

Abstract
Hepatitis C virus (HCV) research is hampered by the use of arbitrary representative isolates in cell culture and immunology. The most replicative isolate in vitro is a subtype 2a virus (JFH-1); however, genotype 1 is more prevalent worldwide and represents about 70% of infections in the United States, and genotypes differ from one another by 31% to 33% at the nucleotide level. For phylogenetic and immunologic analyses, viruses H77 and HCV-1 (both subtype 1a) are commonly used based on their historic importance. In an effort to rationally design a representative subtype 1a virus (Bole1a), we used Bayesian phylogenetics, ancestral sequence reconstruction, and covariance analysis on a curated set of 390 full-length human HCV 1a sequences from GenBank. By design, Bole1a contains variations present in widely circulating strains and matches more epitope-sized peptides in a full-genome comparison to subtype 1a isolates than any other sequence studied. Parallel analyses confirm that selected epitopes from the Bole1a genome were able to elicit a robust T cell response. In a proof of concept for infectivity, the envelope genes (E1 and E2) of Bole1a were expressed in an HIV pseudoparticle system containing HCV envelope genes and HIV nonenvelope genes with luciferase expression. The resulting Bole1a pseudoparticle robustly infected Hep3B cells. In this study, we demonstrate that a rationally designed, fully synthetic HCV genome contains representative epitopes and envelope genes that assemble properly and mediate entry into target cells.
INTRODUCTION
Hepatitis C virus (HCV) affects approximately 170 million people worldwide (47). Approximately 20% to 25% of patients with acute hepatitis C achieve spontaneous clearance of the virus, but 75% to 80% develop chronic infection (23). Approximately 20% of chronic hepatitis C patients develop cirrhosis, and of these, 4% will develop hepatocellular carcinoma and 6% will develop end-stage liver disease (37). There is no available HCV vaccine, and commonly used interferon-based treatment is toxic, prolonged, expensive, not consistently successful, and not effective in the most advanced forms of disease (5).
HCV is a small enveloped Flaviviridae family virus with a 9.6-kb single, positive-stranded RNA genome consisting of a 5′ untranslated region (UTR), a large open reading frame encoding the virus-specific proteins, and a 3′ UTR (29). The 5′ UTR contains an internal ribosome entry site (IRES) that mediates translation of a single polyprotein of approximately 3,000 amino acids. The polyprotein consists of structural proteins (C, E1, and E2) located in the N terminus, followed by p7 and nonstructural proteins (NS2, NS3, NS4A, NS4B, NS5A, and NS5B) encoded in the remainder.
While there is a recognized need for an effective HCV vaccine, selection of the viral strain to be used as an antigen has been arbitrary. Studies in humans and chimpanzees have shown that the host immune system is able to launch an effective response to HCV (15), and people who have cleared infection once are likely to do so again (24, 28), though this effect is potentially attributable to host genetics (38). The genetic diversity of HCV, which is even greater than that of HIV (29), poses a great challenge to the development of an effective vaccine (15). Selection of an appropriate strain as a vaccine candidate is crucial, since even a single amino acid substitution can reduce vaccine effectiveness by eliminating recognition by T cells specific for that epitope (10). Use of an ancestral or consensus sequence as a vaccine candidate has been proposed for HIV-1 (12). Compared to a consensus sequence, a mosaic approach (including sequences of multiple individual epitopes) generated more-vigorous T cell responses to HIV-1 epitopes (1). Mosaic candidates have recently been identified for HCV, although their effectiveness is still unknown (50).
In this paper, we present a synthetic subtype 1a virus genome, and the resulting computationally derived genome is representative of widely circulating strains, has functional envelope genes that mediate entry into hepatoma cells in vitro, and matches more CD8+ T cell epitopes than any other subtype 1a sequence in GenBank, whether comparing all 9-mers or all known common epitopes. We call this sequence “Bole1a” as a metaphorical reference to botany, where bole refers to the portion of a tree's trunk that supports the branches.
MATERIALS AND METHODS
Human subjects.
The Baltimore Before and After Acute Study of Hepatitis (BBAASH) cohort is a prospective study of persons at risk for hepatitis C infection (8). Eligible participants have a history of or ongoing intravenous drug use and are seronegative for anti-HCV antibodies at enrollment. Written consent was obtained from each participant. Once enrolled, participants receive counseling to reduce intravenous drug use and its complications. Blood is drawn for isolation of serum, plasma, and peripheral blood mononuclear cells (PBMCs) in a protocol designed for monthly follow-up (7). Participants with acute HCV infection were referred for evaluation of treatment. The study was approved by the Institutional Review Board at the Johns Hopkins School of Medicine.
Synthetic coding sequence reconstruction.
