

Abstract
The largest family of toxin-antitoxin (TA) modules are encoded by the vapBC operons, but their roles in bacterial physiology remain enigmatic. Microarray analysis in Mycobacterium smegmatis overexpressing VapC/VapBC revealed a high percentage of downregulated genes with annotated roles in carbon transport and metabolism, suggesting that VapC was targeting specific metabolic mRNA transcripts. To validate this hypothesis, purified VapC was used to identify the RNA cleavage site in vitro. VapC had RNase activity that was sequence specific, cleaving single-stranded RNA substrates at AUAU and AUAA in vitro and in vivo (viz., MSMEG_2121 to MSMEG_2124). A bioinformatic analysis of these regions suggested that an RNA hairpin 3′ of the AUA(U/A) motif is also required for efficient cleavage. VapC-mediated regulation in vivo was demonstrated by showing that MSMEG_2124 (dhaF) and MSMEG_2121 (dhaM) were upregulated in a ΔvapBC mutant growing on glycerol. The ΔvapBC mutant had a specific rate of glycerol consumption that was 2.4-fold higher than that of the wild type during exponential growth. This increased rate of glycerol consumption was not used for generating bacterial biomass, suggesting that metabolism by the ΔvapBC mutant was uncoupled from growth. These data suggest a model in which VapC regulates the rate of glycerol utilization to match the anabolic demands of the cell, allowing for fine-tuning of the catabolic rate at a posttranscriptional level.
INTRODUCTION
Bacterial toxin-antitoxin (TA) modules were first reported as factors that protect low-copy-number plasmids in bacteria from postsegregational loss (35). This mechanism of protection is achieved through the production of a stable toxin and an unstable, cognate antitoxin. If the plasmid-borne TA operon is lost from a bacterial cell, the toxin persists and kills the plasmid-free cell. Since this initial report, TA operons have been shown to be widespread and found in nearly every sequenced bacterial and archaeal genome. The presence of TA operons on bacterial chromosomes suggests that these modules may play additional roles other than protection of mobile DNA. For example, TA modules have been shown to be associated with antibiotic resistance/tolerance, virulence and pathogenicity islands, bacterial persistence, and pathogen trafficking (26, 30, 31, 43).
TA modules have been classified into three types according to the molecular identity of each component (25). For type I modules, the antitoxin is a small antisense RNA molecule that prevents toxin translation. Type II modules encode a protein antitoxin that binds its cognate toxin and inhibits its activity. Type III TA modules encode a protein toxin and an RNA antitoxin that interact and form a protein-RNA complex. These types are further subdivided into nine families, and coincidentally, nine potential biological functions for toxin-antitoxin modules have been proposed (41). The toxic components of the TA modules exert their effects in different ways. The CcdB and ParE toxins are DNA gyrase inhibitors; the RelE, Doc, and HigB toxins directly or indirectly cleave ribosome-associated mRNAs; HipA is a protein kinase that targets EF-Tu; and the VapC and MazF families are families of RNases that cleave mRNA transcripts independently of the ribosome (25). The VapC and MazF toxins have been collectively described as mRNA interferases whose biochemical activity results in the inhibition of translation (34, 51). Bacteria often harbor more than one type of TA module, and it remains to be determined whether these systems interact to regulate whole-cell physiology.
The most prevalent TA modules in bacteria and archaea are the VapBC family, and the distribution of vapBC operons across species is intriguing. In many sequenced genomes, there are between one and five copies of vapBC TAs. However, in several unrelated organisms, the vapBC family of TAs is greatly expanded in number. For example, the genome of the important human pathogen Mycobacterium tuberculosis harbors 45 putative vapBC operons (3, 4, 50), and the unrelated archaeon Pyrococcus kodakaraensis contains 29 putative vapBC operons (54). Strikingly, among the mycobacteria, this expansion of toxin-antitoxin operons is confined to just the mammalian pathogens M. tuberculosis and M. bovis. In contrast, Mycobacterium smegmatis and Mycobacterium avium subsp. paratuberculosis have a single vapBC TA operon, and M. leprae has none (4). The role of VapBC in the biology of mycobacteria remains unknown. VapBC modules have been implicated in controlling bacterial growth in the intracellular environment (33) and regulating rates of nitrogen fixation and biomass production during symbiosis in a host plant (8). VapC from the enteric bacteria Shigella flexneri 2a and Salmonella enterica has been shown to cleave tRNAfMet in the anticodon stem-loop in a heterologous host, thus inhibiting translation (62). Overexpression of VapC in a ΔvapBC mutant of M. smegmatis caused growth inhibition via disruption of translation, and this effect was most pronounced in rapidly growing cells (i.e., early exponential phase) (51). Interestingly, the phenotype was reversible in the ΔvapBC mutant, which suggests that the antitoxin was not required for reversing the bacteriostasis and that M. smegmatis may use the VapBC module to trigger a reversible bacteriostasis under conditions where the cell's carbon metabolism is not coupled to growth due to other imbalances (e.g., nitrogen limitation) (51). Using a regulator of protein synthesis (i.e., an RNase) to control metabolic activity when required would seem advantageous when one considers that 70 to 90% of cellular ATP consumption is used for protein synthesis (12).
To address the hypothesis of catabolic and anabolic regulation (51), we measured the genome-wide transcriptional response to VapC overexpression (in comparison to VapBC overexpression) and identified a cohort of genes specifically targeted by VapC (operons for sugar transport). Using purified VapC and RNase assays, we have identified the cut site of VapC in these sugar transport operons, confirming our microarray data. In a ΔvapBC mutant, the specific rate of glycerol consumption was greater than that needed for growth, and therefore, metabolism appeared to be uncoupled from anabolic reactions and metabolic demand. These data support a model for VapC controlling the balance of catabolic and anabolic reactions in the mycobacterial cell to optimize cellular metabolism.
MATERIALS AND METHODS
Bacterial strains, plasmids, and culture conditions.
Bacterial strains and plasmids used in this study are listed in Table 1. Escherichia coli strains for plasmid preparation were grown in Luria-Bertani (LB) medium supplemented with the appropriate antibiotic for plasmid selection. Mycobacterium smegmatis strain mc2155 (56) and ΔvapBC deletion strain (JR121) (51) were grown in LB supplemented with 0.05% (wt/vol) Tween 80 (LBT) and Hartmans de Bont (HdB) (55) minimal medium supplemented with 0.2% glycerol and 0.05% (wt/vol) Tween 80. All M. smegmatis cultures used in growth experiments, for harvesting RNA, and for use in biochemical assays were set up as follows. For initial starter cultures, strains were inoculated into LBT, grown overnight to an optical density at 600 nm (OD600) of below 0.4, and then placed at 4°C. A second starter culture was then inoculated (OD600 of 0.005) from the LBT overnight cultures into appropriate growth medium to be used in the final experiment. This was done to ensure that an optical density (OD600 of >0.2) associated with early-exponential-phase cells would be achieved of this second starter for direct use. The second starter was used to inoculate flasks containing HdB or LBT medium to an OD600 of 0.0025 for use in experiments. All strains were grown at 37°C with agitation (200 rpm). The OD600 was measured in a Jenway 6300 spectrophotometer. Strains harboring pMind constructs were inoculated into appropriate medium containing hygromycin B (50 μg ml−1). For complementation of growth experiments, tetracycline (20 ng ml−1) was included in all media to ensure that expression occurred upon inoculation of cultures. For microarray analysis, ΔvapBC deletion mutant strain JR121 was used harboring tetracycline-inducible expression constructs pMind_vapC and pMind_vapBC for overexpression of toxin VapC and toxin-antitoxin complex VapBC as previously published (51). Western blot analysis was used to detect the levels of VapC protein using VapC polyclonal antibodies as previously described (51).
Table 1.
Bacterial strains and plasmids used in this study
| Bacterial strain or plasmid | Relevant genotype or phenotype or descriptiona | Source or reference |
|---|---|---|
| E. coli DH5α | fhuA2 Δ(argF-lacZ)U169phoAglnV44 ϕ80 Δ(lacZ)M15 gyrA96recA1relA1endA1thi-1hsdR17 | Stratagene |
| M. smegmatis | ||
| mc2155 | Electrocompetent wild-type strain of M. smegmatis | 56 |
| JR121 | mc2155 ΔvapBC::aphA-3; Kmr | 51 |
| Plasmids | ||
| pDEST17 | Gateway expression vector for M. smegmatis; Hygr | Lab stock |
| pcDNA3 | Mammalian expression vector; Apr | Invitrogen |
| pGEM-T Easy | Cloning vector; Apr | Promega |
| pJR29 | pMind harboring vapC with synthetic RBS from kanamycin marker; Kmr Hygr | 51 |
| pJR230 | pMind harboring vapBC with synthetic RBS from kanamycin marker; Kmr Hygr | 51 |
RBS, ribosome binding site.
Cell harvest of batch cultures for RNA extraction.
For use in microarray analysis, M. smegmatis JR121 harboring pMind-vapBC (pJR230) and pMind-vapC (pJR29) were grown in a 400-ml volume of HdB in a 2-liter flask. Cultures were grown to an OD600 of ∼0.1, and protein expression was then induced with tetracycline (20 ng ml−1). Cells were harvested 60 min postinduction by using a cold-glycerol-saline quenching method described previously (6, 61). Four independent experiments were performed. For gene expression analysis comparing mRNA from the ΔvapBC mutant and the wild-type mc2155 strain, cultures were grown in a 200-ml volume of HdB to an OD600 of ∼0.1 before harvesting with glycerol-saline solution.
For extraction of total RNA for microarray and quantitative real-time PCR (qRT-PCR), cells were resuspended in TRIzol reagent (Invitrogen), and RNA was extracted according to the manufacturer's instructions. The RNA was resuspended in water containing 0.1% diethyl pyrocarbonate (DEPC)–Milli-Q and incubated at 55°C for 10 min. To remove any contaminating DNA present in samples, extracted RNA was treated with 2 U of RNase-free DNase using a Turbo RNase-free kit (Ambion) according to the manufacturer's instructions. Alternatively, for total RNA extraction required for use in reverse transcriptase PCR of the 5′ untranslated region (5′UTR) of MSMEG_2124, cells were resuspended in 5 M guanidinium isothiocyanate (GITC) (pH 7), pelleted by centrifugation, resuspended in 0.5 ml fresh GITC, and transferred to a new tube containing approximately 0.3 g of 0.1-mm and 2.5-mm zirconia beads. Beads were mixed in a Fastprep FP120 bead beater (Thermosavant) for 20 s and left to cool for 1 min, and the process was repeated 3 times for increasing periods (i.e., 20, 25, 30, and 30 s). The supernatant was removed, and nucleic acids were twice extracted with phenol-chloroform followed by isopropanol precipitation. The precipitated RNA was washed with 70% ethanol and resuspended in 25 μl of 10 mM Tris-HCl (pH 7.0) and 0.5 mM MnCl2. The extracted RNA was DNase treated by the addition of 1 μl DNase (Promega) per μg RNA present and incubated at 37°C for 30 min with shaking at 600 rpm. The DNase reaction was stopped by the addition of EGTA stop solution and heated to 65°C for 10 min. All RNA concentrations were determined using a Nanodrop ND-1000 spectrophotometer, and RNA integrity was analyzed by agarose gel electrophoresis. RNA samples were stored at −80°C.
Microarray analysis and quantitative real-time PCR.
