

Abstract
At 37°C, the structure of poliovirus is dynamic, and internal polypeptides VP4 and N terminus of VP1 (residues 1 to 53) externalize reversibly. An Fab fragment of a monospecific antibody, which binds to residues 39 to 55 of VP1, was utilized to locate the N termini of VP1 in native (160S) particles in this “breathing” state. Fab and virus were mixed and imaged via cryogenic electron microscopy. The resulting reconstruction showed the capsid expands similarly to the irreversibly altered cell entry intermediate (135S) particle, but the N terminus of VP1 is located near the 2-fold axes, instead of the “propeller tip” as in 135S particles.
TEXT
The viral capsids of many nonenveloped viruses reversibly expose internal, unexposed amino acid sequences under physiological conditions. This dynamic behavior is often termed “breathing.” “Breathing” in nonenveloped viruses has been observed for picornaviruses, e.g., poliovirus (21, 25), human rhinovirus 14 (HRV14) (19, 20), HRV16 (19), swine vesicular disease virus (16), and coxsackievirus (24); nodaviruses, e.g., Flock House virus (3); tetraviruses, e.g., Nudaurelia ω capensis and Helicoverpa armigera stunt virus (4); bromoviruses, e.g., cowpea chlorotic mottle virus (28); tombusviruses, e.g., tomato bushy stunt virus (15, 29); and sobemoviruses, e.g., southern bean mosaic virus (23). Transient, reversible exposition of internal proteins may be an important step for the cell entry process, especially for conformational changes required to insert internal, hydrophobic, membrane-binding peptides into the membrane (10, 12, 17). (This research was conducted by J. Lin in partial fulfillment of the requirements for a Ph.D. from Brigham Young University, Provo, UT, 2011.)
“Breathing” in poliovirus was first discovered when antibodies to normally internal amino acid sequences, VP4 and N terminus of VP1 (residues 1 to 53, [5]), were found to bind to native particles (21, 25). The studies suggested that capsid “breathing” represented large dynamic changes in virion structure at 37°C but not at 25°C. To study this purported structure and mark the exposed N terminus of VP1, we utilized cryogenic electron microscopy (cryo-EM) and three-dimensional (3D) image reconstruction to observe the structure of viruses incubated at 37°C with an Fab of a monospecific (polyclonal) antibody to the designated region (13, 25).
Poliovirus 1/Mahoney was grown and purified as described previously (7, 26). Fab was prepared from a preparation of affinity-purified antisera targeted to amino acids 39 to 55 of poliovirus type 1/Mahoney VP1 (25) by use of the ImmunoPure Fab preparation kit (Pierce, Rockford, IL). Solutions of native poliovirus (sedimentation coefficient, 160S) and Fab were mixed at 37°C with an Fab-to-virus ratio of 95:1 and incubated at 37°C for 2 h.
Frozen-hydrated specimens were prepared and imaged at 300 kV as described previously (27). An FEI Vitrobot (Hillsboro, OR) was used to vitrify the poliovirus-Fab sample and keep it at 37°C until it was plunged into liquid ethane. A 3D reconstruction was computed by the model-based, iterative process described previously (22, 27), with previous reconstructions of native poliovirus (sedimentation coefficient,160S) (D. M. Belnap et al., unpublished data) and the cell entry intermediate (135S) particle (1) as the starting models. Despite differences in the starting models, the capsid structure, the Fab density location and volume, and the center slice of the two resulting maps were nearly identical. In the micrograph, some 160S particles appeared to have few or no Fab bound (data not shown). A 160S poliovirus map (particles at room temperature or lower before plunge freezing) (Belnap et al., unpublished) and the initial 160S-37°-Fab reconstruction served as the starting models for multiple-model-based classification (11, 22). Each particle image was compared to a model projection from each state. Selected particles had a minimum difference of 0.04 in the correlation coefficient value between the correlation coefficients obtained from each form. Finally, 137 particles were selected to reconstruct the structure of the 160S 37°C “breathing” state with Fab bound (160S-37°-Fab) at 25-Å resolution.
Aggregation of particles was observed in cryo-EM micrograph.
A cryo-EM micrograph of 160S (Fig. 1B) showed a relatively smooth outer capsid surface. On the other hand, the cryo-EM micrograph of 160S complexed with Fab incubated at 37°C for 2 h (Fig. 1A) showed that the capsid surface was not smooth and contained additional densities, which indicates bound Fab, externalized VP4, externalized N terminus of VP1, or a combination of bound Fab and externalized polypeptide. These particles were inclined to aggregate (Fig. 1A). Hydrophobic portions of externalized peptides may cause the observed aggregation (17). Only nonaggregated particles were picked to do the reconstruction.