HCV subtype 1a (n = 390) and 1b (n = 296) sequences that included at least the entire open reading frame of the polyprotein, were obtained from human specimens, and were not epidemiologically redundant were downloaded from GenBank (accession numbers AB016785, AB049087-101, AB154177, AB154179, AB154181, AB154183, AB154185, AB154187, AB154189, AB154191, AB154193, AB154195, AB154197, AB154199, AB154201, AB154203, AB154205, AB191333, AB249644, AB429050, AF009606, AF139594, AF165045, AF165047, AF165049, AF165051, AF165053, AF165055, AF165057, AF165059, AF165061, AF165063, AF176573, AF207752-74, AF208024, AF313916, AF356827, AF483269, AF511948-50, AJ000009, AJ132996-97, AJ238799-800, AJ278830, AY045702, AY460204, AY587844, AY615798, AY695437, AY956463-8, D10749, D10934, D11168, D14484, D50480-82, D63857, D85516, D89815, D89872, D90208, DQ071885, DQ838739, EF032883, EF032886, EF032892, EF032900, EF407411-57, EF407458-504, EF621489, EF638081, EU155213-16, EU155217-35, EU155233, EU155236-381, EU234061, EU234063-65, EU239713, EU239714, EU239715-17, EU255927-99, EU255960-2, EU256000-1, EU256002-97, EU256045, EU256054, EU256059, EU256061-2, EU256064-6, EU256075-103, EU256104, EU256106-7, EU260395-6, EU362882, EU362888-901, EU362911, EU482831-2, EU482833, EU482834-89, EU482839, EU482849, EU482859, EU482860, EU482874, EU482875, EU482877, EU482879-81, EU482883, EU482885-6, EU482888, EU529676-81, EU529682, EU569722-23, EU595697-99, EU660383-85, EU660386, EU660387, EU660388, EU677248, EU677253, EU687193-95, EU857431, EU862823-24, EU862826-27, EU862835, FJ024086, FJ024087, FJ024274-76, FJ024277, FJ024278, FJ024279, FJ024280-82, FJ181999-201, FJ205867-69, FJ390394-95, FJ390396-8, FJ390399, FJ410172, L02836, M58335, M84754, U01214, U16362, U45476, U89019, and X61596).
We refer to the data set of the 390 subtype 1a sequences as the “original data set” for the rest of the paper. The sequences were aligned using MUSCLE v3.0 (9) and modified using BioEdit v7.0.5.3 (13). To avoid idiosyncrasies of any individual phylogeny, we constructed 2 independent phylogenetic trees by applying MrBayes v3.2 (31) to nucleotide positions 869 to 1292 (Core/E1) and 8276 to 8615 (NS5B) of the full-genome alignment (position numbers are based on the reference genome H77 [GenBank accession number AF009606]). These segments were chosen because they were shown to be the most phylogenetically informative (33, 34). We refer to them as “Simmonds' regions” in this paper. We ran 30 million iterations of MrBayes v3.2 and confirmed convergence of parameters for phylogenetic trees inferred from both of Simmonds' regions using Tracer v1.5 (Rambaut A, available from the author [http://beast.bio.ed.ac.uk/Tracer]). Simmonds' regions yielded different trees, a result which is expected due to the large number of possible trees (11); nonetheless, analysis of these two data sets converged with similar model parameters. In addition, recombination in HCV is rare (40). Hence, we can assume the same phylogenetic tree or same evolutionary history for the entire length of the genome (17). Using both phylogenetic trees reconstructed with Simmonds' regions, we inferred ancestral sequences for each of the HCV-1a coding regions (31). The ancestral sequence is obtained as a probability distribution for each position such that there is a probability of observing each base. Bole1a is derived in the following manner.
(i) For each nucleotide position i in the genome, if both trees agreed on the maximum posterior probability (MPP) residue, the probability of that position pi was selected to be the greater of the two MPPs. We define these positions as concordant.
(ii) For discordant positions (where the MPP residues did not agree), the joint probabilities of the codon k containing the discordant positions based on both trees were designated pck(core/E1) and pck(NS5B). For concordant residues within such codons, the pi calculated in the previous step was used in calculating the joint probability.
(iii) The codon with the higher joint MPP from the two trees was selected to represent that codon position. This codon-based analysis resolves cases in which more than one position in the codon is discordant and accommodates 6-fold degenerate codons.
(iv) To determine a stringent threshold for codon MPP, the inflection in the distribution of codon MPPs at which the variance in the second derivative was less than 10−6 for MPP values was found to be 0.9837, corresponding to individual residue MPPs of >0.99.
(v) Each codon with an MPP greater than or equal to 0.9837 based on either tree was accepted as ancestral, and its constituent positions were defined as resolved.
(vi) Covariance analysis was used to examine still-unresolved positions. The basic assumption of phylogenetic reconstruction that each site evolves independently ignores covarying and interacting sites. In order to take such sites into consideration, the observed (o) and expected (e) frequencies of pairs of bases were determined and the chi-square metric was calculated as shown in equation 1 and adjusted for multiple comparisons using the Holm-Bonferroni method at an α value of 0.05 (14).
| (1) |
Using the adjusted chi-square metric, all resolved positions j that significantly covaried with unresolved positions i were identified. In the case of a positive interaction (oij > eij), the MPP codon containing the positively interacting residue was selected. For negative interactions (oij < eij), all codons with the negatively interacting base were eliminated and the MPP codon was selected from the remaining codons.