Microarray analysis was performed as previously described (6). Synthesis of cDNA and aminoallyl-dUTP (aa-dUTP) labeling was performed using protocol standard operating procedure (SOP) M007 provided by the Pathogen Functional Genomics Research Center (PFGRC) (http://pfgrc.jcvi.org) established by the National Institute of Allergy and Infectious Diseases (NIAID) and the J. Craig Venter Institute (JCVI). Cy dye-labeled cDNA probes were hybridized to microarray slides supplied by the PFGRC funded by NIAID using protocol SOP M008 provided by the PFGRC. Slides were scanned in a Genepix 4000A scanner at a 10-μm pixel size and autoadjusted photomultiplier gain, and data analysis was performed as previously described (6). All data have been deposited at the Gene Expression Omnibus (GEO, NCBI) under accession number GSE29631. qRT-PCR was performed to validate gene expression changes detected by microarray analysis and detect mRNA differences between the wild-type mc2155 and ΔvapBC mutant strain. The sequences of selected genes were retrieved from the JCVI/Comprehensive Microbial Resource (CMR) database (http://cmr.jcvi.org) for primer design, which was performed using Primer3 (version 0.4.0) (http://frodo.wi.mit.edu/primer3/) or Primer Express software v3.0 (Applied Biosystems). All primers were optimized to determine optimal primer concentration. All oligonucleotides used in this study are listed in Table S1 in the supplemental material and labeled MSMEG_XXXX (gene identification [ID] number) followed by the letter M for use in microarray validation or Q for use in qRT-PCR for the ΔvapBC mutant and strain mc2155. For qRT-PCR, total RNA (1 μg) was obtained and reverse transcribed with random primers (250 ng) and SuperScript III reverse transcriptase (both from Invitrogen) according to the manufacturer's instructions. Quantitative real-time PCR was performed according to the manufacturer's instructions with Platinum SYBR Green qPCR SuperMix-UDG with ROX (Invitrogen) using an ABI Prism 7500 (Applied Biosystems). The qRT-PCR cycle included stage I (1 step of 2 min at 95°C), stage II (40 cycles, with 1 cycle consisting of 15 s at 95°C and 1 min at 60°C), followed by stage III dissociation step (1 cycle consisting of 15 s at 95°C, 1 min at 60°C, 15 s at 95°C, and 15 s at 60°C). Relative gene expression was determined from calculated threshold cycle (CT) values using sigA gene (MSMEG_2758) as an internal normalization standard. Expression ratios were subjected to a two-tailed paired t test (VapC/VapBC) or unpaired t test (ΔvapBC/mc2155) to determine statistical significance.
Reverse transcriptase PCR for determination of MSMEG_2124 5′UTR.
Total RNA was reverse transcribed using SuperScript III reverse transcriptase (RT) (Invitrogen, USA) with random hexamers. Briefly, 1 μg DNase-treated RNA was incubated with a total of 100 ng random primer oligonucleotides (Invitrogen) at 65°C for 5 min and then cooled for 1 min on ice, followed by the addition of 4 μl of 5× buffer (250 mM Tris-HCl, pH 8.3, containing 375 mM KCl and 15 mM MgCl2), 1 μl dithiothreitol (DTT), 1 μl of 10 mM deoxynucleoside triphosphates (dNTPs), and 0.7 μl SuperScript III RT. The reaction was performed at 25°C for 10 min, 50°C for 1 h, and 85°C for 10 min. For each sample, a negative RT-PCR (no reverse transcriptase added) was performed and used as a negative control in subsequent PCRs. Genomic DNA and cDNA were used as the templates in traditional PCRs utilizing Taq DNA polymerase (Invitrogen) and gene (transcript)-specific primers. Two primer sets were used. Primers MSMEG_2124 RT Rev (Rev stands for reverse) and MSMEG_2124 RT Fwd (Fwd stands for forward) were used to verify the annotated MSMEG_2124 gene, and primers MSMEG_2124 (RT) Rev and MSMEG_2124 77-98 RT Fwd were used to determine whether the 5′UTR included up to 98 bp upstream of MSMEG_2124. cDNA or genomic DNA (gDNA) (2.5 μl) was added to a final concentration of around 37.5 ng per 25-μl PCR mixture. The PCR conditions were as follows: (i) an initial denaturation step at 95°C for 5 min; (ii) 39 cycles, with 1 cycle consisting of 30 s at 95°C, 30 s at 65 to 69°C, 55 s at 72°C, and 30 s at 95°C; (iii) a final extension step of 68°C for 7 min. PCR products were analyzed by agarose gel electrophoresis.
Tryptic digestion of the VapBC complex and purification of VapC from M. smegmatis.
The VapBC complex from M. smegmatis was purified by the method of Robson et al. (51) and digested with 0.1 mg ml−1 trypsin (Sigma-Aldrich) for 60 min at room temperature. Reactions were stopped by the addition of 0.1 mg ml−1 trypsin inhibitor from Glycine max (soybean) (Sigma-Aldrich) and incubated for 15 min at room temperature. Trypsin digestion products of the purified VapBC complex inhibited with trypsin inhibitor were dialyzed against 50 mM phosphate buffer (pH 7.4) and 100 mM NaCl overnight and subjected to anion-exchange chromatography to separate the trypsin plus inhibitor from VapC. The trypsin digestion products were loaded onto a 5-ml HiTrap Q anion-exchange column (GE Healthcare) and washed with 25 ml of 50 mM phosphate buffer (pH 7.4) and 100 mM NaCl at a flow rate of 1 ml min−1 to remove the trypsin. Bound proteins were removed by running a gradient of 0 to 100% (vol/vol) elution buffer (50 mM phosphate buffer [pH 7.4], 1 M NaCl) over 50 ml at a flow rate of 1 ml · min−1. Fractions containing the desired protein were then analyzed by SDS-PAGE. VapC was further purified by size exclusion chromatography. VapC was analyzed on an analytical S75 10/300 gel filtration column (GE Healthcare, United Kingdom), and fractions containing VapC protein were analyzed by SDS-PAGE.
Transcription of pentaprobe RNA.
The pentaprobe pack was obtained from Joel McKay, School of Molecular Bioscience, University of Sydney, Australia, and consists of 12 plasmids (38). This included both forward (A922 to A227) and reverse (A928 to A933) pentaprobe strands under a T7 promoter. A922 is complementary to A928, A923 is complementary to A929, and so on. Plasmids were transformed into electrocompetent E. coli DH5α cells. Successful transformants were cultured in LB broth including 50 μg ml−1 ampicillin, and the plasmid was extracted using the Qiagen miniprep kit according to the manufacturer's instructions. Primers for amplification of the pentaprobe insert were designed to flank the T7 promoter in pcDNA3 (pcDNA3 PP Fwd [PP stands for pentaprobe]) and the end of the pentaprobe insert (pcDNA3 PP Rev). The pentaprobe insert was PCR amplified using the Pfx DNA polymerase system (Invitrogen) and gel purified. DNA concentrations were determined using a Nanodrop ND-1000 spectrophotometer. RNA was transcribed from 800 ng of the pentaprobe PCR product using the T7 MEGAscript kit (Ambion) according to the manufacturer's instructions. Transcribed RNA was purified by sodium acetate and ethanol precipitation and resuspended in nuclease-free water. RNA concentrations were determined using a Nanodrop ND-1000 spectrophotometer, and the secondary structure of RNA molecules was predicted using RNAstructure version 4.6 (42).
Primers were designed for the amplification of 50 bases on either side of the ATA(A/T) cut site in the 932 pentaprobe RNA. The 100-bp fragment (termed 932 50-150) was PCR amplified, gel purified, and then used as a template in a further PCR with the same reverse primer (932 150 Rev) as described above and a forward primer including a T7 promoter (T7 932 50 Fwd). The DNA concentration of the template was determined, and RNA was transcribed and purified as described above. Single-stranded pentaprobe RNA was annealed with its reverse complement RNA strand (i.e., 927 with 933) to form double-stranded RNA (dsRNA). Equal molar ratios of each strand were combined with nuclease-free water, heated to 70°C for 15 min, and then cooled slowly to room temperature. Annealed RNA was run on an agarose gel and stained with acridine orange to distinguish between double- and single-stranded RNA. Double-stranded nucleic acids stain bright fluorescent green, whereas single-stranded nucleic acids stain bright reddish-orange. A single strand of pentaprobe DNA was amplified by asymmetric PCR from the template pentaprobe PCR product using the Pfx DNA polymerase system (Invitrogen). However, 1:50 and 1:100 reverse/forward primer ratios were used instead of 1:1 ratios, and 35 cycles were employed to increase amplification. The PCR products were analyzed by agarose gel electrophoresis and staining with acridine orange, and single-stranded DNA products were gel purified.
VapC RNase activity assay.
Mycobacterium smegmatis VapC RNase activity assay mixtures contained 1 μg purified VapC protein for pentaprobe assays and 0.02 μg purified VapC for M. smegmatis transcript assays, 1 μg purified RNA (molar ratios of VapC to RNA varied from 5:1 to 1:1), 12 mM phosphate buffer (sodium phosphate typically used except for matrix-assisted laser desorption ionization mass spectrometry [MALDI MS] when ammonium phosphate was used), 6 mM NaCl, and 6 mM MgCl2. The MgCl2 concentrations used in this study are consistent with previously reported studies (5, 44, 62). Individual assay reaction mixtures were set up for each time point to reduce the possibility of RNase contamination. Negative controls included RNA-only reaction mixtures (no VapC added) or reaction mixtures with 12 mM EDTA added or with VapBC complex added instead of VapC. Assay mixtures were incubated at 37°C, and time point reactions were stopped by the addition of 10 μl formamide loading dye, or for analysis by mass spectrometry, assay mixtures were heat inactivated by incubating at 70°C for 10 min followed by the addition of ammonium acetate and ethanol for precipitation of RNA.
Design of 932 RNA oligonucleotides and cut-site analysis by MALDI MS.
RNA oligonucleotides were designed to cover bases 54 to 242 of pentaprobe 932. Nine overlapping oligonucleotides (932 RNA oligonucleotides 1 to 9) of 30 to 36 bases were designed to cover the 188-base RNA segment. RNAstructure version 4.6 was used to predict secondary structure, and the short oligonucleotides were designed to mimic these regions of the whole 932 pentaprobe RNA molecule. RNA oligonucleotide assays were precipitated to remove contaminants by the addition of ammonium acetate (final concentration of 5 M) (pH 5.2) and three times the volume of 100% ethanol. The samples were precipitated on ice for 30 min, centrifuged at 13,000 × g for 20 min at 4°C, then washed with 70% (vol/vol) ethanol, and centrifuged as described above. The supernatant was then removed, and the RNA pellet was resuspended in nuclease-free water and stored at −80°C. Matrix for MALDI MS of RNA oligonucleotides was prepared fresh daily. The matrix solution consisted of 5 mg of 3-hydroxypicolinic acid (3-HPA), 10 μl of 2.5 M diammonium citrate, 125 μl of acetonitrile (ACN), and 365 μl of DEPC water. The solution was vortexed well until all matrix dissolved and then centrifuged for 5 min at 13,000 × g. A 1-μl aliquot of matrix solution was spotted onto either an Anchorchip or stainless steel target plate (Bruker Daltonics) and left to air dry. A 1-μl sample and 1 μl of the oligonucleotide calibration standard (Bruker Daltonics) were spotted onto the dried matrix and left to air dry.
An Autoflex II MALDI–time of flight (MALDI-TOF) mass spectrometer (Bruker Daltonics) was used to analyze samples. The samples were analyzed in linear mode, with the mass range selector set at “low range,” pulsed ion extraction of 150 ns, gain to 2,500 V, acceleration voltage to 20 kV, and a range of 2 to 12 kDa was collected. Laser power was typically around 60%. The spectra for the oligonucleotide calibration standard (Bruker Daltonics) were collected first, and the spectrometer was calibrated with an automatic polynomial correction. The spectra were saved and exported to DataAnalysis software (Bruker Daltonics) where the baseline was subtracted and peaks were identified and labeled. The online tool Mongo Oligo Mass Calculator version 2.06 (52) was used to determine theoretical masses of RNA oligonucleotide fragments to compare to masses from obtained spectra. The Mongo Oligo Mass Calculator allowed the addition of 5′ and 3′ modifications to the RNA oligonucleotide.