Fig 1.

Electron micrographs of frozen-hydrated poliovirus. (A) A complex of 160S and Fab to the 39to 55 residues of VP1 incubated at 37°C for 2 h. (B) 160S particles. Bar = 30 nm.
Exposed peptides in “breathing” structure are near the 2-fold axis.
The density map of the 160S-Fab complex at 37°C (Fig. 2A) shows considerable extra density near the 2-fold axes of the particle (Fig. 2A and E), suggesting that the Fab-binding site—and by inference the site of externalization of the N termini of VP1 in the “breathing state”—is at or near the 2-fold axes, far from the receptor binding site at the “canyon” (2, 9). The Fab-binding site indicates that, in the poliovirus “breathing” state, the N terminus of VP1 is found at the 2-fold axis. This extra density is likely some combination of Fab and the externalized peptides (VP4 and the N terminus of VP1). But because of its structural rigidity and large mass, the Fab is probably the major contributor to the observed density. Because the Fab density does not show the full shape of an Fab (Fig. 2A and E) (6), we infer that the epitope is flexible, binding regions of the monospecific (polyclonal) Fabs overlap, or both (22).
Fig 2.
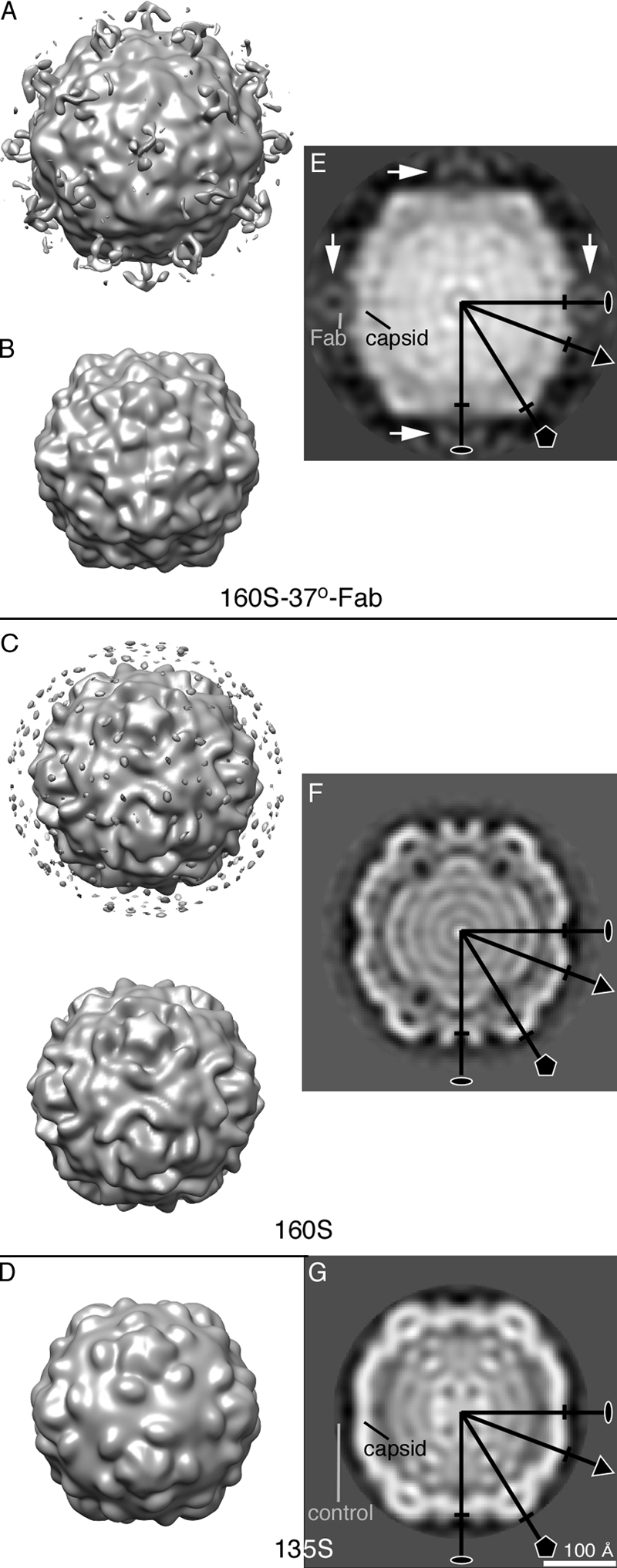
Structure of 160S-37°-Fab complex compared to 160S and 135S particles, viewed along a 2-fold symmetry axis. Surface representations of the 3D structures of 160S-Fab complex at 37°C (A and B). 160S and Fab to the 39 to 55 residues of VP1 were incubated at 37°C for 2 h. 160S-37°-Fab complex reconstructions at resolution 25 Å at contour levels of 0σ (A) and 1.3σ (B). Surface representations of “room-temperature” 160S (C) (1) and 135S (D) (5) maps are shown at contour level 0σ. The 160S and 135S reconstructions were computed from 123 and 137 particle images, respectively, to resolutions comparable to that of the 160S-37°-Fab reconstruction. (The 137 135S particle images were selected from a previously used 8,000-particle data set [5].) In panel C, the layer of noise seen in the top view was removed in the bottom view. Corresponding central sections of the poliovirus 160S-37°-Fab complex (E), 160S (F), and 135S (G) reconstructions. The sections are perpendicular to a 2-fold symmetry axis. Fab densities are centered on 2-fold axes (white arrows). For the 160S-37°-Fab structure (E), relative density was estimated by comparing average densities computed from spheres (radius = 3 pixels) centered on the designated spots (“Fab” and “capsid”). There the Fab contained ∼25% of the density of the capsid, suggesting an Fab occupancy of approximately one-fourth. For 135S (G), a similar calculation was made, but the “control” position contained no positive density, indicating the absence of protein or nucleic acid. Some symmetry axes are labeled. The markers perpendicular to each symmetry axis, 5-fold (pentagon), 3-fold (triangle), and 2-fold (oval), show the radius of the 160S structure. We note that differences in RNA structure between the 160S-37°C-Fab (E) and 160S-25°C (F) states may reflect differences due to temperature rather than differences due to the changed position of the N terminus of VP1. Scale bar for panels E, F, and G = 100 Å.