(vii) At still-unresolved sites, the MPP codon was selected even if the MPP was less than 0.9837 (as noted in Results, this was rarely necessary).
5′ and 3′ UTR sequence reconstruction.
Although the 5′ UTR and 3′ UTR are noncoding regions, they are essential in the replication of the virus. However, of the 390 sequences, only 6 had completely sequenced 5′ UTR regions and 4 had completely sequenced 3′ UTR regions. Hence, we used additional sequences to better design the noncoding regions. The 5′ UTR (n = 257) and 3′ UTR (n = 46) sequences were from clonal sequences generated from acutely infected subjects in the BBAASH cohort (8). We found that our 90% consensus sequence of the 5′ UTR was identical to the consensus sequence derived from the 6 sequences with complete sequences and also to that of the H77 5′ UTR. The 3′ UTR sequence was divided into 4 parts based on classification by Kolykhalov et al. (18). We determined the 90% consensus sequence for the first part as a short sequence with significant variability among genotypes. For the second segment of the 3′ UTR, we determined that the median length of the homopolymeric uracil tract was 51 residues, which is also a favorable length for replication (49). We selected a segment of median length for the third segment, a polypyrimidine tract consisting of mainly U with interspersed C residues. The last (3′ end) part, a conserved sequence of 98 bases for which we used the 90% consensus sequence, was confirmed with 15 additional sequences from an unrelated study (51).
HCV pseudoparticle (HCVpp) system.
A region of Bole1a nucleotide sequences encoding the last 21 amino acids of the core followed by the E1 and E2 regions (E1E2) was synthesized (Blue Heron, Bothell, WA) and then subcloned into the expression vector pcDNA3.2/V5/Dest (Invitrogen, Carlsbad, CA) using Gateway technology. Full-length E1E2 was sequenced after cloning and showed no errors. Pseudoparticles containing the luciferase reporter gene were generated as described elsewhere (16, 22, 26). Briefly, a plasmid expressing Bole1a E1E2 was cotransfected into 293T cells with a vector expressing HIV core proteins and luciferase. Supernatants were collected 48 h after transfection. Pseudoparticles expressing E1E2 from H77, E1E2 from another subtype 1a HCV virus, and no E1E2 (mock) were produced in parallel with pseudoparticles expressing Bole1a E1E2 for a comparison of infectivities. Serial 2-fold dilutions of pseudoparticles were used to infect Hep3B hepatoma cells in duplicate wells of a 96-well plate for 5 h, followed by measurement of luciferase activity 72 h postinfection.
CD81 blocking experiments.
Hep3b cells were incubated with a mouse anti-human CD81 monoclonal antibody (100 μg/ml, clone 1.3.3.22; Santa Cruz Biotechnology) or mouse IgG1 isotype control (Santa Cruz Biotechnology) for 1 h at 37°C, and HCVpp infection was assessed as above.
Neutralization by human plasma.
Heat-inactivated plasma or serum was diluted 1:4 with minimal essential medium (MEM) containing 10% fetal bovine serum (FBS), incubated with each library HCVpp for 1 h at 37°C (final HCVpp dilution, 1:100), added to Hep3b hepatoma cells in duplicate wells of a 96-well plate, and incubated for 5 h at 37°C, followed by replacement of medium. Luciferase activity was measured as above. HCVpp infection was measured in terms of relative light units (RLUs) in the presence of plasma or serum samples (RLUtest) compared to average infection in the presence of normal human serum (Gemini Bio-Products, West Sacramento, CA) and plasma pooled from seronegative BBAASH participants (RLUcontrol). Percent neutralization was calculated as [1 − (RLU test/RLUcontrol)] × 100.
Diversity analysis.
Diversity plots were generated using VarPlot version 1.2 (described in Ray et al. [30] and available from the author at http://sray.med.som.jhmi.edu/scroftware/VarPlot). Plots were generated using a window size of 20 codons (to reflect the upper limit of T cell epitopes) and a step size of 1. Nonsynonymous and synonymous distances were calculated using the models of Nei and Gojobori (25). The E1E2 pixel alignment (see Fig. 2b) was drawn using VisSPA v1.6 (http://sray.med.som.jhmi.edu/SCRoftware/VisSPA/).
Fig 2.

(a) A diversity plot comparing mean pairwise nonsynonymous (dN) and synonymous (dS) diversity among subtype 1a sequences to mean pairwise distances between Bole1a and subtype 1a sequences using a sliding window size of 20 codons. For this comparison, the original data set of 390 full-genome sequences was the source of polyprotein reference sequences. (b) An alignment comparison of E1E2 using Bole1a as the reference sequence, the consensus (of 390 sequences), H77, and HCV-1 sequences, and a 1b (D90208) sequence. Vertical bars indicate positions with amino acid differences between that sequence and Bole1a, and the asterisk indicates the position of HVR1.