Transcription of MSMEG_2124, MSMEG_2123, MSMEG_2122, MSMEG_2121, MSMEG_0650, and MSMEG_1965.
The gene encoding MSMEG_2124 was PCR amplified to include the 5′UTR. MSMEG_2124 plus 150 bp upstream was cloned using primers MS_2124 T7 pGem F and R [F stands for forward, and R stands for reverse]) was cloned into pGEM-T Easy Vector system (Promega) using the 3′-T overhang. The resulting plasmid was sequence verified and used as a DNA template in subsequent PCRs. The Pfx DNA polymerase system (Invitrogen) and pUC/M13 primers were used to amplify MSMEG_2124 plus the 5′UTR from pGEM_MS2124 to include the T7 promoter. PCR products were gel purified, and the DNA concentration was determined using a Nanodrop ND-1000 spectrophotometer.
Primers for amplification of genes in the annotated MSMEG_2124 operon and MSMEG_1965 (tRNA fMet) were designed to flank either end of the gene (termed MSMEG_XXXX RNA F and MSMEG_XXXX RNA R in Table S1 in the supplemental material). MSMEG_2124 (no 5′UTR), MSMEG_2123, MSMEG_2122, MSMEG_2121, and MSMEG_1965 genes were PCR amplified from M. smegmatis mc2155 genomic DNA using the Pfx DNA polymerase system (Invitrogen) and gel purified. A further round of PCR amplification using the purified MSMEG_2124, -2123, -2122, -2121, and -1965 PCR products and a forward primer including the T7 RNA polymerase promoter site (termed MSMEG_XXXX RNA T7 F in Table S1) was performed to add the T7 promoter site immediately upstream of the gene sequence for use in subsequent transcription reactions. MSMEG_0650 was amplified from pDEST-MSMEG_0650 (a kind gift from Shaun Lott, Department of Structural Biology, University of Auckland) using T7 forward and reverse primers and the Pfx DNA polymerase system (Invitrogen). The gel-purified PCR product was used as the template in subsequent transcription reactions. Oligonucleotide sequences can be found in Table S1. DNA concentrations were determined using a Nanodrop ND-1000 spectrophotometer. PCR products were sequenced using a T7 primer to ensure fidelity for RNA transcription.
RNA was transcribed from 800 ng of the PCR product using the T7 MEGAscript kit (Ambion) according to the manufacturer's instructions. Transcribed RNA was purified by sodium acetate and ethanol precipitation and resuspended in nuclease-free water. RNA concentrations were determined using a Nanodrop ND-1000 spectrophotometer, and the secondary structures of RNA molecules were predicted using RNAstructure version 4.6 (42).
RNA bioinformatics.
We employed a local minimum-free energy approach to RNA structure prediction that has been proven to work well in longer sequences (32). This was implemented in the RNALfold algorithm that forms part of the Vienna RNA package. This implementation returns a Z-score for structures that are significantly more stable than expected given their length and nucleotide composition (63). Initial predictions used RNALfold on the three positive exemplars for VapC cleavage (MSMEG_2121, MSMEG_2124 5′UTR, and pentaprobe 932). All three predictions conformed to the same basic structure: a 5′ AUA(U/A) motif followed by a stable hairpin. Given that the three experimentally characterized VapC cleavage targets appeared to share a basic secondary structure, it was possible to build a sequence alignment and consensus secondary structure based on these three substrates. From this, we built a covariance model (CM), which was used for predicting homologous and analogous structures in other sequences (17, 18, 45, 46). The CM was used to search 60 operons and flanking sequences that contained at least one of the 106 genes downregulated by VapC for analogous motifs. The high-scoring matches to this CM were then incorporated into the alignment. The alignment and consensus secondary structure were manually refined, and the CM was then rebuilt and researched against the 60 operons. This process was iterated for several cycles until no further high-scoring matches were found. The CM was then used to search the genome of M. smegmatis.
Molar growth yields and biochemical assays.
Glycerol utilization and dry weights were measured to calculate molar growth yield on glycerol (Yglycerol) (in grams [dry weight] of cells per mole of glycerol consumed) and glycerol consumption (in millimoles of glycerol/h/g [dry weight] of cells). Cultures were grown in HdB. Supernatant samples were taken after the initial inoculation to determine the starting glycerol concentration of each individual flask and at specified time points during growth. All culture samples were centrifuged (5 min, 13,000 rpm) to obtain cell-free supernatant for use in glycerol assay and stored at −20°C. Glycerol concentration was measured by detecting NADH oxidation (absorbance at 340 nm) using previously published methods (49). Briefly, a working solution of assay mix contained 0.5 U ml−1 glycerokinase from E. coli (Sigma-Aldrich), 0.5 U ml−1 pyruvate kinase from rabbit muscle (Sigma-Aldrich), 1 U ml−1 lactic dehydrogenase from rabbit muscle (Roche), 50 mM Tris HCl (pH 8), 2 mM MgCl2, 0.25 mM NADH, 3 mM phosphoenolpyruvate (PEP), and 3 mM ATP. A standard curve was analyzed with each set of supernatant samples, which was prepared from 10 mM glycerol stock. The reaction mixtures were incubated in microcentrifuge tubes for 15 min at 37°C to allow the enzyme assay to reach an endpoint. For the cell weight (dry weight) of each time point, 20 ml of culture was harvested in triplicate by filtration (0.22 μm; Millipore), and filters were dried at 65°C until the same weight (dry weight) was achieved on consecutive days. The growth rate for the log phase of growth was determined by plotting the log10 of the optical density against time. Biological replicates were performed 2 or 3 times for each time point.
RESULTS
VapC overexpression downregulates a large cohort of genes involved in carbon transport and metabolism.
To gain molecular insight into the role of VapC-mediated inhibition of cell growth (51), the transcriptional response of M. smegmatis to the conditional overexpression of VapC was assessed through microarray analysis. Microarray analysis was performed using ΔvapBC deletion strains expressing VapC and VapBC to ensure that the only changes in gene expression would be in response to the presence of the toxin compared with expression of the inactive VapBC complex. As growth inhibition generally occurs during overexpression of toxin components of TA systems, it was important to preempt this inhibitory effect in order to identify the transcriptional response to VapC (RNase activity) and not a general response associated with inhibition of cell growth and/or general cell stress. A time course experiment was performed to determine the level of VapC expression after induction with tetracycline. Western blotting confirmed VapC expression at the time (60 min) of RNA sampling (Fig. 1A). This level of VapC expression was in good agreement with overexpressed VapBC in the same genetic background (51). Under the conditions of VapC and VapBC expression, no difference in the growth rate between the strains expressing VapC and VapBC was observed at 60 min (time of RNA harvest for microarray studies).
Fig 1.

(A) Detection of VapC conditional expression over time. A time course study of VapC protein expression was performed using M. smegmatis ΔvapBC deletion mutant containing the tetracycline-inducible construct pMind-vapC. VapC expression was induced with 20 ng ml−1 tetracycline, and protein samples were taken over time at 0, 30, 60, 90, 120, 150, and 180 min postinduction for Western blot analysis. A positive control using purified VapC (+) was also included. (B) Validation of microarray analysis with quantitative RT-PCR. Microarray data were validated by comparing the gene expression changes of selected genes obtained from microarray analysis with that of qRT-PCR. The bars represent changes in the mRNA levels in the M. smegmatis ΔvapBC mutant (expressing VapC versus VapBC) for 4 (microarray) or 3 (qRT-PCR) independent biological replicates, and the error bars represent standard deviations. Gene expression was normalized to sigA before the transcription levels in the ΔvapBC deletion strain expressing VapC was compared with that of VapBC. With the exception of MSMEG_4182 and MSMEG_0515, the values of gene expression from qRT-PCR were statistically significant (∗, P value < 0.05; ∗∗, P value < 0.005). CRP, cyclic AMP receptor protein.
Comparison of the transcriptional response of the ΔvapBC strain expressing VapC with that of the same strain expressing VapBC identified a total of 205 genes exhibiting a 1.50-fold or greater change in expression (P < 0.05, four biological repeats including two dye swaps). This represents an altered expression of 2.86% of the M. smegmatis genome (total 6,933 genes) (see Table S2 in the supplemental material). Of these differentially expressed genes, 106 genes were downregulated and 99 were upregulated in response to VapC (Table S2). In order to analyze the data, genes were assigned to categories that had previously been used to decipher the genome sequence of Mycobacterium tuberculosis and were based on putative functions of the encoded proteins (10). The microarray data were validated by qRT-PCR analysis of 9 selected genes (Fig. 1B). In response to VapC, 34% of the gene transcripts that were downregulated (≥1.50-fold) were predicted to be involved in the transport and catabolism of carbohydrate sources (Table 2). The majority of these downregulated gene transcripts are organized in operons with other genes that also exhibited downregulation (18 genes are involved in carbohydrate transport, and 18 are required for carbohydrate utilization). The carbon source used in our experiments was glycerol, and analysis of genes involved in glycerol transport and utilization allowed us to construct a metabolic pathway for glycerol metabolism in M. smegmatis (Fig. 2). The transcript levels for two transporters of glycerol, MSMEG_6758 (annotated as glpF) and MSMEG_2124 (annotated as a glycerol uptake facilitator) decreased 1.71- and 2.73-fold, respectively. Moreover, the transcript levels that are involved in aerobic utilization of glycerol also decreased (≥1.50-fold). These genes include MSMEG_6759 annotated as a putative glycerol kinase (glpK) and MSMEG_6761 encoding glycerol-3-phosphate dehydrogenase (glpD2), which leads to the production of dihydroxyacetone phosphate (DHA-P) (Fig. 2, pathway 1). Homologues to genes of known phosphotransferase (PTS) systems were identified in the genome of M. smegmatis (57). The above-mentioned glycerol uptake facilitator (dhaF [MSMEG_2124]) appears in an operon with associated dhaMLK genes (MSMEG_2121 to MSMEG_2123), which were suggested to be components of a PTS system responsible for transport of dihydroxyacetone (DHA) (59) (Fig. 2). M. smegmatis cannot grow on DHA as the sole carbon and energy source, which suggests that dhaF (MSMEG_2124) could be an efflux mechanism for DHA (Fig. 2). Alternatively, DhaF could act as a glycerol transporter and the dhaKLM gene products act as a second pathway for glycerol metabolism as they do in organisms such as Klebsiella pneumoniae, Clostridium butyricum, and Citrobacter freundii (19, 20, 27) and more recently E. coli (15, 26). In this two-branch pathway, glycerol is dehydrogenated to DHA, which is then phosphorylated to DHA-P via DHA kinase (dhaKLM), as well as being dehydrated by a coenzyme B12-dependent glycerol dehydratase enzyme to form 3-hydroxypropionaldehyde (3-HPA) followed by reduction via 1,3-propanediol dehydrogenase to generate propanediol by-products (20, 21, 29). The transcripts encoding enzymes associated with the reductive branch of a putative anaerobic glycerol pathway in M. smegmatis, i.e., glycerol dehydratases, MSMEG_1546 to MSMEG_1549, were also downregulated 2-fold (Fig. 2, pathway 2). Analysis of the amino acid sequences for MSMEG_1546 and MSMEG_1547 suggested that these proteins were homologues of DhaB subunits from K. pneumoniae (BLAST analysis indicated ∼50 to 65% identity and 70 to 80% positive results). Other carbon transport gene transcripts that were decreased belong to the ATP binding cassette (ABC) family whose substrates include xylose (MSMEG_1704 and MSMEG_6018), arabinose (MSMEG_1711 and MSMEG_1712), ribose (MSMEG_3090, -3095, and -3601), sugar alcohol (MSMEG_5571 to -5574) as well as other unknown sugar substrates (MSMEG_3267, -3269, and -5058) (Table 2).
