubMed] [Google Scholar]
- 145.Kress S, Greenlee WF. Cell-specific regulation of human CYP1A1 and CYP1B1 genes. Cancer Research. 1997;57(7):1264–1269. [PubMed] [Google Scholar]
- 146.Naya FJ, Stellrecht CMM, Tsai MJ. Tissue-specific regulation of the insulin gene by a novel basic helix- loop-helix transcription factor. Genes and Development. 1995;9(8):1009–1019. doi: 10.1101/gad.9.8.1009. [DOI] [PubMed] [Google Scholar]
- 147.Lee JE, Hollenberg SM, Snider L, Turner DL, Lipnick N, Weintraub H. Conversion of Xenopus ectoderm into neurons by NeuroD, a basic helix-loop-helix protein. Science. 1995;268(5212):836–844. doi: 10.1126/science.7754368. [DOI] [PubMed] [Google Scholar]
- 148.Kim JY, Chu K, Kim HJ, et al. Orphan nuclear receptor small heterodimer partner, a novel corepressor for a basic helix-loop-helix transcription factor BETA2/NeuroD. Molecular Endocrinology. 2004;18(4):776–790. doi: 10.1210/me.2003-0311. [DOI] [PubMed] [Google Scholar]
- 149.Park MJ, Kong HJ, Kim HY, Kim HH, Kim JH, Cheong JH. Transcriptional repression of the gluconeogenic gene PEPCK by the orphan nuclear receptor SHP through inhibitory interaction with C/EBPα . Biochemical Journal. 2007;402(3):567–574. doi: 10.1042/BJ20061549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Båvner A, Johansson L, Toresson G, Gustafsson JÅ, Treuter E. A transcriptional inhibitor targeted by the atypical orphan nuclear receptor SHP. EMBO Reports. 2002;3(5):478–484. doi: 10.1093/embo-reports/kvf087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Boulias K, Talianidis I. Functional role of G9a-induced histone methylation in small heterodimer partner-mediated transcriptional repression. Nucleic Acids Research. 2004;32(20):6096–6103. doi: 10.1093/nar/gkh947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Fang S, Miao J, Xiang L, Ponugoti B, Treuter E, Kemper JK. Coordinated recruitment of histone methyltransferase G9a and other chromatin-modifying enzymes in SHP-mediated regulation of hepatic bile acid metabolism. Molecular and Cellular Biology. 2007;27(4):1407–1424. doi: 10.1128/MCB.00944-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Song G, Wang L. Nuclear receptor SHP activates miR-206 expression via a cascade dual inhibitory mechanism. PLoS ONE. 2009;4(9) doi: 10.1371/journal.pone.0006880. Article ID e6880. [DOI] [PMC free article] [PubMed] [Google Scholar]