Nucleotide sequence accession numbers.
The Bole1a genomic sequence has been deposited in GenBank under accession number JQ791196.
RESULTS
Trees for the E1 and NS5B regions generated ancestral sequences that agreed at 9,763 (∼98%) of 9,992 nucleotide sites in the alignment (gaps were counted as characters). Applying our codon threshold of an MPP of 0.9837 or higher in either tree left 68/3,012 (2.2%) codons unresolved. Of these 68, 42 were choices between synonymous codons and 26 were choices between nonsynonymous codons. Covariance networks were used to resolve ambiguities.
Covarying positions.
Of the 68 unresolved codons, 4 were determined based on dependence with resolved positions in the genome (H77 positions 1157, 1611, 2120, and 6554). All four of the positions (1157, 1611, 2120, and 6554) led to synonymous changes. Positions 1611 and 6554 were linked to multiple sites across the genome (50 and 3, respectively), whereas positions 1157 and 2120 were each linked to one other resolved position. Because the covariance was detected only statistically, biological interaction is a question for further research.
Representative characteristics of Bole1a.
Once we determined a complete representative sequence for Bole1a, we wanted to ensure that it represents any set of nucleotide or protein HCV subtype 1a sequences and not just the sequences from which it was reconstructed. We used two additional data sets for confirmation. The first data set was from a paper by Yusim et al. (50) and collected from the Los Alamos National Laboratory (LANL) HCV database. This data set contains 143 sequences, 136 of which are present in the original data set; however, the authors of that report curated the data set to avoid resampling linked sequences. We refer to this data set as the “Yusim data set.” The second data set, which we refer to as the “E1E2 data set,” contains 214 E1E2 sequences; these were obtained from our ongoing BBAASH cohort (8). The sequences in the latter data set are unrelated to any full-length sequences in GenBank or from the LANL database. Neighbor-joining trees showed that Bole1a consistently branches from the center, suggesting that it is representative of both the Yusim and E1E2 data sets (Fig. 1).
Fig 1.

Neighbor-joining trees showing Bole1a and the Yusim data set (a) and Bole1a and the E1E2 data set (b). The Bole1a sequence is shown in bold in both figures.
Based on a full-genome pairwise comparison, Bole1a is more similar to subtype 1a sequences than any other sequence in the original data set (average and median reductions in nonsynonymous distance of 39% and 44%, respectively) (Fig. 2a). In sliding windows of 20 codons (approximating the upper limit of the size of T cell epitopes) spanning the genome, the similarity of Bole1a was greater than 98% overall (mean and median similarities of 98.4% and 98.9%, respectively). Not surprisingly, the lowest similarity between Bole1a and subtype 1a circulating genomes was in hypervariable region 1 (HVR1), where similarity was as low as 73% (similarity among subtype 1a isolates at the same position was 64%). A comparison of the Bole1a sequence and the consensus sequence of the original data set, H77, HCV-1, and a 1b sequence demonstrates the high variability in HVR1 (as shown by an asterisk in Fig. 2b).
We compared all 9-mers of the Bole1a amino acid sequence to those of sequences in the Yusim data set to represent potential epitope coverage. The use of 9-mers for this comparison is based on the typical major histocompatibility complex (MHC) class I-restricted epitope length recognized by effector CD8+ T cells that are a crucial component of immune control in spontaneous clearance of HCV infection (32). Bole1a provides 78% exact-match 9-mer coverage for the HCV polyprotein whether compared to the Yusim data set or the original data set (data not shown; method previously described by Yusim [50]). In the highly diverse E1 and E2 regions of the E1E2 data set, Bole1a provides 58% exact-match 9-mer coverage. We then compared Bole1a with the Yusim data set by individual proteins and confirmed that highly heterogeneous regions, such as E1 and E2, have lower coverage by Bole1a than do more-conserved regions, such as the core and NS4B. In all cases, Bole1a provided greater 9-mer coverage than the reference sequence H77. Bole1a matched 99% of all 9-mers in a full-genome comparison when a mismatch at 1 or 2 positions was allowed. In summary, we found that Bole1a matched 95% of all modal (most frequently observed) 9-mers at each position of the genome, whereas individual sequences in the Yusim data set had a median modal 9-mer coverage of 80% (Fig. 3a).
Fig 3.

Bole1a (indicated by an asterisk) is highly representative based on coverage of modal (most commonly observed) 9-mers provided by Bole1a and all other sequences in the Yusim data set (a) and identity to known epitopes, depicted as a histogram showing the percentages of epitope sequences that are identical to those of the known and common 338 T cell epitopes (b).