Table 2.
Gene clusters with purported roles in carbon transport and metabolism downregulated in M. smegmatis in response to VapC
| Predicted carbon substrate(s) | Gene ID or locus tag | Gene name | n-fold changea | P value | Descriptionb |
|---|---|---|---|---|---|
| Fructose | MSMEG_0084 | ptsH | 1.28 | 4.97E−02 | Phosphocarrier protein hpr |
| MSMEG_0085 | fruA | 1.37 | 2.74E−02 | PTS system, Fru family protein, IIABC components | |
| MSMEG_0086 | fruK | 1.24 | 1.35E−01 | 1-Phosphofructokinase | |
| MSMEG_0087 | fruR | 1.06 | 3.34E−01 | Glucitol operon repressor | |
| MSMEG_0088 | ptsI | 1.81 | 2.11E−03 | Phosphoenolpyruvate-protein phosphotransferase | |
| Glycerol | MSMEG_1542 | 1.23 | 2.60E−02 | Transcriptional regulator | |
| MSMEG_1543 | 2.18 | 4.70E−03 | EPTC-inducible aldehyde dehydrogenase | ||
| MSMEG_1544 | 1.35 | 2.08E−01 | PduO protein | ||
| MSMEG_1545 | 1.12 | 1.50E−01 | Conserved hypothetical protein | ||
| MSMEG_1546 | 1.98 | 1.30E−03 | Coenzyme B12-dependent glycerol dehydrogenase small subunit | ||
| MSMEG_1547 | 2.02 | 2.14E−03 | Glycerol dehydratase large subunit | ||
| MSMEG_1548 | 1.97 | 1.01E−02 | Propanediol utilization: dehydratase, medium subunit | ||
| MSMEG_1549 | 1.68 | 1.56E−02 | Glycerol dehydratase reactivation factor large subunit; this gene contains a frameshift which is not the result of sequencing error | ||
| Xylose | MSMEG_1704 | xylF2 | 1.63 | 2.66E−02 | ABC transporter |
| MSMEG_1705 | xylG2 | 1.47 | 2.52E−02 | d-Xylose transport ATP-binding protein XylG | |
| MSMEG_1706 | xylH2 | 1.41 | 1.76E−02 | Xylose transport system permease protein XylH | |
| Arabinose | MSMEG_1707 | pho | (1.23) | 7.29E−02 | Phosphatase YfbT |
| MSMEG_1708 | araR | 1.0 | 7.60E−01 | Ribose operon repressor, putative | |
| MSMEG_1709 | araG | 1.07 | 7.50E−01 | Inner membrane ABC transporter permease protein YjfF | |
| MSMEG_1710 | araF | 1.27 | 6.67E−02 | Ribose transport system permease protein RbsC | |
| MSMEG_1711 | araK | 1.85 | 3.03E−02 | ATP-binding protein of ABC transporter | |
| MSMEG_1712 | araE | 3.28 | 8.23E−04 | ABC transporter periplasmic binding protein YtfQ | |
| MSMEG_1713 | araB | 1.39 | 3.19E−01 | l-Ribulokinase | |
| MSMEG_1714 | araD | 1.30 | 1.84E−01 | l-Ribulose-5-phosphate 4-epimerase UlaF | |
| MSMEG_1715 | araA | 1.48 | 1.80E−01 | l-Ribulose-5-phosphate 4-epimerase UlaF | |
| Glucose, trehalose, N-acetylglucosamine | MSMEG_2114 | 1.25 | 1.57E−02 | Glucose-6-phosphate isomerase, putative | |
| MSMEG_2115 | (1.11) | 6.32E−01 | Conserved hypothetical protein | ||
| MSMEG_2116 | ptsG | 1.33 | 1.21E−02 | PTS system, glucose-specific IIBC component | |
| MSMEG_2117 | crr | 1.32 | 2.54E−02 | b-Glucoside-specific EII permease | |
| MSMEG_2118 | nagB2 | 1.37 | 3.29E−02 | Glucosamine-6-phosphate isomerase | |
| MSMEG_2119 | nagA | 1.38 | 1.07E−02 | N-Acetylglucosamine-6-phosphate deacetylase | |
| MSMEG_2120 | pstR | (1.16) | 3.51E−01 | Tetratricopeptide repeat family protein | |
| Dihydroxyacetone | MSMEG_2121 | ptsT | 2.13 | 3.74E−04 | Multiphosphoryl transfer protein (MTP) |
| MSMEG_2122 | dhaL | 1.96 | 2.03E−03 | Dihydroxyacetone kinase, L subunit | |
| MSMEG_2123 | dhaK | 2.21 | 6.61E−05 | Dihydroxyacetone kinase, DhaK subunit | |
| MSMEG_2124 | dhaF | 2.73 | 6.19E−04 | Glycerol uptake facilitator, MIP channel | |
| MSMEG_2125 | dhaR | 1.02 | 5.76E−01 | Glycerol operon regulatory protein | |
| Ribose, ribonucleosides | MSMEG_3089 | deoC | 1.06 | 6.80E−01 | Deoxyribose-phosphate aldolase |
| MSMEG_3090 | rbsC2 | 1.75 | 1.54E−02 | Ribose transport permease protein RbsC | |
| MSMEG_3091 | rbsA2 | 1.03 | 8.12E−01 | Ribose transport ATP-binding protein RbsA | |
| MSMEG_3092 | rbsR2 | (2.08) | 5.16E−01 | Transcriptional regulator, sugar-binding family protein | |
| MSMEG_3093 | sugK | 1.15 | 5.30E−01 | Putative sugar kinase protein | |
| MSMEG_3094 | sugD | 1.58 | 4.08E−02 | Oxidoreductase, zinc-binding dehydrogenase family protein | |
| MSMEG_3095 | rbsB2 | 1.80 | 2.30E−03 | d-Ribose-binding periplasmic protein | |
| Unknown | MSMEG_3264 | abcR | 1.07 | 3.92E−01 | Transcriptional regulator |
| MSMEG_3265 | sugK | 1.38 | 3.32E−03 | Arabitol-phosphate dehydrogenase | |
| MSMEG_3266 | abcE | 1.42 | 1.78E−02 | Maltose/maltodextrin-binding protein | |
| MSMEG_3267 | abcF | 1.69 | 4.34E−02 | Transporter | |
| MSMEG_3268 | abcG | 1.19 | 5.50E−02 | ABC transporter, permease protein | |
| MSMEG_3269 | abcK1 | 1.69 | 1.58E−02 | Putative sugar ABC transporter ATP-binding protein | |
| MSMEG_3270 | abcK2 | 1.26 | 2.10E−01 | sn-Glycerol-3-phosphate ABC transporter, ATP-binding protein | |
| MSMEG_3271 | sugK | 1.24 | 2.90E−01 | Dihydroxyacetone kinase | |
| Ribose | MSMEG_3598 | rbsB3 | 1.28 | 4.65E−03 | Periplasmic sugar-binding proteins |
| MSMEG_3599 | rbsR3 | 1.42 | 1.01E−02 | Sugar-binding transcriptional regulator, LacI family protein | |
| MSMEG_3600 | (1.02) | 7.80E−01 | Hypothetical protein | ||
| MSMEG_3601 | rbsC3 | 1.53 | 6.94E−03 | Ribose/xylose/arabinose/galactoside ABC-type transport systems, permease components | |
| MSMEG_3602 | rbsA3 | 1.22 | 2.29E−01 | Ribose transport ATP-binding protein RbsA | |
| MSMEG_4170 | rbsA4 | (1.08) | 5.15E−01 | Ribose transport ATP-binding protein RbsA | |
| MSMEG_4171 | rbsC4 | 1.21 | 2.61E−01 | Ribose transport system permease protein RbsC | |
| MSMEG_4172 | rbsB4 | 1.46 | 3.36E−02 | d-Ribose-binding periplasmic protein RbsB | |
| MSMEG_4173 | rbsR4 | (1.13) | 1.63E−01 | MaoC family protein | |
| Unknown sugar | MSMEG_4655 | (1.19) | 1.89E−01 | Transcriptional regulator, AraC family protein | |
| MSMEG_4656 | 1.37 | 9.08E−03 | Sugar ABC transporter ATP-binding protein | ||
| MSMEG_4657 | 1.42 | 1.60E−02 | ABC transporter membrane protein | ||
| MSMEG_4658 | 1.49 | 4.33E−04 | Sugar ABC transporter substrate-binding protein | ||
| MSMEG_4659 | 1.02 | 6.77E−01 | GntR family protein transcriptional regulator | ||
| MSMEG_4660 | 1.14 | 3.38E−01 | Hypothetical protein | ||
| MSMEG_4661 | 1.47 | 1.02E−02 | Sugar kinase, ribokinase family protein | ||
| MSMEG_4662 | 1.27 | 1.67E−01 | Deoxyribose-phosphate aldolase superfamily protein | ||
| MSMEG_4663 | iolB | 1.78 | 1.88E−03 | IolB protein | |
| MSMEG_4664 | iolD | 1.53 | 4.17E−02 | IolD protein | |
| MSMEG_4665 | iolE | 1.30 | 4.81E−03 | IolE protein | |
| MSMEG_4666 | 1.06 | 8.33E−01 | myo-Inositol 2-dehydrogenase | ||
| MSMEG_5057 | 1.36 | 1.46E−03 | Conserved hypothetical protein | ||
| MSMEG_5058 | sugK | 1.52 | 4.32E−04 | ABC transporter, ATP-binding protein SugC | |
| MSMEG_5059 | sugG | 1.46 | 8.71E−05 | ABC transporter, permease protein SugB | |
| MSMEG_5060 | sugF | 1.37 | 1.24E−04 | ABC transporter, permease protein SugA | |
| MSMEG_5061 | sugE | 1.30 | 2.96E−04 | Bacterial extracellular solute-binding protein | |
| MSMEG_5062 | 1.11 | 1.62E−02 | Conserved hypothetical protein | ||
| Succinate | MSMEG_5302 | dctA | 1.73 | 2.57E−02 | Aerobic C4-dicarboxylate transport protein |
| Sugar alcohol | MSMEG_5571 | smoK | 1.80 | 3.06E−03 | ABC transporter ATP-binding protein |
| MSMEG_5572 | smoG | 1.76 | 1.37E−02 | Sugar ABC transporter permease protein | |
| MSMEG_5573 | smoF | 1.99 | 2.01E−02 | Sugar ABC transporter permease protein | |
| MSMEG_5574 | somE | 1.92 | 1.20E−03 | Substrate-binding protein | |
| MSMEG_5575 | smoR | 1.68 | 2.30E−03 | Repressor | |
| Xylose | MSMEG_6018 | xylG1 | 1.60 | 2.39E−02 | Xylose transport permease protein XylH |
| MSMEG_6019 | xylF1 | (1.20) | 7.50E−01 | ABC-type sugar transport system ATPase component | |
| MSMEG_6020 | xylE1 | 1.11 | 5.15E−01 | d-Xylose-binding periplasmic protein | |
| MSMEG_6021 | xylA1 | 1.65 | 2.57E−02 | Xylose isomerase | |
| MSMEG_6022 | xylR1 | (1.08) | 3.13E−01 | Xylose repressor, ROK family protein transcriptional regulator | |
| Glycerol | MSMEG_6238 | 1.70 | 3.49E−02 | Putative two-component system sensor kinase | |
| MSMEG_6239 | 1.77 | 1.44E−02 | 1,3-Propanediol dehydrogenase | ||
| MSMEG_6240 | 1.44 | 1.84E−01 | Conserved hypothetical protein | ||
| MSMEG_6241 | 1.73 | 1.15E−01 | ATPase associated with various cellular activities | ||
| MSMEG_6242 | 2.02 | 3.94E−02 | Alcohol dehydrogenase containing iron | ||
| MSMEG_6243 | 1.27 | 4.33E−02 | Response regulator receiver domain protein | ||
| MSMEG_6756 | glpK2 | 1.19 | 1.88E−02 | Glycerol kinase | |
| MSMEG_6757 | glpR | 1.47 | 5.86E−04 | Glycerol operon regulatory protein | |
| MSMEG_6758 | glpF | 1.71 | 2.11E−04 | Transport integral membrane protein | |
| MSMEG_6759 | glpK1 | 2.10 | 8.12E−04 | Glycerol kinase | |
| MSMEG_6760 | glpD | 1.13 | 9.60E−01 | Conserved hypothetical protein | |
| MSMEG_6761 | glpD2 | 2.10 | 7.42E−04 | Glycerol-3-phosphate dehydrogenase 2 | |
| Ribose | MSMEG_6802 | (1.18) | 7.28E−01 | ABC transporter ATP-binding protein | |
| MSMEG_6803 | 1.41 | 4.11E−02 | Ribose transport system permease protein RbsC | ||
| MSMEG_6804 | 1.45 | 5.03E−03 | Sugar ABC transporter substrate-binding protein |
Boldface values indicate a downregulation of 1.5-fold or greater in gene expression. Shading indicates significant fold changes with P value of <0.05. The values in parentheses are upregulated genes.