The weak Fab density on the 160S-37°-Fab capsid is likely due to the low occupancy of Fab on the capsid (Fig. 2A and E). The low occupancy is consistent with (i) the observation that the antibody used in this study showed a stronger binding affinity with 135S and genome-released (80S) particles than with “breathing” 160S particles (25) and (ii) the presence of Fab-free 160S particles in the micrographs despite the high concentration of 160S and excess Fab used during sample preparation. The weaker binding may occur because of the reversible transient externalization of N terminus of VP1 or the partial exposure of the peptide epitopes in the 160S “breathing” state.
Because 3D reconstructions are averages of the imaged particles, the relatively low resolution of the 160S-37°-Fab map suggests that, in addition to multiple conformations of the Fab and its epitope, multiple capsid conformations were averaged. At 37°C, the “breathing” 160S particle likely has a dynamic structure because of the reversible externalization of the internal peptides. Therefore, particles in the “breathing” state probably have multiple conformations.
Comparison of reversible “breathing” and irreversible 160S-to-135S changes.
During cell entry, poliovirus receptor (CD155) interacts with a 160S particle and induces the formation of the intermediate 135S particle (7, 14). This transition includes the irreversible externalization of VP4 and N terminus of VP1 from the internal side of the capsid (7, 8, 14). Previous authors have asked whether the reversible exposition of these same polypeptides during “breathing” is similar to the irreversible changes that occur in the 160S-to-135S transition (12, 21).
The structure of the 160S-37°-Fab complex (Fig. 2B) differs significantly from the 160S “room-temperature” structure (160S) (Fig. 2C) and is similar to but still different from the 135S structure (Fig. 2D). The shape of the RNA in the 160S-37°-Fab complex appears more similar to that in the 135S particle than the RNA in the 160S structure (Fig. 2E to G), and both Fab-trapped “breathing” particles and 135S particles are expanded with respect to 160S particles. However, comparison of central density slices of 160S-37°-Fab, 160S, and 135S maps (Fig. 2E to G) shows that the 160S-37°-Fab capsid is ∼8% larger than 160S and ∼5% larger than 135S at the 5-fold axis, nearly same as 160S and ∼3% smaller than 135S at the 2-fold axis, and nearly same as 160S and 135S at the 3-fold axis, demonstrating that the expansion in the “breathing” state differs in details from that previously described for 135S particles (1).