The obvious limitation of comparing 9-mer coverage is that not all 9-mers are recognized as T cell epitopes. To focus on epitope coverage, we selected all known subtype 1a T cell epitopes (both CD4 and CD8) associated with a positive result in at least one assay from the Immune Epitope Database (http://www.immuneepitope.org/). Of the 548 epitopes in the database, only 338 were present in at least half of the sequences of the Yusim data set (excluding AF271632 and AX100563 due to their linkage with HCV-1 and H77, respectively). Bole1a had the highest (100%) coverage of these 338 epitopes (Fig. 3b). HCV-1 and H77, which are commonly used as antigens in HCV immunology (6, 19, 43, 45, 46), matched only 317 and 316 (∼93%), respectively, of these 338 epitopes. Figure 3b shows the distribution of epitope coverage for all sequences in the Yusim data set. When epitopes that were present in 80% of the sequences were included, Bole1a provided 94% coverage while H77 and HCV-1 provided 87% and 91% coverage, respectively (data not shown). It should be noted that because HCV-1 and H77 have been the primary isolates used as antigens in many of the studies from which these epitopes are derived, their coverage may be due in part to analytical bias. Lastly, gamma interferon enzyme-linked immunosorbent spot assay (ELISpot) analysis of variant epitopes demonstrated that variants in the Bole1a genome are more consistently immunogenic than those of other sequences, including H77 and a simple consensus (4a).
Bole1a pseudoparticle.
Approximately 75% of individual E1E2 isolates tested have low infectivity (less than 5 standard deviations above background) when used to pseudotype lentiviral particles (Fig. 4). The diversity of the envelope genes is extremely high, with an average nonsynonymous diversity of 36% in 20-codon windows (Fig. 2). As a result of this diversity and our methods, Bole1a has a unique HVR1 sequence (ETHVTGGSAARATAGFAGLFTPGAKQN) among the genomes we have examined, and searches for this peptide sequence using BLAST (http://blast.ncbi.nlm.nih.gov) and HMMER (http://hmmer.janelia.org) did not reveal any identical sequences. To test functionality despite these negative predictors for infectivity, the E1E2 sequence of Bole1a was used to construct a lentiviral pseudoparticle. Surprisingly, HCVpp Bole1a infected Hep3B target cells with high efficiency comparable to that of the highly infectious and well-characterized isolate HCVpp-H77 (Fig. 4). Blocking the Bole1a HCVpp with anti-CD81 antibody led to at least a 10-fold reduction in infectivity (P = 0.0008), whereas the isotype control antibody did not change infectivity (P = 0.85) (Fig. 4). RLU values below the threshold (Fig. 4) are found to be reproducibly low with high variance. Although the comparison panel of subtype 1a HCVpp excluded those that contained stop codons, frameshift mutations, or other obvious defects, it is evident that there are other biological or artifactual characteristics that render many of those clones less infectious. Importantly, the goal of this experiment was to determine whether Bole1a E1E2 would be functional at all in spite of its synthetic origin and the high variability of the HCV envelope; that this E1E2 was infectious in the HCVpp system was highly unexpected. Additionally, the Bole1a HCVpp was readily neutralized by human sera. We observed that 30% of BBAASH subjects inhibited at least 85% of entry and 90% of subjects from the BBAASH cohort (36 out of 40) inhibited at least 50% of Bole1a HCVpp entry, whereas normal human serum and pooled HCV-seronegative sera were nonneutralizing.
Fig 4.
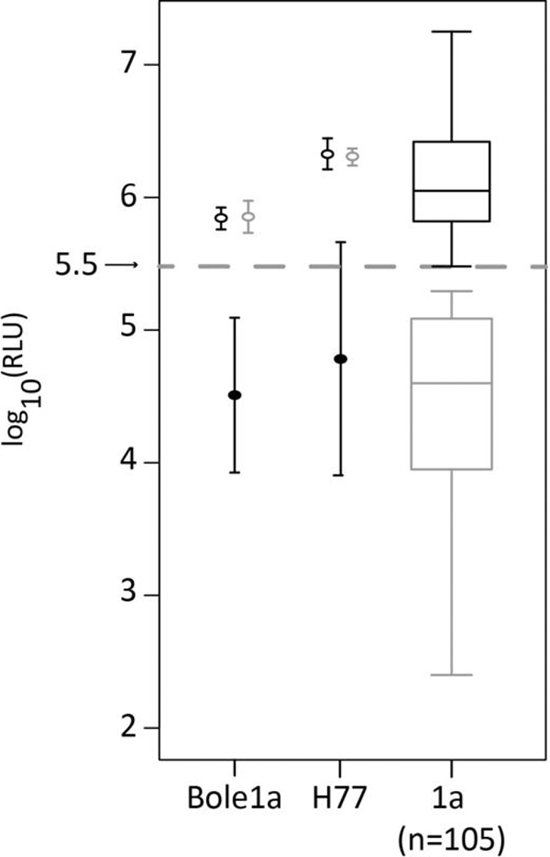
The infectivity of various HCVpp is shown in log10 (RLU). The gray dashed line represents the RLU threshold for infectious HCVpp. The open black circles in the first two columns represent average infectivities without any antibodies. The filled black circles show average infectivities in the presence of anti-CD81, and the open gray circles show infectivity in the presence of an isotype control. The error bars show the standard deviations calculated from 3 separate experiments. The black and gray boxplots on the right show the distributions of infectivity without antibody of all infectious and noninfectious subtype 1a HCVpp, respectively.