PTS, phosphotransferase; EPTC, S-ethyl dipropylcarbamothioate; EII, enzyme II; MIP, major intrinsic protein family.
Fig 2.

VapC regulation of the major glycerol-utilizing pathways identified in M. smegmatis. The schematic diagram indicates the glycerol-utilizing pathways present in M. smegmatis. The numbers in red represent gene expression ratios determined by microarray analysis in response to VapC expression. The black numbers represent gene expression ratios of selected genes involved in glycerol metabolism obtained through comparison of ΔvapBC deletion mutant mRNA levels with M. smegmatis mc2155 as determined by qRT-PCR (see Fig. 8). Two pathways for glycerol metabolism are represented and have been assembled based on putative roles for genes annotated in M. smegmatis genome. Pathway 1 indicates glycerol utilization mediated by an ATP-dependent glycerol kinase and a glycerol dehydrogenase. A second pathway (pathway 2) requires a glycerol dehydrogenase and dihydroxyacetone kinase to convert glycerol to dihydroxyacetone phosphate (DHA-P). A parallel branch in pathway 2 dehydrates glycerol by a coenzyme B12-dependent glycerol dehydratase to synthesize propanediol derivatives. The genes and the proteins they encode are as follows: glpF, glycerol uptake facilitator; glpK, glycerol kinase; glpD, glycerol-3-phosphate dehydrogenase; dhaF, glycerol uptake facilitator; dhaKLM, DHA kinase; dhaB, glycerol dehydratase; dhaT, NADH-dependent 1,3-propanediol dehydrogenase. Abbreviations: G-3-P, glycerol-3-phosphate; DHA, dihydroxyacetone; 3- and 2-HPA, 3- and 2-hydroxypropionaldehyde; TCA, tricarboxylic acid cycle. All gene expression ratios shown for individual genes are statistically significant (P value of <0.05), with the exception of glpF qRT-PCR value 1.58 (P value of 0.17). The average gene expression ratios of the 4 downregulated genes (MSMEG_1546 to MSMEG_1549) are shown for dhaB.
In response to VapC expression, genes involved in transport and scavenging of phosphate were upregulated (see Table S2 in the supplemental material). Genes belonging to the high-affinity phosphate-specific transport systems PstSCAB and PhnDCE in M. smegmatis were upregulated (1.71-, 1.85-, and 1.68-fold for pstS, pstC, and phnD transcripts, respectively) (23, 25). Also upregulated 1.67-fold was the gene that encodes the PitA low-affinity phosphate transporter (24).
VapC is a sequence-specific RNase that targets AUAU and AUAA sites.
The microarray data suggested that VapC was targeting specific mRNA transcripts involved in carbon transport and metabolism, particularly those involved in glycerol metabolism. Overexpression of VapC from M. smegmatis for the purposes of protein purification was unsuccessful due to its toxicity to the cell, but VapC can be expressed in complex with its VapB antitoxin and the VapBC protein complex can be purified (51). It is a well-known feature of toxin-antitoxin systems that the antitoxin is more susceptible to proteolytic degradation than the toxin (27, 44). Thus, functional VapC was obtained by treating the VapBC complex with trypsin and digesting the antitoxin VapB. The difference in the predicted isoelectric points of trypsin (10.5) and VapC (4.9) enabled separation of the two by anion-exchange chromatography (see Fig. S1 in the supplemental material). MALDI MS of purified VapC revealed two different species (16.33 kDa and 14.94 kDa) corresponding to VapC with and without the C-terminal His tag which is in accordance with the theoretical molecular masses of both species (16.30 kDa and 14.91 kDa). There were no MALDI MS peaks for VapB.
VapC from M. smegmatis (MSMEG_1284) belongs to the PIN-domain superfamily (NCBI annotation, PilT-domain-containing protein; Pfam number PF01850), which contains 4,089 proteins from 1,139 different species across the three domains of life (19). PIN-domain proteins are small (∼130 amino acids) and are characterized by three strictly conserved acidic residues proposed to constitute the active site. VapC from M. smegmatis contains these three acidic residues (Asp4, Asp99, and Glu118). Biochemically, the PIN-domain proteins are ribonucleases; in prokaryotes, they are typically found as the toxic member of toxin-antitoxin systems, and in eukaryotes, they are associated with nonsense-mediated decay (NMD) of RNA (58) and preprocessing of rRNA (39). VapC from H. influenzae, Shigella flexneri 2a, and Salmonella enterica and five VapC proteins from M. tuberculosis display RNase activity (1, 14, 44, 50). To assess RNase activity of VapC from M. smegmatis, we produced RNA from a set of 12 plasmids whose overlapping inserts encode every combination of five bases. Collectively, these are called pentaprobes; the first six plasmids (922 to 927) contain sequences in the forward direction, and the last six (928 to 933) contain the reverse complement sequences (i.e., 928 is the reverse complement to 922) (38). RNA was transcribed from a PCR product including a T7 promoter at the 5′ end of the pentaprobe insert. Each pentaprobe RNA was incubated in vitro with purified VapC at 37°C in an assay buffer containing 6 mM MgCl2 at pH 7.4. Negative controls included either EDTA (as PIN-domains are Mg2+/Mn2+-dependent ribonucleases) or VapBC or no VapC (to ensure RNase activity was not due to contaminating ribonucleases). VapC degraded RNA pentaprobes 925, 926, 927, and 932 but did not degrade pentaprobes 922, 923, and 924 (Fig. 3A shows pentaprobes 924, 925, and 932; for other pentaprobes tested, see Fig. S2 in the supplemental material). Different fragmentation patterns were observed on urea-denaturing polyacrylamide gels corresponding to different pentaprobe RNAs (Fig. 3A). Based on these data, VapC is a sequence-specific Mg2+-dependent RNase, as demonstrated by its abilities to cleave a subset of pentaprobe RNAs and show differing fragmentation patterns for the different pentaprobe RNA substrates. Inclusion of EDTA in the reaction mixture inhibited VapC activity consistent with a Mg2+-dependent RNase (Fig. 3A). The VapC from M. smegmatis also appears to not only cleave RNA but also to bind RNA and suggests that binding and cleavage are separable properties, and this warrants further investigation given the fact that VapCs (i.e., Rv0065 and Rv0617) from M. tuberculosis do not show this RNA binding property (1). VapC cleaves pentaprobe 932 RNA most efficiently and demonstrates no nuclease activity against its single-stranded DNA (ssDNA), dsDNA, and dsRNA equivalents (Fig. 3B). Secondary structures in the dsRNA are present (Fig. 3B, dsRNA, top band) that VapC appears to bind to, as shown by the disappearance of the top band and a slight shift in the bottom band upon the addition of VapC. Intriguingly, the cleavage of the 932 RNA substrate is far more efficient than cleavage of corresponding short oligonucleotides, and this suggests that additional features such as RNA secondary structure play a role in VapC-mediated mRNA degradation.
Fig 3.

in vitro RNase activity of VapC. (A) VapC displays sequence-specific RNase activity against pentaprobe RNA molecules 925 and 932 but not 924 in the presence of 6 mM MgCl2. Negative controls show no nonspecific degradation of RNA due to contaminating ribonucleases and include the following: RNA-only controls (−VapC) and EDTA, addition of 12 mM EDTA to the assay reaction mixture and VapBC, and the addition of purified VapBC complex instead of VapC to the assay reaction mixture. The addition of VapC (+VapC) results in degradation of the 925 and 932 pentaprobe RNA molecules over a period of 5 to 60 min (time indicated in minutes across the top of gels) but not the 924 RNA molecule, indicating that VapC is a sequence-specific RNase. A molecular size marker at the left side of the gel shows molecular sizes of single-stranded RNA (ssRNA) in bases. (B) VapC does not cleave dsDNA, ssDNA, or dsRNA. VapC does not cleave 932 dsRNA, 927 ssDNA, or 927 dsDNA (+VapC) but cleaves ssRNA of the same sequence. Negative controls (−VapC) show no nuclease contamination.
To determine the sequence specificity of VapC, short overlapping RNA oligonucleotides were designed to cover the sequence of the parent 932 pentaprobe RNA substrate. VapC cleaves four of the nine short RNA oligonucleotides, and MALDI MS was used to identify the VapC cleavage sites in three of the four short oligonucleotides (Fig. 4A). VapC cleaves RNA oligonucleotides at AUAA and AUAU sequences yielding a 5′ phosphate on the 3′ cleavage product. Less efficient cleavage at other sites (for example GAUG) was also observed (Fig. 4B and C). Three bases on either side of the cleavage sites in 932 RNA oligonucleotides 3, 4, and 5 were analyzed using the online program WebLogo (13), and the VapC consensus sequence was determined (Fig. 4C). Thus, the optimal sequences for VapC cleavage from M. smegmatis is AUAU and AUAA leaving a 5′ phosphate on the 3′ cleavage product consistent with a metal-dependent RNase (2). The “wobble” in the target sequence indicates that other “suboptimal” sequences will also be cleaved by VapC (albeit much less efficiently). Indeed, it is likely that the optimal substrate is a combination of the target sequence with a particular RNA secondary structure.
Fig 4.

VapC from M. smegmatis targets AU-rich sequences. (A) MALDI-TOF MS spectra for 932 RNA oligonucleotide 3 RNase assay. Negative controls show no contaminating ribonucleases in the protein preparation (EDTA) or the assay buffer (−VapC). The m/z values for each RNA fragment are shown above each peak. Identification of VapC cleavage sites (+VapC 15, 30, and 60 min) in 932 RNA oligonucleotide 3 are shown in panel B. (B) The 932 RNA oligonucleotide 3 is shown by colored bases, and the cut sites (arrows) at AUAU, AUAA, and GAUG are shown with the corresponding m/z values. (C) Analysis of VapC cleavage sites across 932 RNA oligonucleotides 3, 4, and 5 as calculated by WebLogo. VapC cleaves after position 3. The height of the letter is proportional to the frequency of that base at that particular position. The positions are labeled 1 to 6.
VapC cleaves RNA transcripts in vitro that are downregulated by VapC expression in vivo.