VP4 and the N terminus of VP1 were observed to externalize from the internal poliovirus capsid in both the 160S “breathing” and 160S-to-135S transitions (8, 21, 25). The expansion of the capsid may facilitate externalization of the internal peptides (12) in both processes. However, in the “breathing” state, the internal polypeptides exit the capsid reversibly (21) (and are likely trapped in the externalized state by bound Fab in the structure reported here), whereas in the 160S-to-135S transition, an irreversible conformational change leads to permanent externalization. Additionally, antiviral drugs block the formation of 135S poliovirus particles but not “breathing” (21). Moreover, the externalized N terminus of VP1 is located in very different positions in the 135S particle and in the “breathing” state. Thus, we have shown in this study of the 160S-37°-Fab map (Fig. 2A) that the N terminus of VP1 is near the 2-fold axis in the poliovirus “breathing” state. In contrast, in the 135S cryo-EM model (5), residues 42 to 52 were tentatively located at the canyon between the mesa and propeller tip, and the V8-cleaved 135S (5) and 135S-Fab complex (with an Fab that binds residues 24 to 40 of VP1) (22) showed that the N terminus of VP1 is located at the propeller tip. Curiously, in complexes of 80S “empty” particles (which have been shown to be derived from 135S particles during infection or during heating at 50°C [8]) with an Fab against VP1 residues 24 to 40, the externalized N terminus of VP1 partitions between two sites, one at the tips of the propellers (like the 135S particles) and the other near the 2-fold axes (as in the “breathing state,” 160S-37°-Fab complex) (22).
These observations suggest two models for externalization of VP4 and the N terminus of VP1: (i) separate processes and exit points or (ii) a two-stage, single-exit process. In both models, the reversible and irreversible changes may involve temporary expansion of the capsid beyond that observed for the metastable “breathing” (Fig. 2) and 135S (1, 5) states.
In the first model, the different sites for the externalized VP1 peptide are consistent with two different exit channels and externalization mechanisms for “breathing” and the 160S-to-135S transition. Thus, in the transformation to the 135S particle via the poliovirus-receptor interaction (18) or by heating to 50°C (7), the N terminus may be directed to a different exit site (i.e., canyon [5]) than that observed in the reversible exposure observed at 37°C (Fig. 2). In this model, the exit of the N-terminal peptides via the second site in the canyon but not the site at the 2-fold is inhibited by the antiviral drugs at 37°C.
In the second model, the reversible (“breathing”) externalization may represent only the beginning of the irreversible externalization process. The irreversible process (leading to the 135S particle) begins with “breathing” particles in which the N terminus of VP1 is reversibly externalized from a unique exit site and placed near the 2-fold axis. Structural changes induced by receptor binding (in vivo) or heating (in vitro) then irreversibly move the externalized peptide to the canyon by enlarging the exit hole or extending the peptide. In this model, antiviral drugs might inhibit either the structural change allowing the peptide to move from the 2-fold to the base of the canyon or the expansion of the hole and relaxation into the 135S form. Following or concurrent with RNA release, similar structural changes allow the peptides to move back to the 2-fold axis (22).
ACKNOWLEDGMENTS
This work was supported by BYU institutional funds (to D.M.B.). Jun Lin was partially supported by grant R15AI084085 from the National Institute of Allergy and Infectious Diseases (to D.M.B.) and a fellowship from the BYU Department of Chemistry and Biochemistry. This work was also supported by NIH grant R01AI020566 (to J.M.H.).
We thank Jeffrey Farrer and Michael Standing for microscopy support, Eduardo Sanz-Garcia and Bernard Heymann for computer help, Peter Shen and Mark Hazel for helpful discussions, and our peer reviewers for helpful suggestions on the manuscript.
The 160S-37°-Fab map was deposited in the EM Data Bank (entry 5387).
Footnotes
Published ahead of print 7 March 2012
REFERENCES
- 1. Belnap DM, et al. 2000. Molecular tectonic model of virus structural transitions: the putative cell entry states of poliovirus. J. Virol. 74:1342–1354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Belnap DM, et al. 2000. Three-dimensional structure of poliovirus receptor bound to poliovirus. Proc. Natl. Acad. Sci. U. S. A. 97:73–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bothner B, Dong XF, Bibbs L, Johnson JE, Siuzdak G. 1998. Evidence of viral capsid dynamics using limited proteolysis and mass spectrometry. J. Biol. Chem. 273:673–676 [DOI] [PubMed] [Google Scholar]
- 4. Bothner B, et al. 2005. Maturation of a tetravirus capsid alters the dynamic properties and creates a metastable complex. Virology 334:17–27 [DOI] [PubMed] [Google Scholar]