DISCUSSION
In conducting and communicating research related to a virus like HCV with extremely high genetic diversity, representative genomes are an essential reference tool. Reference genomes are used as a basis for numbering positions of important landmarks, such as primers, probes, epitopes, and catalytic sites (20), as antigens in peptide-based (6, 19, 43, 45, 46) and lentiviral pseudoparticle-based (2, 22, 26, 41) assays, and as reference standards in replication assays (4, 42, 48, 52). To date, no reference HCV genome has been optimal for all of these applications due to idiosyncrasies of the isolates and assays involved. Empiricism and tradition, rather than rational design, have guided the selection of HCV reference genomes.
We report the rational design of a synthetic representative HCV subtype 1a genome, dubbed Bole1a, using Bayesian phylogenetic tree methods, ancestral sequence reconstruction, and covariance analysis. Bole1a branches centrally among 390 full-genome sequences used in its design (tree not shown), a carefully curated 143-sequence full-genome data set, and separate genomic regions, including an independent set of 214 E1E2 sequences from a Baltimore cohort (Fig. 1). Thus, Bole1a is phylogenetically representative of widely circulating strains. A full-genome nonsynonymous diversity comparison and a 9-mer peptide coverage analysis showed that Bole1a is able to provide more coverage (94% and 78%, respectively) than any other sequence in the data set, including H77, a traditional reference sequence (Fig. 2 and 3a). Bole1a also provides unsurpassed epitope coverage when compared to all known T cell epitopes (Fig. 3b). Two strains that nearly matched Bole1a's epitope coverage were H77 (AF009606) and HCV-1 (M62321), on which the overlapping peptides used to discover T cell epitopes were based; therefore, they are the strains expected to have the highest epitope coverage, and further research using Bole1a as an antigen is likely to reveal previously unrecognized epitopes. In this light, it is remarkable that Bole1a was even comparable to reference antigens that have been used to date.
Preliminary analyses have shown that epitopes from Bole1a are the most immunogenic of any isolate tested. In those cases in which Bole1a epitopes differed from the traditional consensus (2 out of 15 tested), T cells from chronically infected patients recognized Bole1a epitopes better than the corresponding epitopes from circulating and consensus sequences (Burke et al., submitted). Since Bole1a is representative of circulating strains, it is unlikely to contain escape mutations that hinder viral fitness. For example, the Bole1a sequence has a Y at position 1444, whereas an F at the position is believed to be an escape mutation causing the NS3 1436-1444 epitope to elicit a less robust T cell response (27). Additionally, Bole1a contains the KLVALGINAV sequence at NS3 1406-1416. Three variants of this epitope have been shown to have diminished T cell responses without a change in MHC-binding ability, making escape the most likely explanation for these variants (35).
Previous studies in HIV-1 have suggested that using artificial representative sequences, such as consensus or ancestral sequences, is an effective way to minimize the sequence dissimilarity between a vaccine strain and circulating viruses and addresses biological and artifactual defects in individual clones (12). Our study is distinct in that our representative sequence is not simply a reconstructed ancestor or a pure consensus. We used ideas from both schools of thought to most accurately reconstruct a representative sequence that has unescaped T cell epitopes and can be used in vaccine design. Although a global vaccine is desirable, it is likely that the immense global diversity of HCV cannot be captured efficiently in one vaccine strain. Genotypes differ at 31% to 33% of nucleotide sites, while subtypes differ at 20% to 25% of sites. Mixed-subtype infections cannot be ignored, but they are observed rarely, with a reported prevalence of less than 5% (39). Toyoda et al. also found that the number of HCV variants was larger in patients with mixed-HCV-subtype infection than in patients with single-subtype infection, suggesting that a global vaccine candidate may not be able to provide effective protection.
As an indication of potential utility in studies of HCV replication, infection, and neutralization, we expressed the Bole1a envelope genes in an HCV/HIV pseudoparticle system, demonstrating infection of target Hep3B cells with high efficiency (Fig. 4). This proof-of-concept study demonstrates that the Bole1a envelope E1E2 heterodimer is able to fold and assemble correctly. Because HCV E1 and E2 genes are critical for host cell entry, they are also important targets for antibody-mediated virus neutralization (36). Because it lacks evident immunologically driven escape mutations, Bole1a may represent the ideal platform to study determinants of HCV fitness.
Here we describe Bole1a, the first rationally designed and representative HCV genome. It is clear that synthetic viral genomes provide a powerful platform for virologic investigation (44). Bole1a is phylogenetically and immunologically representative, and its envelope genes are functional on lentiviral pseudoparticles. The remarkable immunogenicity of Bole1a is in agreement with computational studies of HIV (3), focused cross-sectional studies of HCV (27), and evolutionary studies during acute HCV infection (21) that indicate that immune escape is dominated by centrifugal substitutions. Vaccine development for HCV and similarly variable viruses may benefit from a more rational, less idiosyncratic approach to antigen selection.