A cohort of genes downregulated in response to conditional VapC expression in the M. smegmatis ΔvapBC mutant were identified by microarray analysis in Table S2 in the supplemental material). An annotated operon encoding genes involved in glycerol uptake and metabolism (MSMEG_2124, -2123, -2122, and -2121) (Fig. 2) included the ATAA target sequence in the 5′UTR of MSMEG_2124 and the ATAT target sequence in the 3′ end of MSMEG_2121, the last gene in the operon. All genes in this operon were downregulated in response to overexpression of VapC (Table 2), suggesting that the operon mRNA is destabilized by cleavage at each end, resulting in degradation of the entire mRNA. Reverse transcriptase PCR (RT-PCR) confirmed that the 5′UTR for the operon included the ATAA target sequence 95 bases upstream of the MSMEG_2124 start codon (data not shown). The genes encoding MSMEG_2124 (with and without the 5′UTR), MSMEG_2123, MSMEG_2122, and MSMEG_2121 were PCR amplified, and the addition of a T7 promoter to the 5′ end allowed for production of the RNA transcript for each gene (Fig. 5). The gene encoding MSMEG_0650 (phnF) that exhibited no change in the microarray data (Table S2) and does not contain the cleavage site was also transcribed to produce mRNA. VapC shows RNase activity against MSMEG_2121 mRNA in vitro and to a lesser extent toward MSMEG_2124 that includes the 5′UTR (Fig. 5). Removal of the MSMEG_2124 5′UTR that includes the ATAA cut site greatly reduces the RNase activity of VapC against this transcript, indicating that VapC is cleaving at ATAA. Other transcripts in the operon that do not contain the cut site exhibit a small amount of RNA degradation by VapC (RNA transcripts MSMEG_2122 and MSMEG_2123 due to cleavage of suboptimal sequences) (Fig. 5). The RNase activity of VapC was higher against these biologically relevant substrates (M. smegmatis mRNA transcripts) compared to the synthetic pentaprobe RNAs tested above (61.35 pmol of VapC was used in the pentaprobe assays, whereas 1.23 pmol of VapC was used in the M. smegmatis mRNA transcript assays). VapC does not cleave MSMEG_0650, suggesting that VapC is directly targeting the downregulated transcripts by cleaving at AUAA or AUAU sites (Fig. 5, bottom panel) to destabilize the mRNA, resulting in its degradation and subsequent downregulation of all genes in the operon. This mechanism of mRNA destabilization by cleavage at 5′ and 3′ ends has been well studied in E. coli and Bacillus subtilis (11, 37). In addition, VapC displayed no activity against tRNAfMet (MSMEG_1965) from M. smegmatis at high (61.35 pmol) and low (1.23 pmol) concentrations of VapC (see Fig. S3 in the supplemental material). The VapC cut site AUAA is present in tRNAfMet but is not cut, strengthening the idea that the optimal substrate is a combination of sequence and RNA secondary structure.
Fig 5.

VapC cleaves mRNA transcripts in vitro that are downregulated by conditional expression of VapC in vivo. The schematic diagram at the top of the figure represents the MSMEG_2124 operon downregulated in vivo upon VapC induction. The VapC cut site ATAA is present in a conserved stem-loop in the known 5′UTR, and ATAT is also present in a stem-loop in MSMEG_2121 at the end of the operon (cut sites are labeled and indicated by scissors). VapC displays sequence-specific RNase activity against MSMEG_2121 and MSMEG_2124 including 5′UTR, but not MSMEG_2123 and MSMEG_2122 in the presence of 6 mM MgCl2 (time points are indicated along the top of the gels in minutes). MSMEG_0650 does not contain the VapC cut site and is not cleaved by VapC in vitro in the presence of 6 mM MgCl2 (MSMEG_0650 exhibits no change upon VapC induction in vivo). Negative controls show no nonspecific degradation of RNA due to contaminating ribonucleases and include RNA-only controls (−VapC) and 20 mM EDTA added to the assay reaction. A molecular size marker at the left side of the gel shows the molecular sizes of ssRNA in bases.
Optimal mRNA substrate for M. smegmatis VapC.
The experimental data for RNA cleavage by VapC pointed strongly to the involvement of RNA secondary structure contributing to the VapC recognition motif. We took the three substrates that had been experimentally shown to be efficiently cut by M. smegmatis VapC and used the local RNA prediction algorithm RNALfold to look for secondary structures associated with the three experimentally cut sequences (32). All three RNA substrates showed a predicted stable hairpin immediately downstream of the AUA(U/A) sequence (Fig. 6A). Following manual alignment of these three sequences along with their secondary structure motifs, we built a covariance model (CM) (46). CMs are probabilistic models that can be used to score related sequences incorporating both sequence and structure similarity. By using independent benchmarks, the accuracy of CMs is greater than alternative methods (22). The substrate CM was then used to search 39 genes and flanking sequences [containing 62 AUA(U/A) sites] for analogous motifs. The high-scoring matches to this CM were incorporated into the alignment. The alignment and consensus secondary structure were manually refined, and the CM was then rebuilt and researched against the 39 genes. This process was iterated for several cycles until no further high-scoring matches were found. The resulting annotated, consensus structure is shown in Fig. 6B.
Fig 6.

Bioinformatic characterization of the VapC target mRNAs. (A) Predicted RNA secondary structures for the 3 experimentally validated VapC targets found in the MSMEG_2124 5′ UTR, MSMEG_2121 mRNA, and the artificial pentaprobe 932 sequence. (B) Consensus model of predicted VapC targets. (C) Cumulative distribution functions (CDFs) of covariance scores for sequences matching the consensus model (CM). The top panel shows the operon sequences containing at least one downregulated gene and corresponding controls. The bottom panel shows the distribution of scores matching tRNA sequences and a shuffled control. The numbers of searched sequences (N) and matches (M) are indicated in the legends. (D) Schematic diagrams showing the presence or absence of the optimal RNA substrate motif for various RNA substrates (oligo, oligonucleotide). RNase activity was scored for substrates tested. RNase activity is indicated as follows: +, weak activity; ++++, strong activity; X, no activity. Substrates with the predicted optimal structure are cleaved more efficiently than those with other secondary structures.
This refined CM model was then tested against several sets of RNA sequences: genes downregulated in response to VapC and genes upregulated in response to VapC and shuffled control sequences for each set. The distribution of CM bit-scores shows a significant difference in the downregulated operon sequences versus the two different control sets of operons containing upregulated genes and shuffled control sequences (Fig. 6C). A two-sample Kolmogorov-Smirnov test of the scores from the downregulated operons gives a P value of 6.631E−05. We also tested whether the M. smegmatis tRNAs contained matches to the CM. We found that in contrast to earlier work (62), the tRNAs were deficient in predicted VapC targets relative to shuffled control sequences. This predicted RNA motif of -AUAW-hairpin-G- sheds light on previously observed cleavage patterns for a range of substrates tested in the laboratory (Fig. 6D). These experimental results add weight to the predicted optimal substrate for M. smegmatis VapC. The precise role that each position in the RNA motif plays in efficient cleavage is the subject of ongoing work.
VapC is a posttranscriptional regulator of glycerol metabolism in mycobacteria.
The microarray and VapC RNA cleavage assays demonstrated that VapC was targeting mRNA transcripts involved in glycerol transport and metabolism. We hypothesize that this could be a mechanism to control the rate of carbon utilization in response to the requirements of the cells (e.g., energy production to drive anabolic reactions). To test this hypothesis, the wild type and the ΔvapBC mutant were grown in minimal medium containing glycerol, and several parameters of energy were measured (glycerol utilization and the molar growth yield on glycerol). When grown in either LBT (Fig. 7A) or HdB minimal medium with glycerol (0.2% vol/vol) as the sole carbon source (Fig. 7B), the ΔvapBC mutant consistently grew faster (2.7 h doubling time) than the wild-type cells (3.2-h doubling time). When the ΔvapBC mutant was complemented with VapBC, the mutant grew slower (3.5-h doubling time) than the wild-type cells, demonstrating that VapBC was having an effect on the growth rate of M. smegmatis (Fig. 7C) and showing an overcompensation of VapBC levels in the complementing strain. This growth rate phenomenon was subtle, and we had previously discounted its significance (49). However, we revisited and remeasured these data in light of the emerging hypothesis herein.
Fig 7.
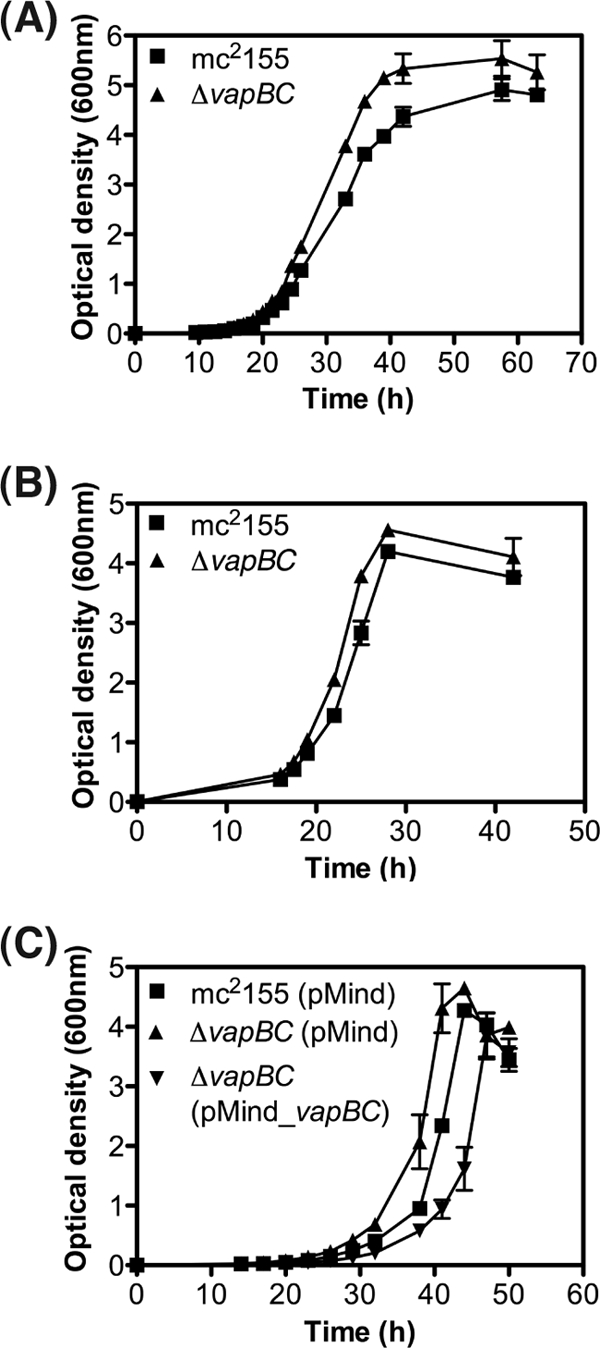
Comparison of growth of the wild-type M. smegmatis mc2155 and ΔvapBC deletion mutant. (A and B) Comparative growth analysis of wild-type M. smegmatis mc2155 and ΔvapBC deletion strain were performed using rich medium LBT (A) and minimal medium HdB containing 0.2% glycerol (B). Growth was determined by optical density at 600 nm over time (hours). (C) Complementation of the ΔvapBC mutant was performed using tetracycline-inducible pMind_vapBC construct. For complementation, empty vector (pMind) controls were used in the wild-type mc2155 strain and the ΔvapBC deletion mutant. Complementation was performed in HdB medium containing 0.2% glycerol. Results for all growth experiments are from three biological replicates, and the error bars represent standard deviations.