- 5. Bubeck D, et al. 2005. The structure of the poliovirus 135S cell entry intermediate at 10-angstrom resolution reveals the location of an externalized polypeptide that binds to membranes. J. Virol. 79:7745–7755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Conway JF, et al. 2003. Characterization of a conformational epitope on hepatitis B virus core antigen and quasiequivalent variations in antibody binding. J. Virol. 77:6466–6473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Curry S, Chow M, Hogle JM. 1996. The poliovirus 135S particle is infectious. J. Virol. 70:7125–7131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fricks CE, Hogle JM. 1990. Cell-induced conformational change in poliovirus: externalization of the amino terminus of VP1 is responsible for liposome binding. J. Virol. 64:1934–1945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. He Y, et al. 2000. Interaction of the poliovirus receptor with poliovirus. Proc. Natl. Acad. Sci. U. S. A. 97:79–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hendry E, et al. 1999. The crystal structure of coxsackievirus A9: new insights into the uncoating mechanisms of enteroviruses. Structure 7:1527–1538 [DOI] [PubMed] [Google Scholar]
- 11. Heymann JB, Conway JF, Steven AC. 2004. Molecular dynamics of protein complexes from four-dimensional cryo-electron microscopy. J. Struct. Biol. 147:291–301 [DOI] [PubMed] [Google Scholar]
- 12. Hogle JM. 2002. Poliovirus cell entry: common structural themes in viral cell entry pathways. Annu. Rev. of Microbiology. 56:677–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hovi T, Roivainen M. 1993. Peptide antisera targeted to a conserved sequence in poliovirus capsid VP1 cross-react widely with members of the genus Enterovirus. J. Clin. Microbiol. 31:1083–1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Huang Y, Hogle JM, Chow M. 2000. Is the 135S poliovirus particle an intermediate during cell entry? J. Virol. 74:8757–8761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jaegle M, Briand JP, Burckard J, Van Regenmortel MH. 1988. Accessibility of three continuous epitopes in tomato bushy stunt virus. Ann. Inst. Pasteur Virol. 139:39–50 [DOI] [PubMed] [Google Scholar]
- 16. Jiménez-Clavero MA, Douglas A, Lavery T, Garcia-Ranea JA, Ley V. 2000. Immune recognition of swine vesicular disease virus structural proteins: novel antigenic regions that are not exposed in the capsid. Virology 270:76–83 [DOI] [PubMed] [Google Scholar]
- 17. Johnson JE. 2003. Virus particle dynamics. Adv. Protein Chem. 64:197–218 [DOI] [PubMed] [Google Scholar]
- 18. Kaplan G, Freistadt MS, Racaniello VR. 1990. Neutralization of poliovirus by cell receptors expressed in insect cells. J. Virol. 64:4697–4702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Katpally U, Fu TM, Freed DC, Casimiro DR, Smith TJ. 2009. Antibodies to the buried N terminus of rhinovirus VP4 exhibit cross-serotypic neutralization. J. Virol. 83:7040–7048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lewis JK, Bothner B, Smith TJ, Siuzdak G. 1998. Antiviral agent blocks breathing of the common cold virus. Proc. Natl. Acad. Sci. U. S. A. 95:6774–6778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li Q, Yafal AG, Lee YM, Hogle J, Chow M. 1994. Poliovirus neutralization by antibodies to internal epitopes of VP4 and VP1 results from reversible exposure of these sequences at physiological temperature. J. Virol. 68:3965–3970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lin J, et al. 2011. An externalized polypeptide partitions between two distinct sites on genome-released poliovirus particles. J. Virol. 85:9974–9983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mackenzie DJ, Tremaine JH. 1986. The use of a monoclonal antibody specific for the N-terminal region of southern bean mosaic virus as a probe of virus structure. J. Gen. Virol. 67:727–735 [Google Scholar]
- 24. Pulli T, Lankinen H, Roivainen M, Hyypia T. 1998. Antigenic sites of coxsackievirus A9. Virology 240:202–212 [DOI] [PubMed] [Google Scholar]
- 25. Roivainen M, Piirainen L, Rysa T, Narvanen A, Hovi T. 1993. An immunodominant N-terminal region of VP1 protein of poliovirion that is buried in crystal structure can be exposed in solution. Virology 195:762–765 [DOI] [PubMed] [Google Scholar]
- 26. Rueckert RR, Pallansch MA. 1981. Preparation and characterization of encephalomyocarditis (EMC) virus, p 315–325 In Pestka S. (ed), Interferons, vol 78 Academic Press, New York, NY: [PubMed] [Google Scholar]
- 27. Shen PS, et al. 2011. The structure of avian polyomavirus reveals variably sized capsids, non-conserved inter-capsomere interactions, and a possible location of the minor capsid protein VP4. Virology 411:142–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Speir JA, et al. 2006. Enhanced local symmetry interactions globally stabilize a mutant virus capsid that maintains infectivity and capsid dynamics. J. Virol. 80:3582–3591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Witz J, Brown F. 2001. Structural dynamics, an intrinsic property of viral capsids. Arch. Virol. 146:2263–2274 [DOI] [PubMed] [Google Scholar]