ACKNOWLEDGMENTS
This study was supported by NIH R01 DA024565 and the SRRS Foundation.
We thank Anna Snider for her help with the HCVpp infection studies and BBAASH participants and study personnel for their essential roles in this work.
Footnotes
Published ahead of print 21 March 2012
REFERENCES
- 1. Barouch DH, et al. 2010. Mosaic HIV-1 vaccines expand the breadth and depth of cellular immune responses in rhesus monkeys. Nat. Med. 16:319–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bartosch B, et al. 2003. In vitro assay for neutralizing antibody to hepatitis C virus: evidence for broadly conserved neutralization epitopes. Proc. Natl. Acad. Sci. U. S. A. 100:14199–14204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bhattacharya T, et al. 2007. Founder effects in the assessment of HIV polymorphisms and HLA allele associations. Science 315:1583–1586 [DOI] [PubMed] [Google Scholar]
- 4. Blight KJ, Kolykhalov AA, Rice CM. 2000. Efficient initiation of HCV RNA replication in cell culture. Science 290:1972–1974 [DOI] [PubMed] [Google Scholar]
- 4a. Burke KP, et al. 2012. Immunogenicity and cross-reactivity of a representative ancestral sequence in HCV infection. J. Immunol., in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ciesek S, Manns MP. 2011. Hepatitis in 2010: the dawn of a new era in HCV therapy. Nat. Rev. Gastroenterol. Hepatol. 8:69–71 [DOI] [PubMed] [Google Scholar]
- 6. Cox AL, et al. 2005. Comprehensive analyses of CD8+ T cell responses during longitudinal study of acute human hepatitis C. Hepatology 42:104–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cox AL, et al. 2005. Cellular immune selection with hepatitis C virus persistence in humans. J. Exp. Med. 201:1741–1752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cox AL, et al. 2009. Rare birds in North America: acute hepatitis C cohorts. Gastroenterology 136:26–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Erickson AL, et al. 2001. The outcome of hepatitis C virus infection is predicted by escape mutations in epitopes targeted by cytotoxic T lymphocytes. Immunity 15:883–895 [DOI] [PubMed] [Google Scholar]
- 11. Felsenstein J. 1978. The number of phylogenetic trees. Syst. Zool. 27:27–33 [Google Scholar]
- 12. Gaschen B, et al. 2002. Diversity considerations in HIV-1 vaccine selection. Science 296:2354–2360 [DOI] [PubMed] [Google Scholar]
- 13. Hall TA. 1997–2011. BioEdit: biological sequence alignment editor for Win95/98/NT/2K/XP/7, version 7.1.3. Ibis Biosciences, Carlsbad, CA: http://www.mbio.ncsu.edu/bioedit/bioedit.html Accessed 26 March 2012 [Google Scholar]
- 14. Holm S. 1979. A simple sequentially rejective multiple test procedure. Scand. J. Stat. 6:65–70 [Google Scholar]
- 15. Houghton M, Abrignani S. 2005. Prospects for a vaccine against the hepatitis C virus. Nature 436:961–966 [DOI] [PubMed] [Google Scholar]
- 16. Hsu M, et al. 2003. Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc. Natl. Acad. Sci. U. S. A. 100:7271–7276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hudson RR. 1983. Properties of a neutral allele model with intragenic recombination. Theor. Popul. Biol. 23:183–201 [DOI] [PubMed] [Google Scholar]
- 18. Kolykhalov AA, Feinstone SM, Rice CM. 1996. Identification of a highly conserved sequence element at the 3′ terminus of hepatitis C virus genome RNA. J. Virol. 70:3363–3371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Koziel MJ, et al. 1995. HLA class I-restricted cytotoxic T lymphocytes specific for hepatitis C virus. Identification of multiple epitopes and characterization of patterns of cytokine release. J. Clin. Invest. 96:2311–2321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kuiken C, et al. 2006. A comprehensive system for consistent numbering of HCV sequences, proteins and epitopes. Hepatology 44:1355–1361 [DOI] [PubMed] [Google Scholar]
- 21. Liu L, et al. 2010. Acceleration of hepatitis C virus envelope evolution in humans is consistent with progressive humoral immune selection during the transition from acute to chronic infection. J. Virol. 84:5067–5077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Logvinoff C, et al. 2004. Neutralizing antibody response during acute and chronic hepatitis C virus infection. Proc. Natl. Acad. Sci. U. S. A. 101:10149–10154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Maheshwari A, Ray S, Thuluvath PJ. 2008. Acute hepatitis C. Lancet 372:321–332 [DOI] [PubMed] [Google Scholar]
- 24. Mehta SH, et al. 2002. Protection against persistence of hepatitis C. Lancet 359:1478–1483 [DOI] [PubMed] [Google Scholar]
- 25. Nei M, Gojobori T. 1986. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 3:418–426 [DOI] [PubMed] [Google Scholar]
- 26. Netski DM, et al. 2005. Humoral immune response in acute hepatitis C virus infection. Clin. Infect. Dis. 41:667–675 [DOI] [PubMed] [Google Scholar]