To determine the metabolic basis for this higher growth rate in the ΔvapBC mutant, we measured the specific rate of glycerol consumption required for growth by the wild type and ΔvapBC mutant during early exponential growth (Fig. 8A, time points T1 and T2). The specific rate of glycerol consumption by the ΔvapBC mutant was 2.4-fold higher (3.4 ± 0.6 mmol glycerol/h/g [dry weight] cells) than that of the wild type (1.4 ± 0.3 mmol glycerol/h/g [dry weight] cells) (time point 1), and this was consistent for two sampling points taken during exponential growth (Fig. 8B). To determine whether this increased rate of glycerol consumption was coupled to growth, we measured the molar growth yield on glycerol (Yglycerol) (Fig. 8C). This analysis revealed that the increased rate of glycerol consumption was not used for generating bacterial biomass, as the Yglycerol was 76 ± 19 g [dry weight] cells/mol glycerol utilized, for the ΔvapBC mutant, and it was 182 ± 21 g [dry weight] cells/mol glycerol utilized for the wild type, suggesting that glycerol consumption was uncoupled from biomass generation in the ΔvapBC mutant (time point 1 [Fig. 8C]). These data suggest that genes involved in glycerol utilization, controlled by VapC, could be upregulated in a ΔvapBC mutant relative to the wild type. To test this hypothesis, quantitative RT-PCR was performed to determine the levels of VapC-targeted mRNA transcripts in the ΔvapBC mutant compared with the wild-type M. smegmatis strain mc2155 during exponential growth on glycerol (Fig. 9). Genes involved in glycerol and dihydroxyacetone metabolism were all significantly upregulated in the ΔvapBC mutant relative to the wild type, which is consistent with this hypothesis (Fig. 9 and summarized in Fig. 2).
Fig 8.

Comparative analysis of glycerol consumption and molar growth yield on glycerol for the wild type and the ΔvapBC mutant. (A) Supernatant samples and dry weights were taken for strain mc2155 (wild type) and the ΔvapBC deletion mutant at two time points during the early stages of growth (T1 and T2). (B) Specific rate of glycerol consumption (mmol glycerol consumed/h/g [dry weight] cells). (C) Molar growth yield on glycerol (Yglycerol) (g [dry weight] cells/mol glycerol utilized) was calculated for each strain. The glycerol consumption rate and Yglycerol were determined for three replicate samples, and the error bars represent standard deviations.
Fig 9.

Comparison of VapC-targeted mRNA transcripts in M. smegmatis mc2155 and the ΔvapBC deletion strain. Quantitative RT-PCR was performed to determine the levels of VapC-targeted mRNA transcripts in the ΔvapBC mutant compared with the wild-type M. smegmatis strain mc2155. The cells were grown to an OD600 of 0.1 in batch culture in HdB medium containing 0.2% glycerol and then harvested for RNA extraction. Differential gene expression changes between the ΔvapBC mutant and mc2155 were determined (∗, P value of <0.05; ∗∗, P value of <0.005). Error bars represent standard deviations from three biological replicates.
DISCUSSION
Efficient growth of a bacterial cell is achieved through the fine-tuning of anabolic (energy-consuming) and catabolic reactions (energy-producing) to achieve maximum growth rate and carbon source utilization (53). This is particularly pertinent to bacterial cells that inhabit natural environments where nutrients are often limiting for growth and coupling of substrate utilization to the production of cells is a major determinant of ecological success. In the laboratory, bacteria are generally grown with an excess of nutrients (carbon and energy rich), and therefore, bacteria may instigate mechanisms to uncouple catabolism from anabolism, enabling them to consume the carbon and energy source faster than it is required for growth. Such a strategy is exemplified by Escherichia coli, which excretes acetate at high rates of glucose consumption even in the presence of high oxygen (60). During growth on glycerol, mycobacteria are proposed to exhibit unregulated growth. In both of these cases, the molecular mechanisms controlling the rate of uncoupling in bacterial growth and the mechanisms whereby excess energy is spilled remain to be elucidated. For many bacteria, one of the main by-products associated with unregulated carbon dissimilation is methylglyoxal, a strong electrophile. Mycobacteria have recently been proposed to produce methylgloxal and are inhibited by excess sugar phosphates (36, 48). Based on these observations, mycobacteria must have evolved control mechanisms that balance the rate of carbon utilization with growth to prevent the buildup of excess sugar phosphates and methylgloxal in the cell.
In this communication, we propose a new mechanism for the control of carbon (sugar) utilization in mycobacteria mediated by the toxin-antitoxin module VapBC. When VapC, a RNase, was overexpressed in Mycobacterium smegmatis, the transcriptional response suggested that VapC was targeting (downregulating) specific mRNA transcripts involved in carbon transport and metabolism, particularly those involved in glycerol metabolism. In response to VapC overexpression, we also observed a number of upregulated genes involved in phosphate uptake. When bacterial cells have an excess of sugar phosphates, methylgloxal is produced, leading to a release of phosphate. Indeed, methylgloxal production has been proposed as a mechanism to replenish intracellular phosphate. These observations suggest a potential link between VapC and phosphate: if VapC downregulates sugar metabolism and hence sugar phosphates do not accumulate, the mycobacterial cell may upregulate phosphate transporters to compensate for this apparent phosphate limitation.
Mycobacteria are generally cultured using glycerol as the standard carbon source, but detailed knowledge on the biochemical processes associated with glycerol uptake, metabolism, and regulation are poorly understood (47, 48). Beste et al. (7) have reported that glycerol kinase is essential for growth of M. tuberculosis in minimal medium containing glycerol, giving validity to the pathway proposed in Fig. 2. On the basis of our microarray analysis, we propose that M. smegmatis has two pathways for the utilization of glycerol (Fig. 2). We base this scheme on the following observations; aerobic bacteria generally utilize a glycerol metabolic pathway that involves an ATP-dependent glycerol kinase and glycerol 3-phosphate dehydrogenases (16). This pathway was downregulated in M. smegmatis along with an associated glycerol facilitator in response to VapC overexpression. A second pathway for glycerol metabolism has been reported to operate in E. coli under fermentative (microaerobic) conditions (16, 28) and uses a type II glycerol dehydrogenase and dihydroxyacetone kinase (DhaKLM) to synthesize dihydroxyacetone phosphate. We could not identify a type II glycerol dehydrogenase homologue, but dhaKLM is present (Fig. 2). We were able to identify a number of genes that were downregulated in response to VapC, which fed into pathways leading to the production of dihydroxyacetone phosphate, the major source of the toxic intermediate for methylglyoxal synthesis.
To test the role of VapC-mediated regulation of glycerol metabolism in vivo, we measured the kinetics and energetics of glycerol utilization by the wild type and an isogenic ΔvapBC mutant. We rationalized that a ΔvapBC mutant should consume glycerol at a higher rate than wild-type cells, and because of this faster utilization rate, uncoupled growth would occur, leading to an overall lower molar growth yield on glycerol. Moreover, genes involved in glycerol utilization should be upregulated in a ΔvapBC mutant relative to the wild type. Both predictions were validated experimentally with glycerol-grown cells in batch culture and qPCR showing that in the ΔvapBC mutant, the levels of transcripts involved in glycerol utilization were indeed elevated during growth on glycerol compared to transcript levels in the wild type. In support of this model, we demonstrated that purified VapC cleaved the dhaKLM operon in vitro, and we identified the specific RNA motif that is the target of VapC RNase activity. We propose that VapBC provides the cell with an elegant mechanism to regulate sugar metabolism in M. smegmatis by targeting specific genes at a posttranscriptional level involved in carbon source utilization in response to the anabolic demands of the cell (e.g., fast or slow growth), thus acting as a link between energy-producing and energy-consuming reactions in the cell. The targeting of mRNA allows the cell to fine-tune its rate of glycerol utilization without the need for complicated transcriptional scenarios allowing for rapid adjustments of catabolic flux to prevent toxic intermediates accumulating. The question remained as to what the cells do with the glycerol consumption that is in excess of growth requirements? At this point, we can only speculate that this could be associated with either increased ATP production and spilling by a futile cycle and/or directed into storage compounds (53). in vitro RNase activity has been reported for VapC proteins previously (1, 14, 44, 50). Two recent papers have connected this biochemical activity to a biological response in diverse microorganisms. vapBC6 from Sulfolobus solfataricus has been shown to be upregulated in response to heat shock, and the RNase activity of VapC6 was shown to directly target transcripts encoding oligopeptide permease (dppB-1) and a tetR transcription factor (40). VapCs from the enteric bacteria Shigella flexneri and Salmonella enterica have also been shown to be sequence-specific RNases cleaving initiator tRNA at the anticodon stem-loop and thus inhibiting translation (62). These results are consistent with our findings for VapC in M. smegmatis insofar as the RNase activity of VapC targets specific RNA substrates in vivo. Our data show that VapC in M. smegmatis is a posttranscriptional regulator of carbon metabolism, whereas the data from the enteric bacteria suggest a role for VapC in the global control of translation. These alternative hypotheses are compatible, as there is no expectation that the biochemical RNase activity of VapC should play equivalent roles in bacteria from such diverse branches of the prokaryotic tree.
The results presented here rule out tRNA as the target for M. smegmatis VapC. The optimal target sequence for M. smegmatis VapC is a sequence and structure motif characterized by an AUA(U/A) head, a short 3′ RNA hairpin, followed by a G tail. This is present in 75% (45/60 operons) of the operons containing downregulated genes and is not present in tRNA (Fig. 8C). The results of our in vitro assays presented here and previously (1) show that the VapC RNases target these optimal sites with fast kinetics (<1 min). However, under conditions where VapC is at relatively high concentrations, suboptimal sites will be cleaved with slow kinetics (over 5 to 60 min). In a biological setting where the vapBC operon is constitutively expressed at very low levels (51), only the optimal target sites (with inherently fast cleavage kinetics) will be relevant.
The general mechanism for mRNA degradation in mycobacteria has not been well studied; however, it has been well characterized in E. coli (37) and Bacillus subtilis (11). A major determinant of mRNA stability in both organisms is the 5′UTR and 3′ end of the transcript (9, 11). These transcript ends often contain stem-loops and other secondary structures that provide a barrier to exoribonuclease degradation (57). Many genes downregulated by VapC include the target sequence in the 5′UTR region and/or the 3′ end of the transcript. Where whole operons are downregulated, the target sequences are often found in the first and last genes of the operon (e.g., MSMEG_2124 to MSMEG_2121). The propensity for VapC from M. smegmatis to target a combination of secondary structure and specific sequences at 5′ and 3′ transcript ends adds validity to our hypothesis.
M. smegmatis harbors a single VapBC module, whereas slowly growing M. tuberculosis harbors 45 vapBC TA systems with very few sugar transporters (59). No glycerol-specific uptake systems have been identified in M. tuberculosis, and it has been suggested that facilitated diffusion of glycerol might be sufficient given the low growth rate (47). This suggests that M. tuberculosis has no mechanism for regulating sugar metabolism at the level of transport, and therefore, tighter control of catabolic pathways might be required. Recent work has shown that M. tuberculosis does not exhibit substrate preferences, i.e., they are able to cometabolize several substrates simultaneously (15). This is achieved by a compartmentalized scheme for metabolism, and therefore, the need for more than one VapBC module may be anticipated. The genetic requirement in M. tuberculosis for two putative VapC proteins (Rv2103c and Rv2494) for strain fitness at a low growth rate and an antitoxin gene (Rv0596c) for fitness at a high growth rate (7) lend support to this argument and show obvious routes to testing VapC-mediated regulation and its biological consequences in M. tuberculosis.
Supplementary Material
ACKNOWLEDGMENTS
This research was funded by the Health Research Council New Zealand and Marsden Fund, Royal Society of New Zealand. J.R. and J.L.M. were funded by Ph.D. scholarships from the Tertiary Education Commission and the University of Waikato, respectively.