- 27. Neumann-Haefelin C, et al. 2008. Analysis of the evolutionary forces in an immunodominant CD8 epitope in hepatitis C virus at a population level. J. Virol. 82:3438–3451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Osburn WO, et al. 2010. Spontaneous control of primary hepatitis C virus infection and immunity against persistent reinfection. Gastroenterology 138:315–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ray SC, Thomas DL. 2010. Hepatitis C, p 2157–2185 In Mandell GL, Bennett JE, Dolin R. (ed), Mandell, Douglas, and Bennett's principles and practice of infectious diseases, 7th ed Churchill Livingstone, Philadelphia, PA [Google Scholar]
- 30. Ray SC, et al. 1999. Acute hepatitis C virus structural gene sequences as predictors of persistent viremia: hypervariable region 1 as decoy. J. Virol. 73:2938–2946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ronquist F, Huelsenbeck JP. 2003. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574 [DOI] [PubMed] [Google Scholar]
- 32. Shoukry NH, et al. 2003. Memory CD8+ T cells are required for protection from persistent hepatitis C virus infection. J. Exp. Med. 197:1645–1655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Simmonds P, et al. 2005. Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes. Hepatology 42:962–973 [DOI] [PubMed] [Google Scholar]
- 34. Simmonds P, et al. 1994. Identification of genotypes of hepatitis C virus by sequence comparisons in the core, E1 and NS-5 regions. J. Gen. Virol. 75:1053–1061 [DOI] [PubMed] [Google Scholar]
- 35. Spangenberg HC, et al. 2005. Intrahepatic CD8+ T-cell failure during chronic hepatitis C virus infection. Hepatology 42:828–837 [DOI] [PubMed] [Google Scholar]
- 36. Stamataki Z, Grove J, Balfe P, McKeating JA. 2008. Hepatitis C virus entry and neutralization. Clin. Liver Dis. 12:693–712 [DOI] [PubMed] [Google Scholar]
- 37. Thomas DL, Seeff LB. 2005. Natural history of hepatitis C. Clin. Liver Dis. 9:383–398 [DOI] [PubMed] [Google Scholar]
- 38. Thomas DL, et al. 2009. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature 461:798–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Toyoda H, et al. 1998. Quasispecies nature of hepatitis C virus (HCV) in patients with chronic hepatitis C with mixed HCV subtypes. J. Med. Virol. 54:80–85 [PubMed] [Google Scholar]
- 40. Viazov S, et al. 2010. Hepatitis C virus recombinants are rare even among intravenous drug users. J. Med. Virol. 82:232–238 [DOI] [PubMed] [Google Scholar]
- 41. von Hahn T, et al. 2007. Hepatitis C virus continuously escapes from neutralizing antibody and T-cell responses during chronic infection in vivo. Gastroenterology 132:667–678 [DOI] [PubMed] [Google Scholar]
- 42. Wakita T, et al. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11:791–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ward S, Lauer G, Isba R, Walker B, Klenerman P. 2002. Cellular immune responses against hepatitis C virus: the evidence base 2002. Clin. Exp. Immunol. 128:195–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wimmer E, Paul AV. 2011. Synthetic poliovirus and other designer viruses: what have we learned from them? Annu. Rev. Microbiol. 65:583–609 [DOI] [PubMed] [Google Scholar]
- 45. Wong DK, et al. 1998. Liver-derived CTL in hepatitis C virus infection: breadth and specificity of responses in a cohort of persons with chronic infection. J. Immunol. 160:1479–1488 [PubMed] [Google Scholar]
- 46. Wong DK, et al. 2001. Detection of diverse hepatitis C virus (HCV)-specific cytotoxic T lymphocytes in peripheral blood of infected persons by screening for responses to all translated proteins of HCV. J. Virol. 75:1229–1235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. World Health Organization 1997. Hepatitis C: global prevalence. Wkly. Epidemiol. Rec. 72:341–348 [PubMed] [Google Scholar]
- 48. Yi M, Villanueva RA, Thomas DL, Wakita T, Lemon SM. 2006. Production of infectious genotype 1a hepatitis C virus (Hutchinson strain) in cultured human hepatoma cells. Proc. Natl. Acad. Sci. U. S. A. 103:2310–2315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. You S, Rice CM. 2008. 3′ RNA elements in hepatitis C virus replication: kissing partners and long poly(U). J. Virol. 82:184–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yusim K, et al. 2010. Genotype 1 and global hepatitis C T-cell vaccines designed to optimize coverage of genetic diversity. J. Gen. Virol. 91:1194–1206 [DOI] [PubMed] [Google Scholar]
- 51. Zhang X, Fan X, Xu Y, Di Bisceglie AM. 2010. Enhanced protocol for determining the 3′ terminus of hepatitis C virus. J. Virol. Methods 167:158–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhong J, et al. 2005. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. U. S. A. 102:9294–9299 [DOI] [PMC free article] [PubMed] [Google Scholar]