Footnotes
Published ahead of print 24 February 2012
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1. Ahidjo BA, et al. 2011. VapC toxins from Mycobacterium tuberculosis are ribonucleases that differentially inhibit growth and are neutralized by cognate VapB antitoxins. PLoS One 6:e21738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Anantharaman V, Aravind L. 2006. The NYN domains: novel predicted RNAses with a PIN domain-like fold. RNA Biol. 3:18–27 [DOI] [PubMed] [Google Scholar]
- 3. Arcus VL, McKenzie JL, Robson J, Cook GM. 2011. The PIN-domain ribonucleases and the prokaryotic VapBC toxin-antitoxin array. Protein Eng. Des. Sel. 24:33–40 [DOI] [PubMed] [Google Scholar]
- 4. Arcus VL, Rainey PB, Turner SJ. 2005. The PIN-domain toxin-antitoxin array in mycobacteria. Trends Microbiol. 13:360–365 [DOI] [PubMed] [Google Scholar]
- 5. Audoly G, et al. 2011. Effect of rickettsial toxin VapC on its eukaryotic host. PLoS One 6:e26528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Berney M, Cook GM. 2010. Unique flexibility in energy metabolism allows mycobacteria to combat starvation and hypoxia. PLoS One 5:e8614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Beste DJ, et al. 2009. The genetic requirements for fast and slow growth in mycobacteria. PLoS One 4:e5349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bodogai M, et al. 2006. The ntrPR operon of Sinorhizobium meliloti is organised and functions as a toxin-antitoxin module. Mol. Plant Microbe Interact. 19:811–822 [DOI] [PubMed] [Google Scholar]
- 9. Bricker AL, Belasco JG. 1999. Importance of a 5′-stem-loop for longevity of papA mRNA in Escherichia coli. J. Bacteriol. 181:3587–3590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cole ST, et al. 1998. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393:537–544 [DOI] [PubMed] [Google Scholar]
- 11. Condon C. 2003. RNA processing and degradation in Bacillus subtilis. Microbiol. Mol. Biol. Rev. 67:157–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cox RA, Cook GM. 2007. Growth regulation in the mycobacterial cell. Curr. Mol. Med. 7:231–245 [DOI] [PubMed] [Google Scholar]
- 13. Crooks GE, Hon G, Chandonia JM, Brenner SE. 2004. WebLogo: a sequence logo generator. Genome Res. 14:1188–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Daines DA, Wu MH, Yuan SY. 2007. VapC-1 of nontypeable Haemophilus influenzae is a ribonuclease. J. Bacteriol. 189:5041–5048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. de Carvalho LP, et al. 2010. Metabolomics of Mycobacterium tuberculosis reveals compartmentalized co-catabolism of carbon substrates. Chem. Biol. 17:1122–1131 [DOI] [PubMed] [Google Scholar]
- 16. Durnin G, et al. 2009. Understanding and harnessing the microaerobic metabolism of glycerol in Escherichia coli. Biotechnol. Bioeng. 103:148–161 [DOI] [PubMed] [Google Scholar]
- 17. Eddy SR. 2002. A memory-efficient dynamic programming algorithm for optimal alignment of a sequence to an RNA secondary structure. BMC Bioinformatics 3:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Eddy SR, Durbin R. 1994. RNA sequence analysis using covariance models. Nucleic Acids Res. 22:2079–2088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Finn RD, et al. 2010. The Pfam protein families database. Nucleic Acids Res. 38:D211–D222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Forage RG, Foster MA. 1982. Glycerol fermentation in Klebsiella pneumoniae: functions of the coenzyme B12-dependent glycerol and diol dehydratases. J. Bacteriol. 149:413–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Forage RG, Lin EC. 1982. DHA system mediating aerobic and anaerobic dissimilation of glycerol in Klebsiella pneumoniae NCIB 418. J. Bacteriol. 151:591–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Freyhult EK, Bollback JP, Gardner PP. 2007. Exploring genomic dark matter: a critical assessment of the performance of homology search methods on noncoding RNA. Genome Res. 17:117–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gebhard S, Cook GM. 2008. Differential regulation of high-affinity phosphate transport systems of Mycobacterium smegmatis: identification of PhnF, a repressor of the phnDCE operon. J. Bacteriol. 190:1335–1343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gebhard S, Ekanayaka N, Cook GM. 2009. The low-affinity phosphate transporter PitA is dispensable for in vitro growth of Mycobacterium smegmatis. BMC Microbiol. 9:254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gebhard S, Tran SL, Cook GM. 2006. The Phn system of Mycobacterium smegmatis: a second high-affinity ABC-transporter for phosphate. Microbiology 152:3453–3465 [DOI] [PubMed] [Google Scholar]
- 26. Gerdes K. 2000. Toxin-antitoxin modules may regulate synthesis of macromolecules during nutritional stress. J. Bacteriol. 182:561–572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gerdes K, Christensen SK, Lobner-Olesen A. 2005. Prokaryotic toxin-antitoxin stress response loci. Nat. Rev. Microbiol. 3:371–382 [DOI] [PubMed] [Google Scholar]
- 28. Gonzalez R, Murarka A, Dharmadi Y, Yazdani SS. 2008. A new model for the anaerobic fermentation of glycerol in enteric bacteria: trunk and auxiliary pathways in Escherichia coli. Metab. Eng. 10:234–245 [DOI] [PubMed] [Google Scholar]
- 29. Gonzalez-Pajuelo M, Meynial-Salles I, Mendes F, Soucaille P, Vasconcelos I. 2006. Microbial conversion of glycerol to 1,3-propanediol: physiological comparison of a natural producer, Clostridium butyricum VPI 3266, and an engineered strain, Clostridium acetobutylicum DG1(pSPD5). Appl. Environ. Microbiol. 72:96–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hayes CS, Sauer RT. 2003. Toxin-antitoxin pairs in bacteria: killers or stress regulators? Cell 112:2–4 [DOI] [PubMed] [Google Scholar]
- 31. Hayes F. 2003. Toxins-antitoxins: plasmid maintenance, programmed cell death, and cell cycle arrest. Science 301:1496–1499 [DOI] [PubMed] [Google Scholar]
- 32. Hofacker IL, Priwitzer B, Stadler PF. 2004. Prediction of locally stable RNA secondary structures for genome-wide surveys. Bioinformatics 20:186–190 [DOI] [PubMed] [Google Scholar]
- 33. Hopper S, et al. 2000. Isolation of Neisseria gonorrhoeae mutants that show enhanced trafficking across polarized T84 epithelial monolayers. Infect. Immun. 68:896–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Inouye M. 2006. The discovery of mRNA interferases: implication in bacterial physiology and application to biotechnology. J. Cell. Physiol. 209:670–676 [DOI] [PubMed] [Google Scholar]
- 35. Jaffe A, Ogura T, Hiraga S. 1985. Effects of the ccd function of the F plasmid on bacterial growth. J. Bacteriol. 163:841–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kalscheuer R, et al. 2010. Self-poisoning of Mycobacterium tuberculosis by targeting GlgE in an alpha-glucan pathway. Nat. Chem. Biol. 6:376–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kushner SR. 2002. mRNA decay in Escherichia coli comes of age. J. Bacteriol. 184:4658–4665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kwan AH, Czolij R, MacKay JP, Crossley M. 2003. Pentaprobe: a comprehensive sequence for the one-step detection of DNA-binding activities. Nucleic Acids Res. 31:e124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lamanna AC, Karbstein K. 2009. Nob1 binds the single-stranded cleavage site D at the 3′end of 18S rRNA with its PIN domain. Proc. Natl. Acad. Sci. U. S. A. 106:14259–14264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Maezato Y, et al. 2011. VapC6, a ribonucleolytic toxin regulates thermophilicity in the crenarchaeote Sulfolobus solfataricus. RNA 17:1381–1392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Magnuson RD. 2007. Hypothetical functions of toxin-antitoxin systems. J. Bacteriol. 189:6089–6092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mathews D. 1 March 2006. RNA secondary structure analysis using RNAstructure. Curr. Protoc. Bioinformatics Chapter 12, Unit 12.6. doi:10.1002/0471250953.bi1206s13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mattison K, Wilbur JS, So M, Brennan RG. 2006. Structure of FitAB from Neisseria gonorrhoeae bound to DNA reveals a tetramer of toxin-antitoxin heterodimers containing PIN domains and ribbon-helix-helix motifs. J. Biol. Chem. 281:37942–37951 [DOI] [PubMed] [Google Scholar]
- 44. Miallau L, et al. 2009. Structure and proposed activity of a member of the VapBC family of toxin-antitoxin systems. VapBC-5 from Mycobacterium tuberculosis. J. Biol. Chem. 284:276–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nawrocki EP, Eddy SR. 2007. Query-dependent banding (QDB) for faster RNA similarity searches. PLoS Comput. Biol. 3:e56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nawrocki EP, Kolbe DL, Eddy SR. 2009. Infernal 1.0: inference of RNA alignments. Bioinformatics 25:1335–1337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Niederweis M. 2008. Nutrient acquisition by mycobacteria. Microbiology 154:679–692 [DOI] [PubMed] [Google Scholar]
- 48. Pethe K, et al. 2010. A chemical genetic screen in Mycobacterium tuberculosis identifies carbon-source-dependent growth inhibitors devoid of in vivo efficacy. Nat. Commun. 1:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pinter JK, Hayashi JA, Watson JA. 1967. Enzymic assay of glycerol, dihydroxyacetone, and glyceraldehyde. Arch. Biochem. Biophys. 121:404–414 [DOI] [PubMed] [Google Scholar]
- 50. Ramage HR, Connolly LE, Cox JS. 2009. Comprehensive functional analysis of Mycobacterium tuberculosis toxin-antitoxin systems: implications for pathogenesis, stress responses, and evolution. PLoS Genet. 5:e1000767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Robson J, McKenzie JL, Cursons R, Cook GM, Arcus VL. 2009. The vapBC operon from Mycobacterium smegmatis is an autoregulated toxin-antitoxin module that controls growth via inhibition of translation. J. Mol. Biol. 390:353–367 [DOI] [PubMed] [Google Scholar]
- 52. Rozenski J. 1999. Mongo Oligo Mass Calculator v2.06. http://library.med.utah.edu/masspec/mongo.htm
- 53. Russell JB, Cook GM. 1995. Energetics of bacterial growth: balance of anabolic and catabolic reactions. Microbiol. Rev. 59:48–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sevin EW, Barloy-Hubler F. 2007. RASTA-Bacteria: a web-based tool for identifying toxin-antitoxin loci in prokaryotes. Genome Biol. 8:R155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Smeulders MJ, Keer J, Speight RA, Williams HD. 1999. Adaptation of Mycobacterium smegmatis to stationary phase. J. Bacteriol. 181:270–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Snapper SB, Melton RE, Mustafa S, Kieser T, Jacobs WR., Jr 1990. Isolation and characterization of efficient plasmid transformation mutants of Mycobacterium smegmatis. Mol. Microbiol. 4:1911–1919 [DOI] [PubMed] [Google Scholar]
- 57. Steege DA. 2000. Emerging features of mRNA decay in bacteria. RNA 6:1078–1090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Takeshita D, Zenno S, Lee WC, Saigo K, Tanokura M. 2007. Crystal structure of the PIN domain of human telomerase-associated protein EST1A. Proteins 68:980–989 [DOI] [PubMed] [Google Scholar]
- 59. Titgemeyer F, et al. 2007. A genomic view of sugar transport in Mycobacterium smegmatis and Mycobacterium tuberculosis. J. Bacteriol. 189:5903–5915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Vemuri GN, Altman E, Sangurdekar DP, Khodursky AB, Eiteman MA. 2006. Overflow metabolism in Escherichia coli during steady-state growth: transcriptional regulation and effect of the redox ratio. Appl. Environ. Microbiol. 72:3653–3661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Villas-Boas SG, Bruheim P. 2007. Cold glycerol-saline: the promising quenching solution for accurate intracellular metabolite analysis of microbial cells. Anal. Biochem. 370:87–97 [DOI] [PubMed] [Google Scholar]
- 62. Winther KS, Gerdes K. 2011. Enteric virulence associated protein VapC inhibits translation by cleavage of initiator tRNA. Proc. Natl. Acad. Sci. U. S. A. 108:7403–7407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Workman C, Krogh A. 1999. No evidence that mRNAs have lower folding free energies than random sequences with the same dinucleotide distribution. Nucleic Acids Res. 27:4816–4822 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.